Bifidobacterial Distribution Across Italian Cheeses Produced from Raw Milk


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Sample Collection
2.2. Bacterial DNA Extraction, 16S rRNA Gene PCR Amplification, and Sequencing
2.3. 16S rRNA/ITS Microbial Profiling
2.4. 16S rRNA/ITS Microbial Profiling Analysis
2.5. Statistical Analysis
3. Results and Discussion
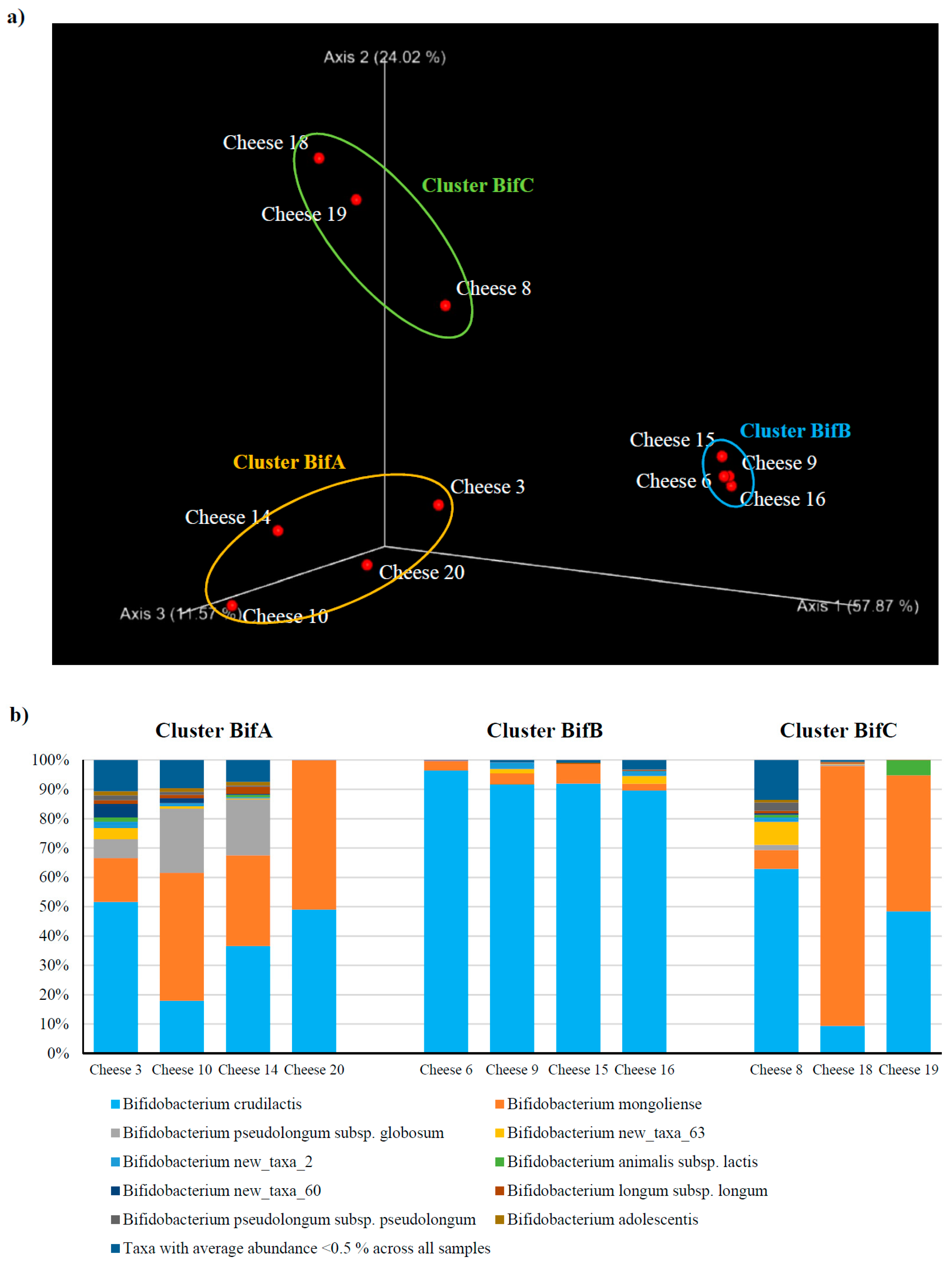
3.1. Dissecting the Distribution of Bifidobacteria Across Italian Cheese
3.2. Assessment of The Bifidobacterial Population at The Subspecies Level
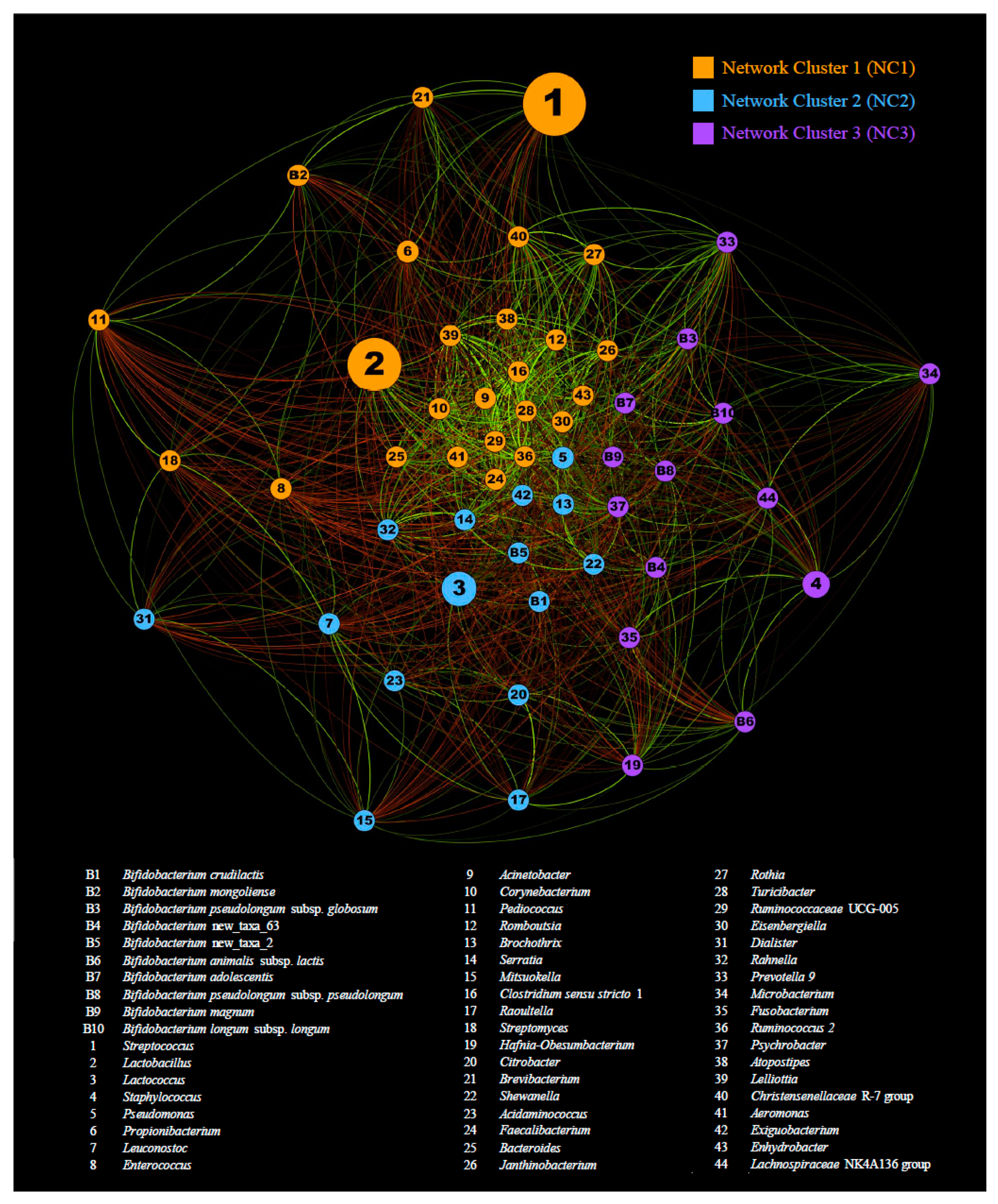
3.3. Co-Variances between Bifidobacteria and Other Cheese Colonizers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Availability of Data and Materials
Conflicts of Interest
References
- Ceugniez, A.; Taminiau, B.; Coucheney, F.; Jacques, P.; Delcenserie, V.; Daube, G.; Drider, D. Use of a metagenetic approach to monitor the bacterial microbiota of “tomme d’orchies” cheese during the ripening process. Int. J. Food Microbiol. 2017, 247, 65–69. [Google Scholar] [CrossRef]
- Marino, M.; Dubsky de Wittenau, G.; Sacca, E.; Cattonaro, F.; Spadotto, A.; Innocente, N.; Radovic, S.; Piasentier, E.; Marroni, F. Metagenomic profiles of different types of italian high-moisture mozzarella cheese. Food Microbiol. 2019, 79, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, S.; Reyes-Mata, M.P.; Nirmalkar, K.; Chavez-Carbajal, A.; Juarez-Hernandez, J.I.; Torres-Gomez, R.E.; Pina-Escobedo, A.; Maya, O.; Hoyo-Vadillo, C.; Ramos-Ramirez, E.G.; et al. Profiling of bacterial and fungal communities of mexican cheeses by high throughput DNA sequencing. Food Res. Int. 2018, 113, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Alegria, A.; Szczesny, P.; Mayo, B.; Bardowski, J.; Kowalczyk, M. Biodiversity in oscypek, a traditional polish cheese, determined by culture-dependent and -independent approaches. Appl. Environ. Microbiol. 2012, 78, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Fuka, M.M.; Wallisch, S.; Engel, M.; Welzl, G.; Havranek, J.; Schloter, M. Dynamics of bacterial communities during the ripening process of different croatian cheese types derived from raw ewe’s milk cheeses. PLoS ONE 2013, 8, e80734. [Google Scholar] [CrossRef] [PubMed]
- Delcenserie, V.; Taminiau, B.; Delhalle, L.; Nezer, C.; Doyen, P.; Crevecoeur, S.; Roussey, D.; Korsak, N.; Daube, G. Microbiota characterization of a belgian protected designation of origin cheese, herve cheese, using metagenomic analysis. J. Dairy Sci. 2014, 97, 6046–6056. [Google Scholar] [CrossRef]
- Riquelme, C.; Camara, S.; Dapkevicius Mde, L.; Vinuesa, P.; da Silva, C.C.; Malcata, F.X.; Rego, O.A. Characterization of the bacterial biodiversity in pico cheese (an artisanal azorean food). Int. J. Food Microbiol. 2015, 192, 86–94. [Google Scholar] [CrossRef]
- Giello, M.; La Storia, A.; Masucci, F.; Di Francia, A.; Ercolini, D.; Villani, F. Dynamics of bacterial communities during manufacture and ripening of traditional caciocavallo of castelfranco cheese in relation to cows’ feeding. Food Microbiol. 2017, 63, 170–177. [Google Scholar] [CrossRef]
- Dalmasso, A.; del Rio, M.D.S.; Civera, T.; Pattono, D.; Cardazzo, B.; Bottero, M.T. Characterization of microbiota in plaisentif cheese by high-throughput sequencing. Food Sci. Technol. 2016, 69, 490–496. [Google Scholar] [CrossRef]
- Alessandria, V.; Ferrocino, I.; De Filippis, F.; Fontana, M.; Rantsiou, K.; Ercolini, D.; Cocolin, L. Microbiota of an italian grana-like cheese during manufacture and ripening, unraveled by 16s rrna-based approaches. Appl. Environ. Microbiol. 2016, 82, 3988–3995. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Duranti, S.; Viappiani, A.; Mangifesta, M.; Segata, N.; van Sinderen, D.; Ventura, M. Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (its) protocol. FEMS Microbiol. Ecol. 2014, 90, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Mangifesta, M.; Mancabelli, L.; Lugli, G.A.; James, K.; Duranti, S.; Turroni, F.; Ferrario, C.; Ossiprandi, M.C.; van Sinderen, D.; et al. Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J. 2017, 11, 2834–2847. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Lugli, G.A.; Milani, C.; James, K.; Mancabelli, L.; Turroni, F.; Alessandri, G.; Mangifesta, M.; Mancino, W.; Ossiprandi, M.C.; et al. Bifidobacterium bifidum and the infant gut microbiota: An intriguing case of microbe-host co-evolution. Environ. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17. [Google Scholar] [CrossRef] [PubMed]
- Turroni, F.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Viappiani, A.; Lugli, G.A.; Ferrario, C.; Gioiosa, L.; Ferrarini, A.; et al. Deciphering bifidobacterial-mediated metabolic interactions and their impact on gut microbiota by a multi-omics approach. ISME J. 2016, 10, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Bunesova, V.; Vlkova, E.; Rada, V.; Killer, J.; Musilova, S. Bifidobacteria from the gastrointestinal tract of animals: Differences and similarities. Benef. Microbes 2014, 5, 377–388. [Google Scholar] [CrossRef]
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Bottacini, F.; Mangifesta, M.; Sanchez, B.; Viappiani, A.; Mancabelli, L.; Taminiau, B.; et al. Genomic encyclopedia of type strains of the genus bifidobacterium. Appl. Environ. Microbiol. 2014, 80, 6290–6302. [Google Scholar] [CrossRef]
- Avershina, E.; Lundgard, K.; Sekelja, M.; Dotterud, C.; Storro, O.; Oien, T.; Johnsen, R.; Rudi, K. Transition from infant- to adult-like gut microbiota. Environ. Microbiol. 2016, 18, 2226–2236. [Google Scholar] [CrossRef]
- Milani, C.; Turroni, F.; Duranti, S.; Lugli, G.A.; Mancabelli, L.; Ferrario, C.; van Sinderen, D.; Ventura, M. Genomics of the genus bifidobacterium reveals species-specific adaptation to the glycan-rich gut environment. Appl. Environ. Microbiol. 2016, 82, 980–991. [Google Scholar] [CrossRef]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut bifidobacteria populations in human health and aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [PubMed]
- Delcenserie, V.; Taminiau, B.; Gavini, F.; de Schaetzen, M.A.; Cleenwerck, I.; Theves, M.; Mahieu, M.; Daube, G. Detection and characterization of Bifidobacterium crudilactis and B. mongoliense able to grow during the manufacturing process of french raw milk cheeses. BMC Microbiol. 2013, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Napoli, S.; Alessandri, G.; Mancabelli, L.; Anzalone, R.; Longhi, G.; Viappiani, A.; Mangifesta, M.; Lugli, G.A.; et al. Colonization of the human gut by bovine bacteria present in parmesan cheese. Nat. Commun. 2019, 10, 1286. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Hevia, A.; Foroni, E.; Duranti, S.; Turroni, F.; Lugli, G.A.; Sanchez, B.; Martin, R.; Gueimonde, M.; van Sinderen, D.; et al. Assessing the fecal microbiota: An optimized ion torrent 16s rrna gene-based analysis protocol. PLoS ONE 2013, 8, e68739. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with qiime 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The silva ribosomal rna gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Place, R.B.; Hiestand, D.; Gallmann, H.R.; Teuber, M. Staphylococcus equorum subsp. Linens, subsp. Nov., a starter culture component for surface ripened semi-hard cheeses. Syst. Appl. Microbiol. 2003, 26, 30–37. [Google Scholar] [CrossRef]
- Watanabe, K.; Makino, H.; Sasamoto, M.; Kudo, Y.; Fujimoto, J.; Demberel, S. Bifidobacterium mongoliense sp. Nov., from airag, a traditional fermented mare’s milk product from mongolia. Int. J. Syst. Evol. Microbiol. 2009, 59, 1535–1540. [Google Scholar] [CrossRef]
- Delcenserie, V.; Gavini, F.; Beerens, H.; Tresse, O.; Franssen, C.; Daube, G. Description of a new species, Bifidobacterium crudilactis sp. Nov., isolated from raw milk and raw milk cheeses. Syst. Appl. Microbiol. 2007, 30, 381–389. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milani, C.; Alessandri, G.; Mancabelli, L.; Lugli, G.A.; Longhi, G.; Anzalone, R.; Viappiani, A.; Duranti, S.; Turroni, F.; Ossiprandi, M.C.; et al. Bifidobacterial Distribution Across Italian Cheeses Produced from Raw Milk. Microorganisms 2019, 7, 599. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120599
Milani C, Alessandri G, Mancabelli L, Lugli GA, Longhi G, Anzalone R, Viappiani A, Duranti S, Turroni F, Ossiprandi MC, et al. Bifidobacterial Distribution Across Italian Cheeses Produced from Raw Milk. Microorganisms. 2019; 7(12):599. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120599
Chicago/Turabian StyleMilani, Christian, Giulia Alessandri, Leonardo Mancabelli, Gabriele Andrea Lugli, Giulia Longhi, Rosaria Anzalone, Alice Viappiani, Sabrina Duranti, Francesca Turroni, Maria Cristina Ossiprandi, and et al. 2019. "Bifidobacterial Distribution Across Italian Cheeses Produced from Raw Milk" Microorganisms 7, no. 12: 599. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120599