Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. IS Elements Identification
2.3. Bifidoprophages Identification
2.4. Prediction of the Antibiotic Resistance Genes
2.5. Phylogenomic Analyses
2.6. Bacitracin A Antibiotic Susceptibility Tests
2.7. Statistical Analyses
3. Results and Discussion
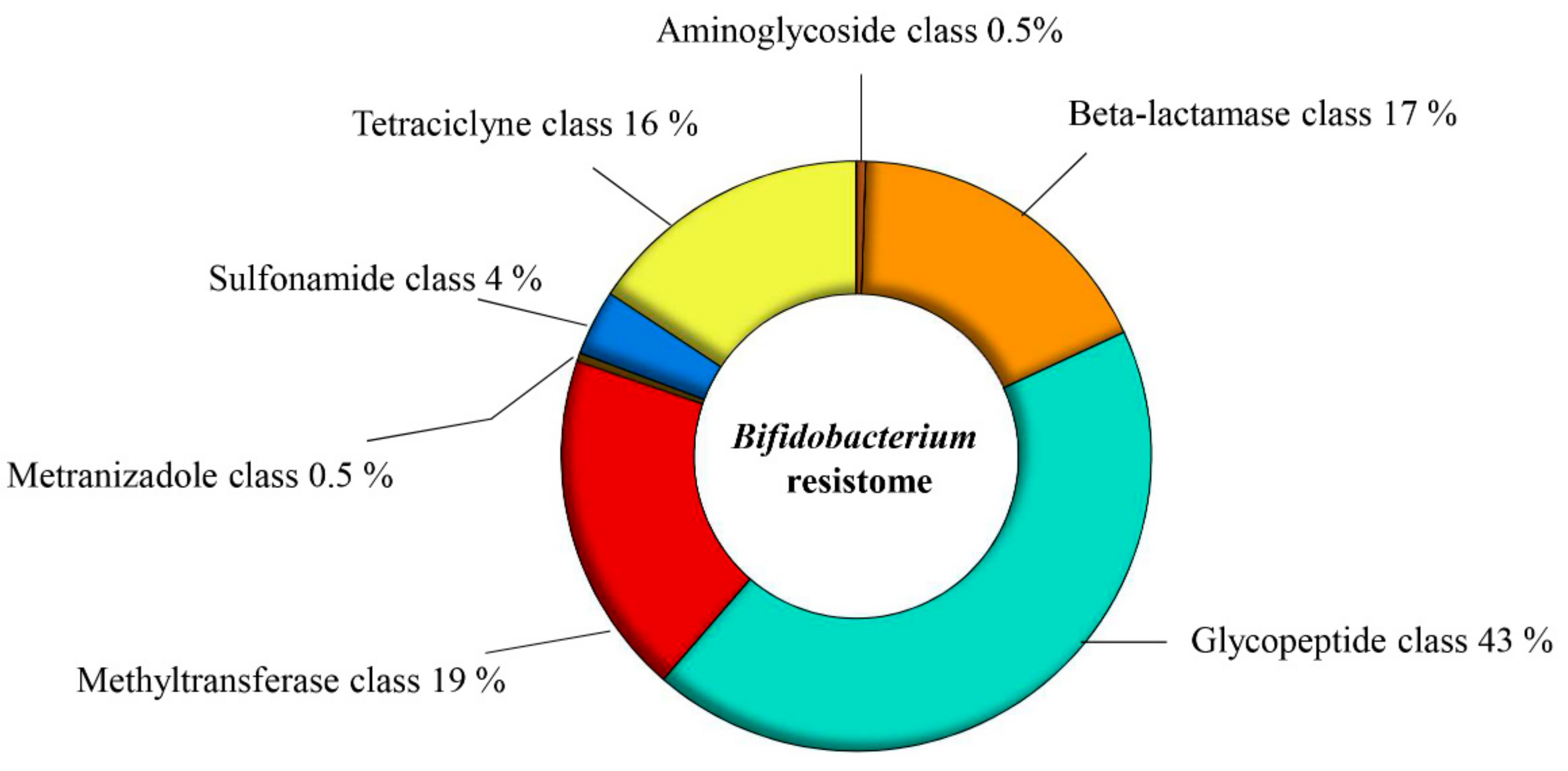
3.1. The Putative Resistome of the Genus Bifidobacterium
3.2. The Predicted Mobilome of the Bifidobacterium Genus
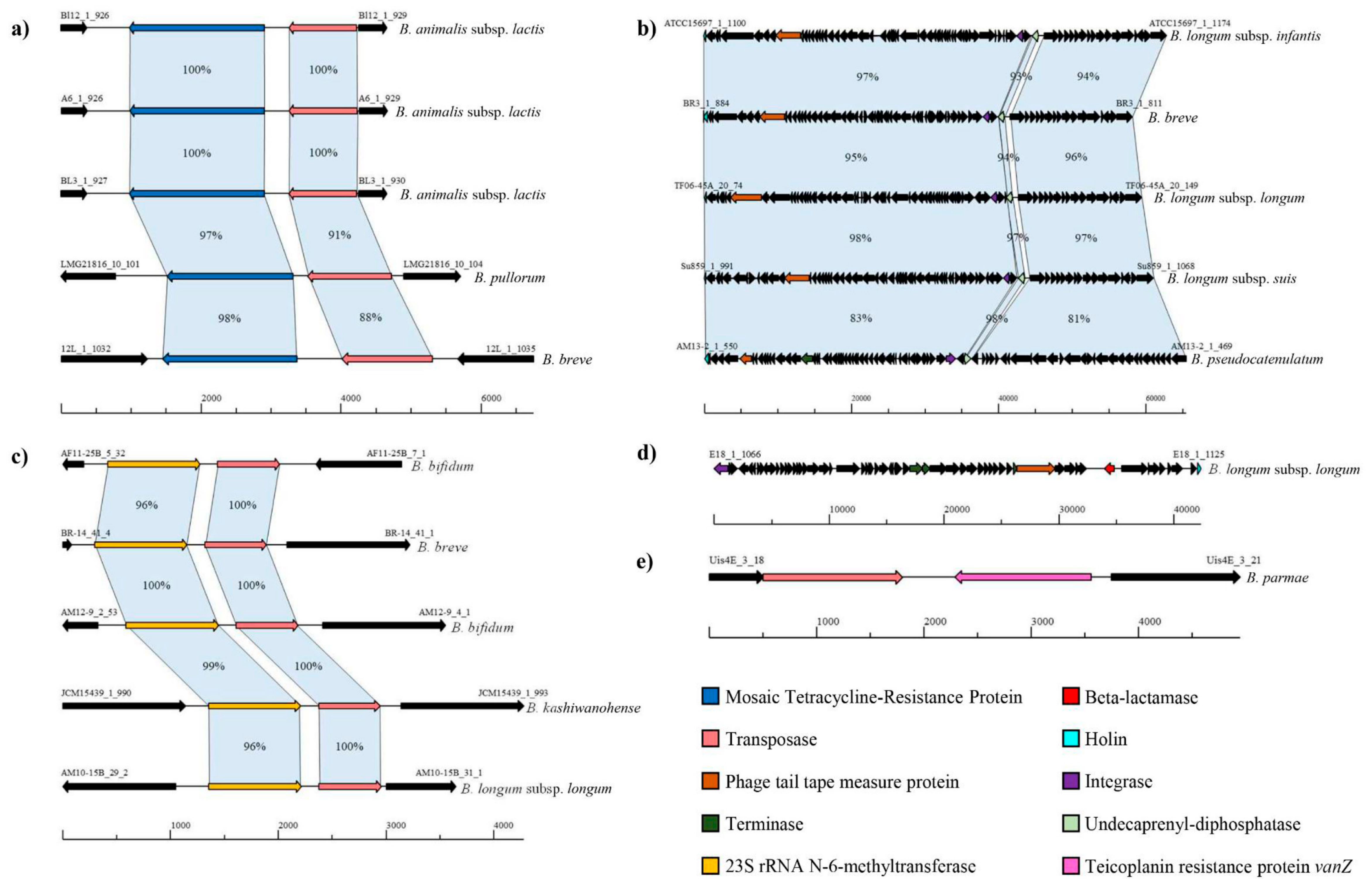
3.3. Identification of the Putative Mobile Resistome of the Bifidobacterium Genus
3.4. Assessment of Bacitracin A Resistance of Bifidobacterium spp
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Milani, C.; Lugli, G.A.; Duranti, S.; Turroni, F.; Bottacini, F.; Mangifesta, M.; Sanchez, B.; Viappiani, A.; Mancabelli, L.; Taminiau, B.; et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Appl. Environ. Microbiol. 2014, 80, 6290–6302. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; van Sinderen, D.; Fitzgerald, G.F.; Zink, R. Insights into the taxonomy, genetics and physiology of bifidobacteria. Antonie Van Leeuwenhoek 2004, 86, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Turroni, F.; Lugli, G.A.; van Sinderen, D. Bifidobacteria and humans: Our special friends, from ecological to genomics perspectives. J. Sci. Food Agric. 2014, 94, 163–168. [Google Scholar] [CrossRef]
- Turroni, F.; Peano, C.; Pass, D.A.; Foroni, E.; Severgnini, M.; Claesson, M.J.; Kerr, C.; Hourihane, J.; Murray, D.; Fuligni, F.; et al. Diversity of bifidobacteria within the infant gut microbiota. PLoS ONE 2012, 7, e36957. [Google Scholar] [CrossRef]
- Verani, J.R.; McGee, L.; Schrag, S.J. Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases Prevention of perinatal group B streptococcal disease—revised guidelines from CDC, 2010. MMWR Recomm. Rep. 2010, 59, 1–36. [Google Scholar]
- Cox, L.M.; Blaser, M.J. Antibiotics in early life and obesity. Nat. Rev. Endocrinol. 2015, 11, 182–190. [Google Scholar] [CrossRef]
- Gibson, M.K.; Crofts, T.S.; Dantas, G. Antibiotics and the developing infant gut microbiota and resistome. Curr. Opin. Microbiol. 2015, 27, 51–56. [Google Scholar] [CrossRef]
- Zou, Z.H.; Liu, D.; Li, H.D.; Zhu, D.P.; He, Y.; Hou, T.; Yu, J.L. Prenatal and postnatal antibiotic exposure influences the gut microbiota of preterm infants in neonatal intensive care units. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 9. [Google Scholar] [CrossRef]
- Marshall, B.M.; Levy, S.B. Food animals and antimicrobials: Impacts on human health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef]
- Ouwehand, A.C.; Forssten, S.; Hibberd, A.A.; Lyra, A.; Stahl, B. Probiotic approach to prevent antibiotic resistance. Ann. Med. 2016, 48, 246–255. [Google Scholar] [CrossRef]
- Arboleya, S.; Sanchez, B.; Milani, C.; Duranti, S.; Solis, G.; Fernandez, N.; de los Reyes-Gavilan, C.G.; Ventura, M.; Margolles, A.; Gueimonde, M. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. J. Pediatr. 2015, 166, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Guinane, C.M.; Hussey, S.; Wall, R.; Ryan, C.A.; Dempsey, E.M.; Murphy, B.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 2012, 56, 5811–5820. [Google Scholar] [CrossRef]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Turroni, F.; Milani, C.; Mangifesta, M.; Ferrario, C.; Anzalone, R.; Viappiani, A.; van Sinderen, D.; et al. Prevalence of Antibiotic Resistance Genes among Human Gut-Derived Bifidobacteria. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Wiedenbeck, J.; Cohan, F.M. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011, 35, 957–976. [Google Scholar] [CrossRef]
- Brito, I.L.; Yilmaz, S.; Huang, K.; Xu, L.; Jupiter, S.D.; Jenkins, A.P.; Naisilisili, W.; Tamminen, M.; Smillie, C.S.; Wortman, J.R.; et al. Mobile genes in the human microbiome are structured from global to individual scales. Nature 2016, 535, 435–439. [Google Scholar] [CrossRef]
- Hagbo, M.; Ravi, A.; Angell, I.L.; Sunde, M.; Ludvigsen, J.; Diep, D.B.; Foley, S.L.; Vento, M.; Collado, M.C.; Perez-Martinez, G.; et al. Experimental support for multidrug resistance transfer potential in the preterm infant gut microbiota. Pediatr. Res. 2019, 231. [Google Scholar] [CrossRef]
- Martinez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef]
- Siefert, J.L. Defining the mobilome. Methods Mol. Biol. 2009, 532, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Smets, B.F.; Barkay, T. Horizontal gene transfer: Perspectives at a crossroads of scientific disciplines. Nat. Rev. Microbiol. 2005, 3, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Guglielmetti, S.; Mayo, B.; Alvarez-Martin, P. Mobilome and genetic modification of bifidobacteria. Benef. Microbes. 2013, 4, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Tremblay, D.; Ferrario, C.; Mancabelli, L.; Duranti, S.; Ward, D.V.; Ossiprandi, M.C.; Moineau, S.; et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ. Microbiol. 2016, 18, 2196–2213. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Turroni, F.; Viappiani, A.; van Sinderen, D.; Ventura, M. Tracking the Taxonomy of the Genus Bifidobacterium Based on a Phylogenomic Approach. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Ferrario, C.; Viappiani, A.; Mancabelli, L.; Mangifesta, M.; Taminiau, B.; Delcenserie, V.; et al. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl. Environ. Microbiol. 2014, 80, 6383–6394. [Google Scholar] [CrossRef]
- Lugli, G.A.; Milani, C.; Turroni, F.; Duranti, S.; Mancabelli, L.; Mangifesta, M.; Ferrario, C.; Modesto, M.; Mattarelli, P.; Jiri, K.; et al. Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genom. 2017, 18, 568. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [Green Version]
- Authority, E.F.S. Guidance on the assessment of bacterial susceptibility to antimicrobials of human and veterinary importance. EFSA J. 2012, 10, 2740. [Google Scholar]
- Arthur, M.; Depardieu, F.; Molinas, C.; Reynolds, P.; Courvalin, P. The vanZ gene of Tn1546 from Enterococcus faecium BM4147 confers resistance to teicoplanin. Gene 1995, 154, 87–92. [Google Scholar] [CrossRef]
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [Google Scholar] [CrossRef] [PubMed]
- Evers, S.; Courvalin, P. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J. Bacteriol. 1996, 178, 1302–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovanetti, E.; Brenciani, A.; Lupidi, R.; Roberts, M.C.; Varaldo, P.E. Presence of the tet(O) gene in erythromycin- and tetracycline-resistant strains of Streptococcus pyogenes and linkage with either the mef(A) or the erm(A) gene. Antimicrob. Agents Chemother. 2003, 47, 2844–2849. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.C. Tetracycline resistance determinants: Mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 1996, 19, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hedayatianfard, K.; Akhlaghi, M.; Sharifiyazdi, H. Detection of tetracycline resistance genes in bacteria isolated from fish farms using polymerase chain reaction. Vet. Res. Forum Int. Q. J. 2014, 5, 269–275. [Google Scholar]
- Wang, N.; Hang, X.; Zhang, M.; Liu, X.; Yang, H. Analysis of newly detected tetracycline resistance genes and their flanking sequences in human intestinal bifidobacteria. Sci. Rep. 2017, 7, 6267. [Google Scholar] [CrossRef]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef]
- Martinez, N.; Luque, R.; Milani, C.; Ventura, M.; Banuelos, O.; Margolles, A. A Gene Homologous to rRNA Methylase Genes Confers Erythromycin and Clindamycin Resistance in Bifidobacterium breve. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Skold, O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Phuong Hoa, P.T.; Nonaka, L.; Hung Viet, P.; Suzuki, S. Detection of the sul1, sul2, and sul3 genes in sulfonamide-resistant bacteria from wastewater and shrimp ponds of north Vietnam. Sci. Total Environ. 2008, 405, 377–384. [Google Scholar] [CrossRef]
- Vetting, M.W.; Hegde, S.S.; Fajardo, J.E.; Fiser, A.; Roderick, S.L.; Takiff, H.E.; Blanchard, J.S. Pentapeptide repeat proteins. Biochemistry 2006, 45, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merens, A.; Matrat, S.; Aubry, A.; Lascols, C.; Jarlier, V.; Soussy, C.J.; Cavallo, J.D.; Cambau, E. The pentapeptide repeat proteins MfpAMt and QnrB4 exhibit opposite effects on DNA gyrase catalytic reactions and on the ternary gyrase-DNA-quinolone complex. J. Bacteriol. 2009, 191, 1587–1594. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Lee, J.H.; Jeong, D.U.; Lee, J.J.; Wu, X.; Jeong, B.C.; Kang, C.M.; Lee, S.H. Determination of pentapeptide repeat units in Qnr proteins by the structure-based alignment approach. Antimicrob. Agents Chemother. 2011, 55, 4475–4478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gueimonde, M.; Sanchez, B.; de Los Reyes-Gavilán, C.G.; Margolles, A. Antibiotic resistance in probiotic bacteria. Front. Microbiol. 2013, 4, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcour, J.; Ferain, T.; Deghorain, M.; Palumbo, E.; Hols, P. The biosynthesis and functionality of the cell-wall of lactic acid bacteria. Antonie Van Leeuwenhoek 1999, 76, 159–184. [Google Scholar] [CrossRef]
- Florez, A.B.; Ladero, V.; Alvarez-Martin, P.; Ammor, M.S.; Alvarez, M.A.; Mayo, B. Acquired macrolide resistance in the human intestinal strain Lactobacillus rhamnosus E41 associated with a transition mutation in 23S rRNA genes. Int. J. Antimicrob. Agents 2007, 30, 341–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, A.S.; Hertel, C.; Holzapfel, W.H.; Franz, C.M. Antibiotic resistances of starter and probiotic strains of lactic acid bacteria. Appl. Environ. Microbiol. 2007, 73, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammor, M.S.; Gueimonde, M.; Danielsen, M.; Zagorec, M.; van Hoek, A.H.; de Los Reyes-Gavilan, C.G.; Mayo, B.; Margolles, A. Two different tetracycline resistance mechanisms, plasmid-carried tet(L) and chromosomally located transposon-associated tet(M), coexist in Lactobacillus sakei Rits 9. Appl. Environ. Microbiol. 2008, 74, 1394–1401. [Google Scholar] [CrossRef] [Green Version]
- Rojo-Bezares, B.; Saenz, Y.; Poeta, P.; Zarazaga, M.; Ruiz-Larrea, F.; Torres, C. Assessment of antibiotic susceptibility within lactic acid bacteria strains isolated from wine. Int. J. Food Microbiol. 2006, 111, 234–240. [Google Scholar] [CrossRef]
- Johnning, A.; Karami, N.; Tang Hallback, E.; Muller, V.; Nyberg, L.; Buongermino Pereira, M.; Stewart, C.; Ambjornsson, T.; Westerlund, F.; Adlerberth, I.; et al. The resistomes of six carbapenem-resistant pathogens—a critical genotype-phenotype analysis. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Dagher, C.; Salloum, T.; Alousi, S.; Arabaghian, H.; Araj, G.F.; Tokajian, S. Molecular characterization of Carbapenem resistant Escherichia coli recovered from a tertiary hospital in Lebanon. PLoS ONE 2018, 13, e0203323. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Yasir, M.; Farman, M.; Kumosani, T.; AlBasri, S.F.; Bajouh, O.S.; Azhar, E.I. Evaluation of gut bacterial community composition and antimicrobial resistome in pregnant and non-pregnant women from Saudi population. Infect. Drug Resist. 2019, 12, 1749–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.A.; Duc le, H.; Cutting, S.M. The use of bacterial spore formers as probiotics. Fems Microbiol. Rev. 2005, 29, 813–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monod, M.; Denoya, C.; Dubnau, D. Sequence and properties of pIM13, a macrolide-lincosamide-streptogramin B resistance plasmid from Bacillus subtilis. J. Bacteriol. 1986, 167, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Phelan, R.W.; Clarke, C.; Morrissey, J.P.; Dobson, A.D.; O’Gara, F.; Barbosa, T.M. Tetracycline resistance-encoding plasmid from Bacillus sp. strain #24, isolated from the marine sponge Haliclona simulans. Appl. Environ. Microbiol. 2011, 77, 327–329. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Wu, C.M.; Wang, M.G.; Wang, Y.; Wang, Y.; Huang, S.Y.; Xia, L.N.; Li, B.B.; Shen, J.Z. First report of the multidrug resistance gene cfr and the phenicol resistance gene fexA in a Bacillus strain from swine feces. Antimicrob. Agents Chemother. 2010, 54, 3953–3955. [Google Scholar] [CrossRef] [Green Version]
- Mahony, J.; Lugli, G.A.; van Sinderen, D.; Ventura, M. Impact of gut-associated bifidobacteria and their phages on health: Two sides of the same coin? Appl. Microbiol. Biotechnol. 2018, 102, 2091–2099. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Fitzgerald, G.F.; Gupta, R.S.; van Sinderen, D. Genomics as a means to understand bacterial phylogeny and ecological adaptation: The case of bifidobacteria. Antonie Van Leeuwenhoek 2007, 91, 351–372. [Google Scholar] [CrossRef]
- Siguier, P.; Varani, A.; Perochon, J.; Chandler, M. Exploring bacterial insertion sequences with ISfinder: Objectives, uses, and future developments. Methods Mol. Biol. 2012, 859, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Casey, E.; Oliveira, J.; Bottacini, F.; James, K.; Franz, C.; Lugli, G.A.; Neve, H.; Ventura, M.; Hatfull, G.F.; et al. Characterization and induction of prophages in human gut-associated Bifidobacterium hosts. Sci. Rep. 2018, 8, 12772. [Google Scholar] [CrossRef]
- Bottacini, F.; Medini, D.; Pavesi, A.; Turroni, F.; Foroni, E.; Riley, D.; Giubellini, V.; Tettelin, H.; van Sinderen, D.; Ventura, M. Comparative genomics of the genus Bifidobacterium. Microbiology 2010, 156, 3243–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menouni, R.; Hutinet, G.; Petit, M.A.; Ansaldi, M. Bacterial genome remodeling through bacteriophage recombination. Fems Microbiol. Lett. 2015, 362, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy-Denomy, J.; Davidson, A.R. When a virus is not a parasite: The beneficial effects of prophages on bacterial fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Milani, C.; Duranti, S.; Lugli, G.A.; Bottacini, F.; Strati, F.; Arioli, S.; Foroni, E.; Turroni, F.; van Sinderen, D.; Ventura, M. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl. Environ. Microbiol. 2013, 79, 4304–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammor, M.S.; Florez, A.B.; Alvarez-Martin, P.; Margolles, A.; Mayo, B. Analysis of tetracycline resistance tet(W) genes and their flanking sequences in intestinal Bifidobacterium species. J. Antimicrob. Chemother. 2008, 62, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Florez, A.B.; Ammor, M.S.; Alvarez-Martin, P.; Margolles, A.; Mayo, B. Molecular analysis of tet(W) gene-mediated tetracycline resistance in dominant intestinal Bifidobacterium species from healthy humans. Appl. Environ. Microbiol. 2006, 72, 7377–7379. [Google Scholar] [CrossRef] [Green Version]
- Gueimonde, M.; Florez, A.B.; van Hoek, A.H.; Stuer-Lauridsen, B.; Stroman, P.; de los Reyes-Gavilan, C.G.; Margolles, A. Genetic basis of tetracycline resistance in Bifidobacterium animalis subsp. lactis. Appl. Environ. Microbiol. 2010, 76, 3364–3369. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.P.; Barbosa, T.M.; Forbes, K.J.; Flint, H.J. High-frequency transfer of a naturally occurring chromosomal tetracycline resistance element in the ruminal anaerobe Butyrivibrio fibrisolvens. Appl. Environ. Microbiol. 1997, 63, 3405–3411. [Google Scholar]
- Cain, B.D.; Norton, P.J.; Eubanks, W.; Nick, H.S.; Allen, C.M. Amplification of the bacA gene confers bacitracin resistance to Escherichia coli. J. Bacteriol. 1993, 175, 3784–3789. [Google Scholar] [CrossRef] [Green Version]
- El Ghachi, M.; Bouhss, A.; Blanot, D.; Mengin-Lecreulx, D. The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J. Biol. Chem. 2004, 279, 30106–30113. [Google Scholar] [CrossRef] [Green Version]
- Zhurina, D.; Dudnik, A.; Waidmann, M.S.; Grimm, V.; Westermann, C.; Breitinger, K.J.; Yuan, J.; van Sinderen, D.; Riedel, C.U. High-Quality Draft Genome Sequence of Bifidobacterium longum E18, Isolated from a Healthy Adult. Genome Announc. 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, L.D.; Grossman, A.D. Autonomous Replication of the Conjugative Transposon Tn916. J. Bacteriol. 2016, 198, 3355–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cury, J.; Touchon, M.; Rocha, E.P.C. Integrative and conjugative elements and their hosts: Composition, distribution and organization. Nucleic Acids Res. 2017, 45, 8943–8956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradigaravand, D.; Palm, M.; Farewell, A.; Mustonen, V.; Warringer, J.; Parts, L. Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 2018, 14, e1006258. [Google Scholar] [CrossRef] [Green Version]
- Mayrhofer, S.; van Hoek, A.H.; Mair, C.; Huys, G.; Aarts, H.J.; Kneifel, W.; Domig, K.J. Antibiotic susceptibility of members of the Lactobacillus acidophilus group using broth microdilution and molecular identification of their resistance determinants. Int. J. Food Microbiol. 2010, 144, 81–87. [Google Scholar] [CrossRef]
- Lin, C.F.; Fung, Z.F.; Wu, C.L.; Chung, T.C. Molecular characterization of a plasmid-borne (pTC82) chloramphenicol resistance determinant (cat-TC) from Lactobacillus reuteri G4. Plasmid 1996, 36, 116–124. [Google Scholar] [CrossRef]
- Parnanen, K.; Karkman, A.; Hultman, J.; Lyra, C.; Bengtsson-Palme, J.; Larsson, D.G.J.; Rautava, S.; Isolauri, E.; Salminen, S.; Kumar, H.; et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 2018, 9, 3891. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancino, W.; Lugli, G.A.; van Sinderen, D.; Ventura, M.; Turroni, F. Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus. Microorganisms 2019, 7, 638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120638
Mancino W, Lugli GA, van Sinderen D, Ventura M, Turroni F. Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus. Microorganisms. 2019; 7(12):638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120638
Chicago/Turabian StyleMancino, Walter, Gabriele Andrea Lugli, Douwe van Sinderen, Marco Ventura, and Francesca Turroni. 2019. "Mobilome and Resistome Reconstruction from Genomes Belonging to Members of the Bifidobacterium Genus" Microorganisms 7, no. 12: 638. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7120638