Microbiota Composition and Functional Profiling Throughout the Gastrointestinal Tract of Commercial Weaning Piglets

 ,
,
Abstract
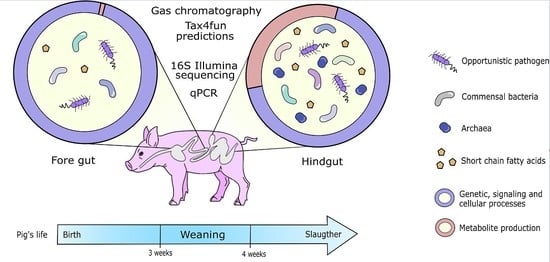
:
1. Introduction
2. Materials and Methods
2.1. Animal and Sample Collections
2.2. DNA Extraction from Digestive Contents and Colon Mucosal Scrapings
2.3. Quantification of Bacteria and Methanogenic Archaea Populations by QPCR
2.4. Standard Curve Assessments for Quantitative PCR Analyses
2.5. MiSeq 16S RDNA Sequencing and Bioinformatic Analysis
2.6. Statistical Analysis of Sequencing Data
2.7. Function Prediction with Tax4Fun
3. Results
3.1. Physiological Parameters of the GIT of Piglets
3.2. Concentration and Proportion of SCFAs Detected in the Hindgut
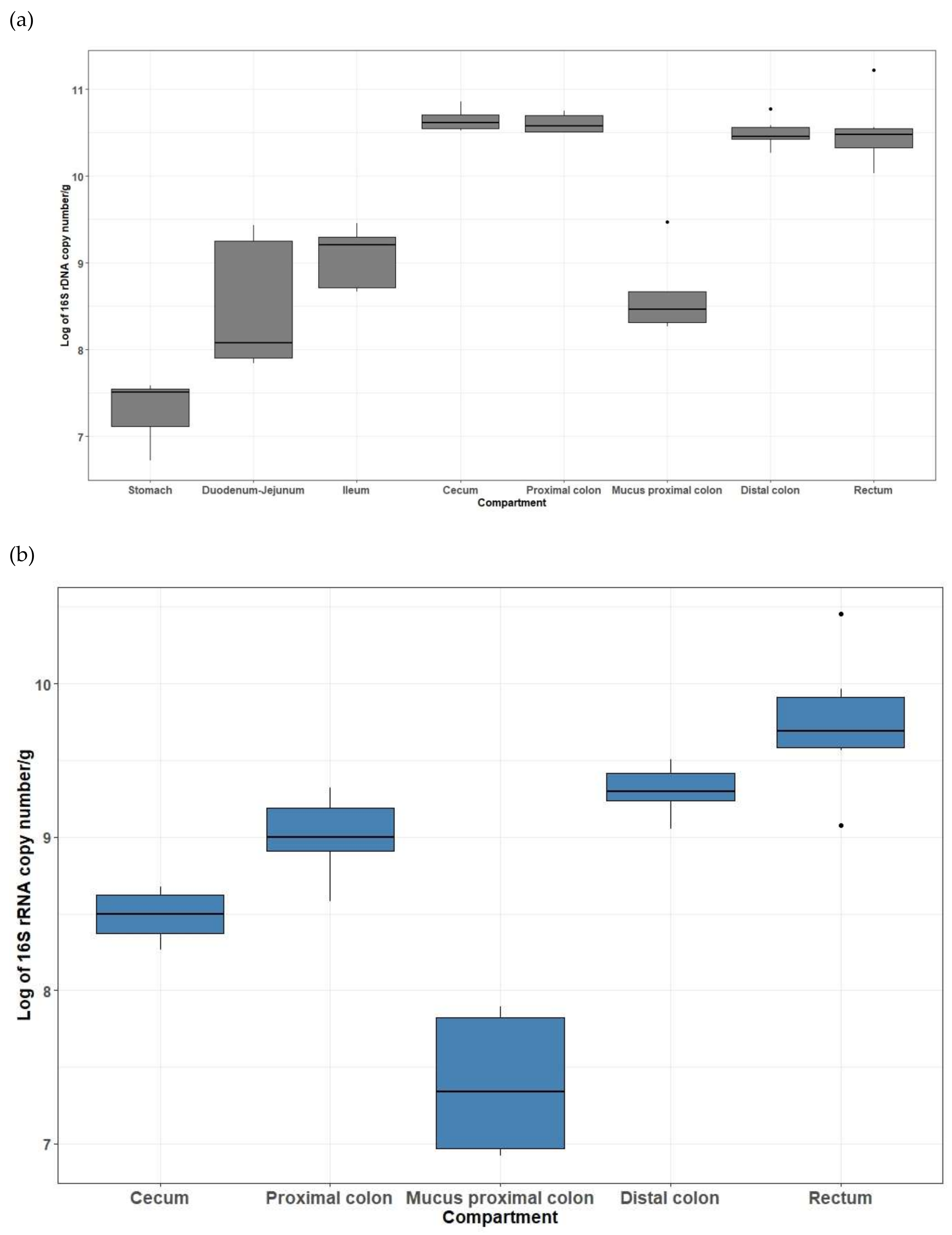
3.3. QPCR Quantification of Total Bacteria and Methanogenic Archaea
3.4. MiSeq Sequencing Data
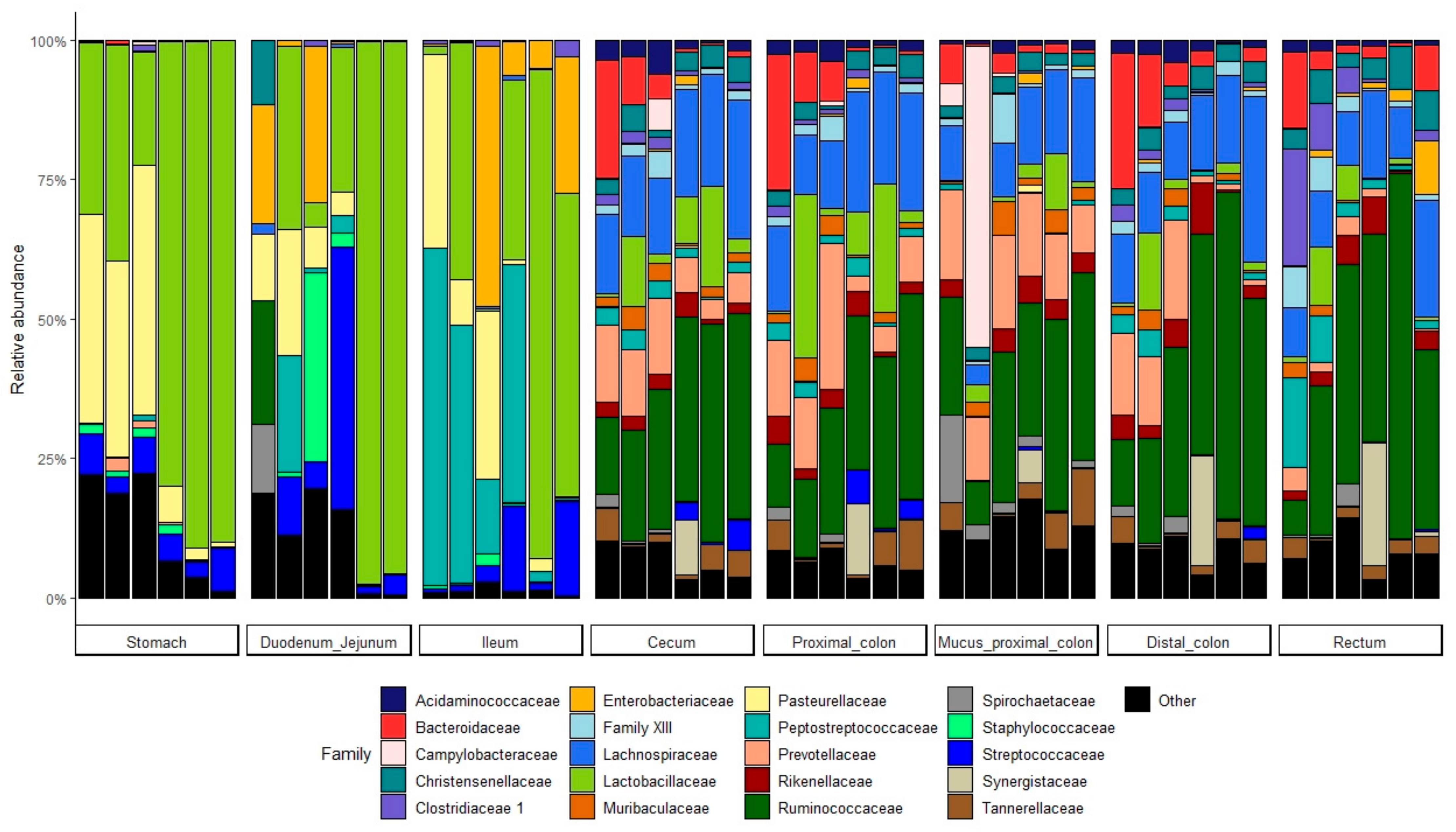
3.5. Bacterial Communities All Along the GIT
3.5.1. At the Phylum Level
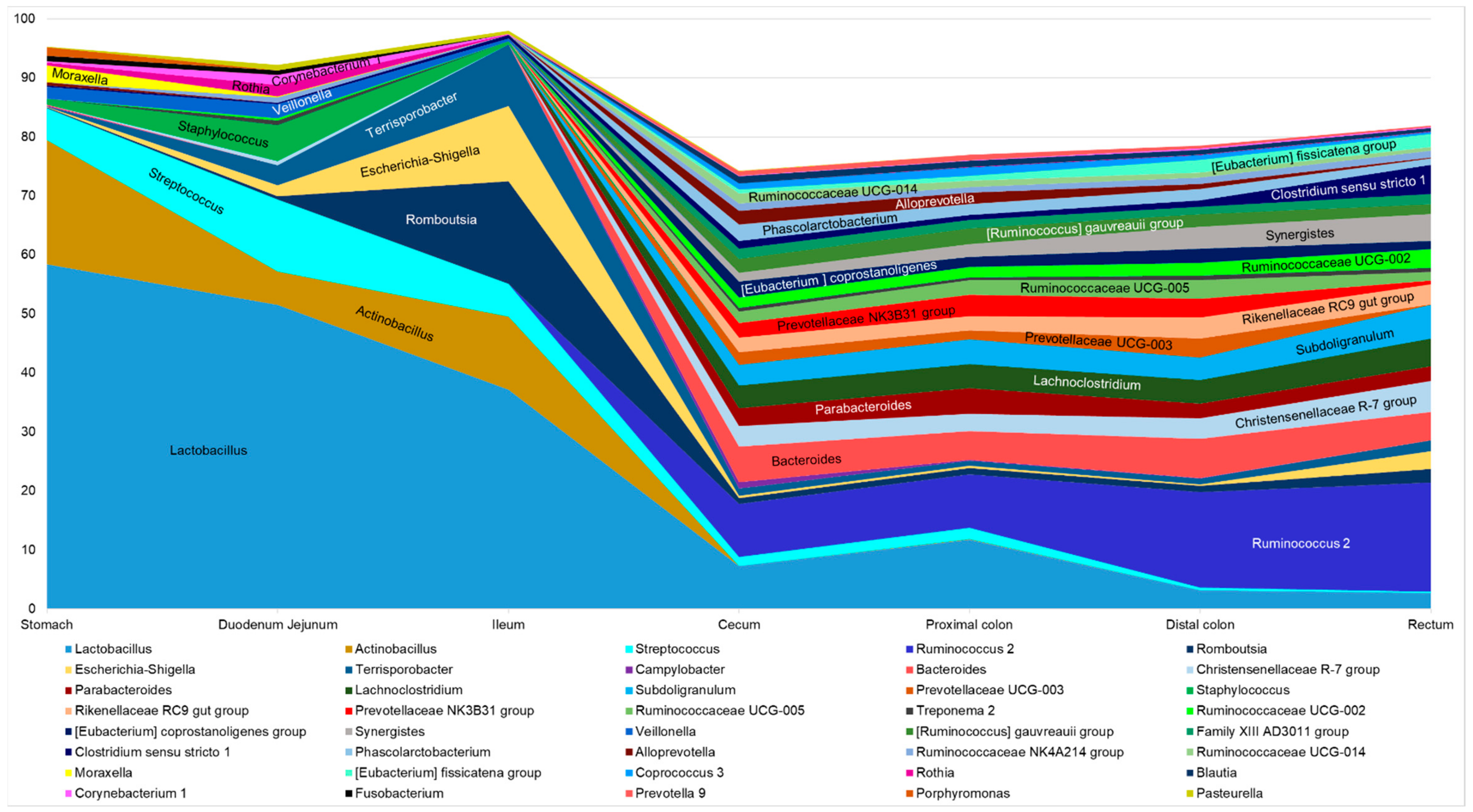
3.5.2. At the Lower Taxonomic Level
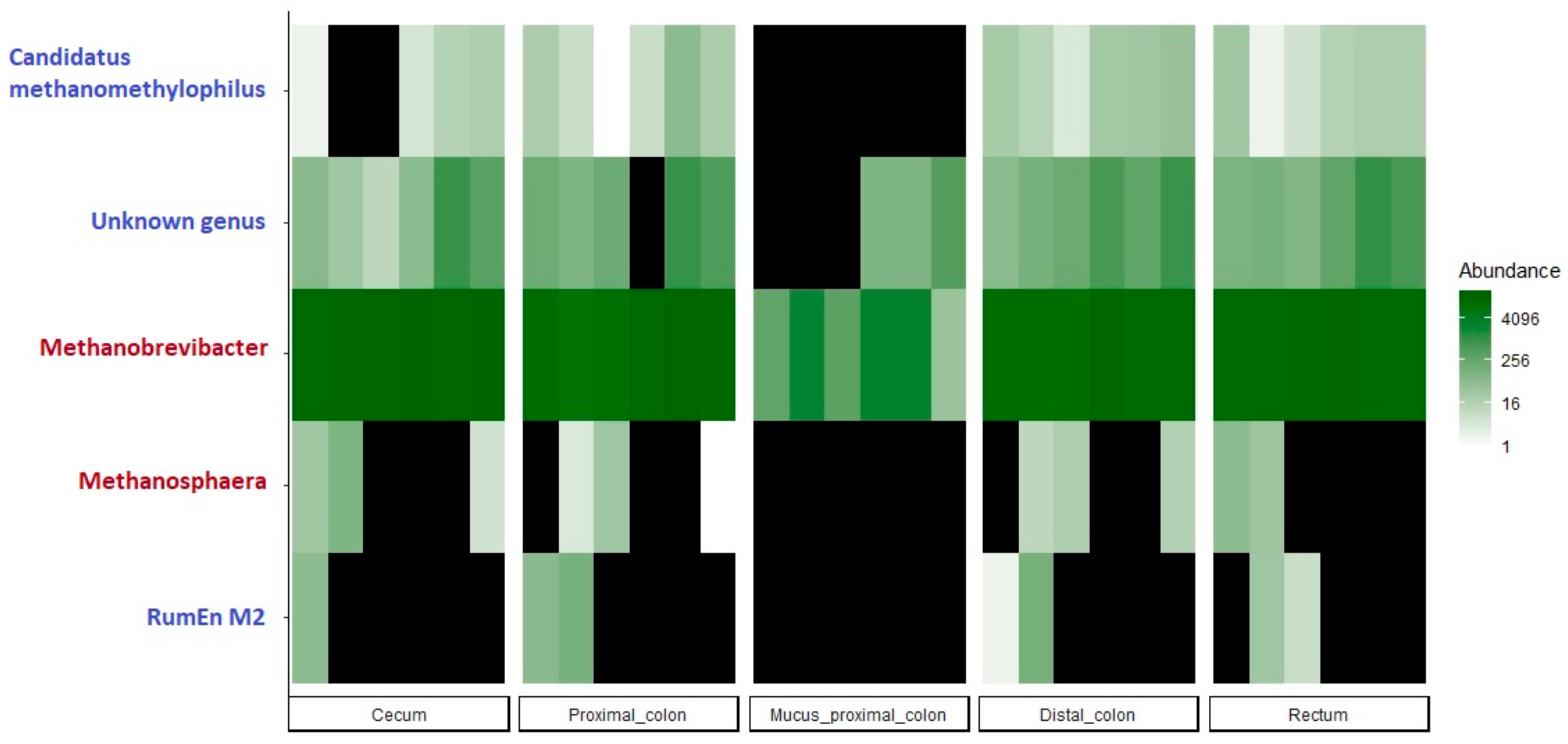
3.6. Archaeal Microbiota Throughout the GIT
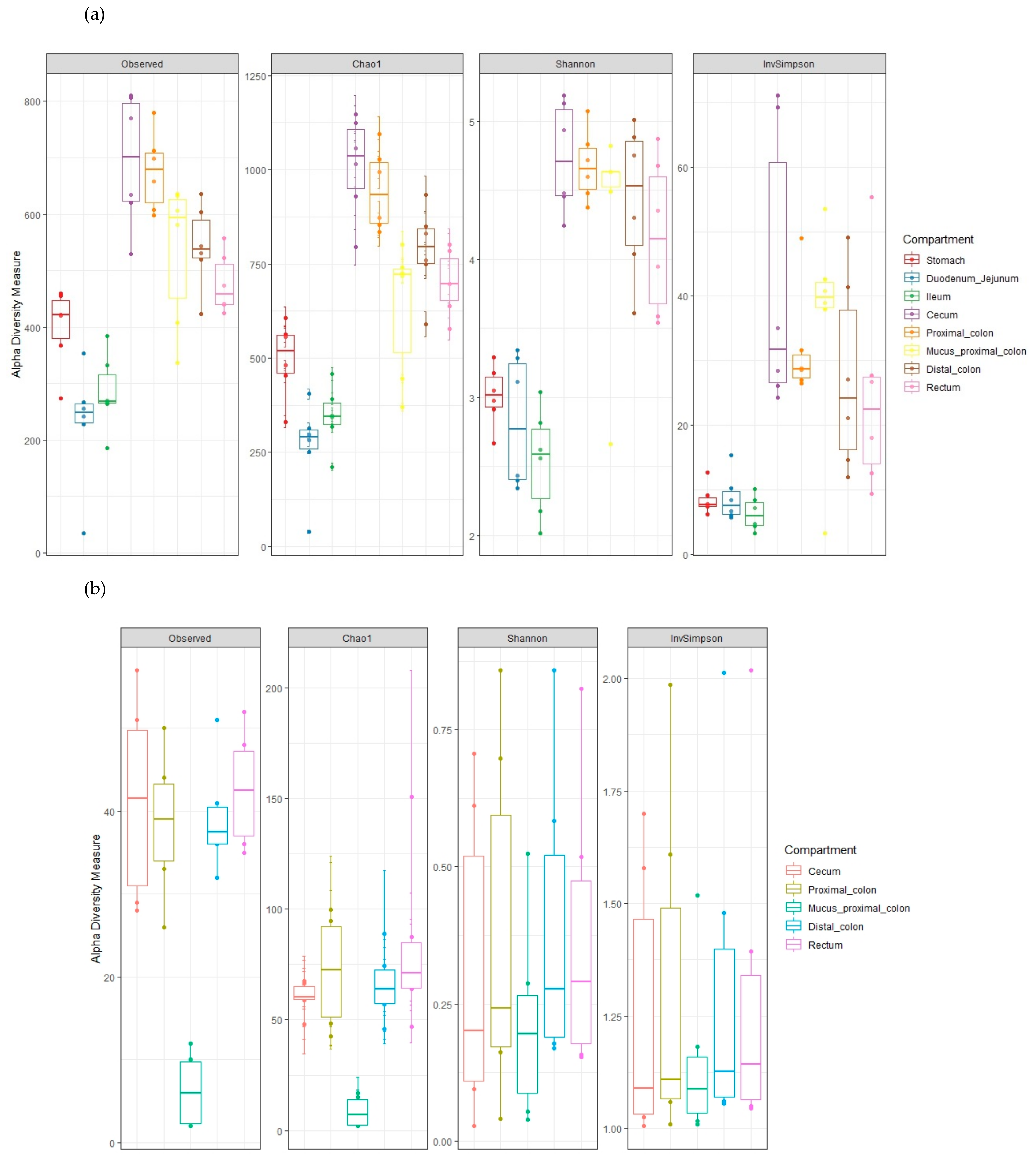
3.7. Diversity and Richness Throughout the GIT Segments
3.7.1. Alpha Diversity and Species Richness
3.7.2. Microbial Community Analysis
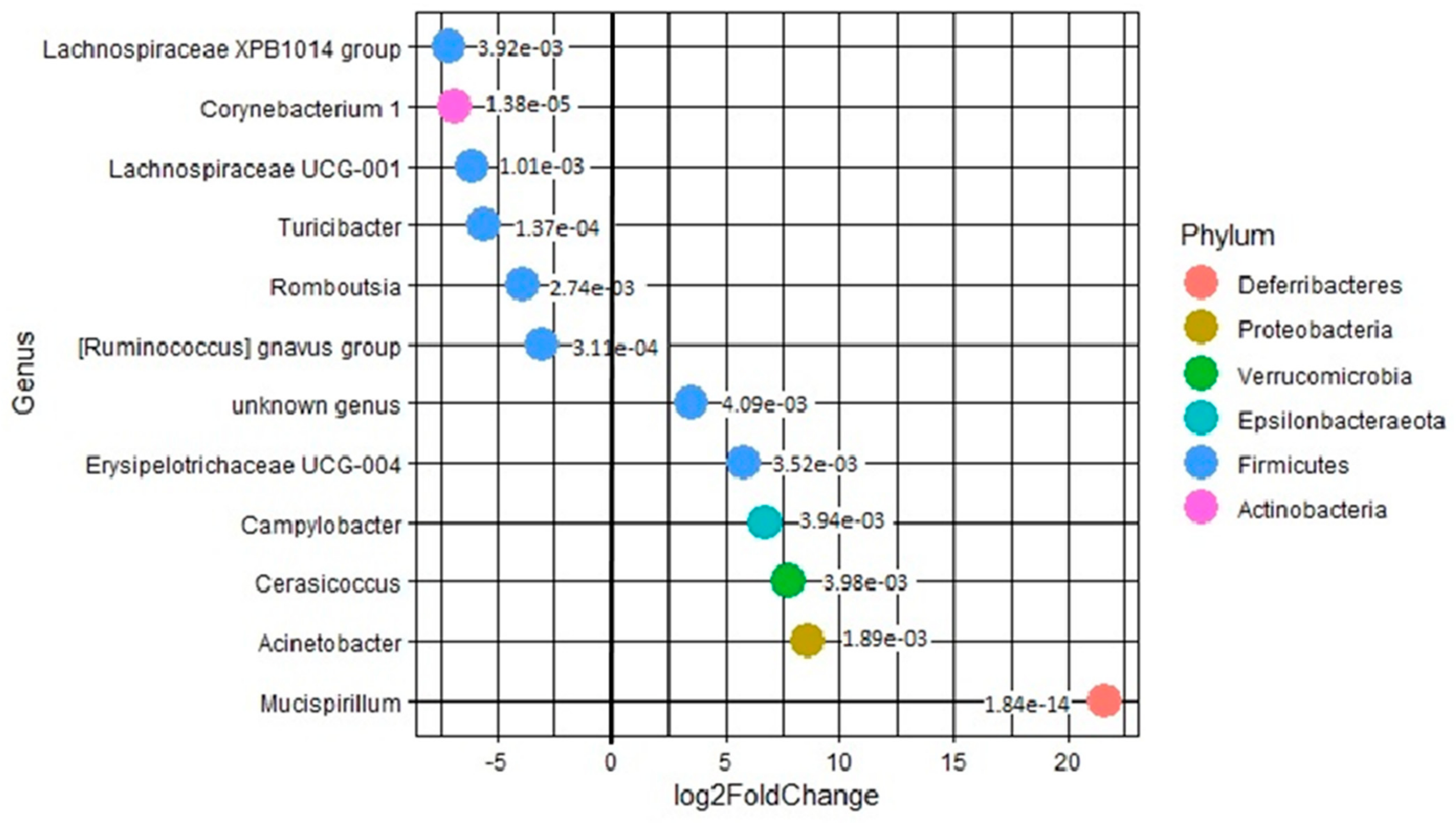
3.8. Differential Analysis of Mucosal Versus Luminal Proximal Colon Samples
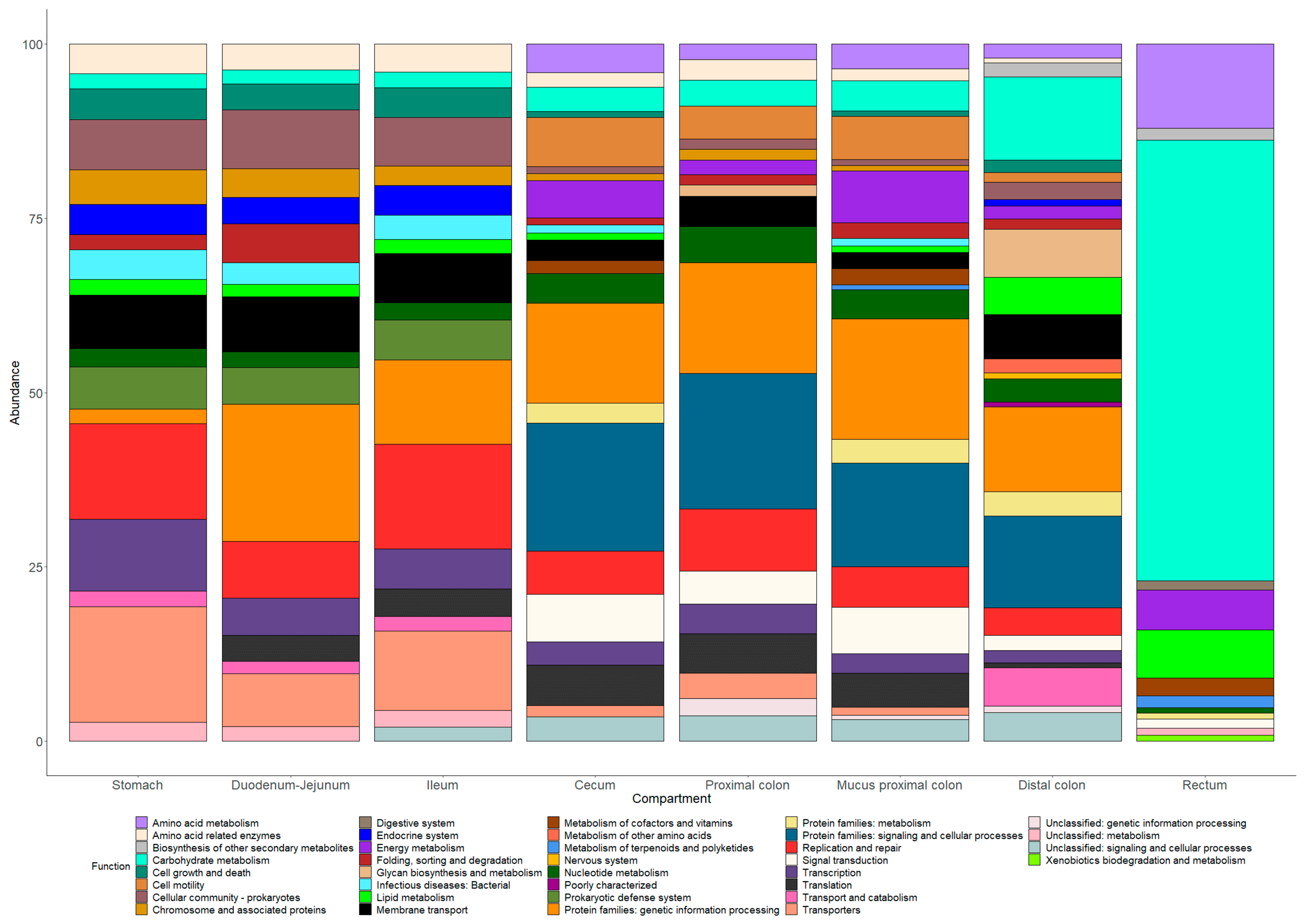
3.9. Prediction of Microbiota Functional Capacity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campbell, J.M.; Crenshaw, J.D.; Polo, J. The biological stress of early weaned piglets. J. Anim. Sci. Biotechnol. 2013, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Gresse, R.; Chaucheyras-Durand, F.; Fleury, M.A.; Van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Gut Microbiota Dysbiosis in Postweaning Piglets: Understanding the Keys to Health. Trends Microbiol. 2017, 25, 851–873. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Pamer, E.G. Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Bin, P.; Tang, Z.; Liu, S.; Chen, S.; Xia, Y.; Liu, J.; Wu, H.; Zhu, G. Intestinal microbiota mediates Enterotoxigenic Escherichia coli-induced diarrhea in piglets. BMC Vet. Res. 2018, 14, 385. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.D. Impact of antibiotic use in the swine industry. Curr. Opin. Microbiol. 2014, 19, 9–15. [Google Scholar] [CrossRef]
- Pajarillo, E.A.B.; Chae, J.P.; Balolong, M.P.; Kim, H.B.; Kang, D.K. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 2014, 60, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.-J.; Chevaleyre, C.; Billon, Y.; Doré, J.; et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes: Role of early-life gut microbiome on pigs’ health. Environ. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 28. [Google Scholar] [CrossRef]
- Hu, J.; Nie, Y.; Chen, J.; Zhang, Y.; Wang, Z.; Fan, Q.; Yan, X. Gradual Changes of Gut Microbiota in Weaned Miniature Piglets. Front. Microbiol. 2016, 7, 1727. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The Maturing Development of Gut Microbiota in Commercial Piglets during the Weaning Transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Han, Y.; Meng, F.; Zhao, J.; Zhou, Z.; Fan, H. Fecal Microbiota Succession of Piglets from Birth to Post-weaning by 454 Pyrosequencing Analysis. Trans. Tianjin Univ. 2017, 23, 211–220. [Google Scholar] [CrossRef]
- Guevarra, R.B.; Hong, S.H.; Cho, J.H.; Kim, B.-R.; Shin, J.; Lee, J.H.; Kang, B.N.; Kim, Y.H.; Wattanaphansak, S.; Isaacson, R.E.; et al. The dynamics of the piglet gut microbiome during the weaning transition in association with health and nutrition. J. Anim. Sci. Biotechnol. 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Kraler, M.; Ghanbari, M.; Domig, K.J.; Schedle, K.; Kneifel, W. The intestinal microbiota of piglets fed with wheat bran variants as characterised by 16S rRNA next-generation amplicon sequencing. Arch. Anim. Nutr. 2016, 70, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Daly, K.; Moran, A.W.; Ryan, S.; Bravo, D.; Shirazi-Beechey, S.P. Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences: Swine mucosa-associated gut microbiota. Environ. Microbiol. 2017, 19, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A.-S.R.; de Jonge, N.; Nielsen, J.L.; Højberg, O.; Lauridsen, C.; Cutting, S.M.; Canibe, N. Impact of Bacillus spp. spores and gentamicin on the gastrointestinal microbiota of suckling and newly weaned piglets. PLoS ONE 2018, 13, e0207382. [Google Scholar] [CrossRef]
- De Rodas, B.; Youmans, B.P.; Danzeisen, J.L.; Tran, H.; Johnson, T.J. Microbiome profiling of commercial pigs from farrow to finish. J. Anim. Sci. 2018, 96, 1778–1794. [Google Scholar] [CrossRef]
- Adhikari, B.; Kim, S.W.; Kwon, Y.M. Characterization of Microbiota Associated with Digesta and Mucosa in Different Regions of Gastrointestinal Tract of Nursery Pigs. Int. J. Mol. Sci. 2019, 20, 1630. [Google Scholar] [CrossRef]
- Mu, C.; Yang, Y.; Su, Y.; Zoetendal, E.G.; Zhu, W. Differences in Microbiota Membership along the Gastrointestinal Tract of Piglets and Their Differential Alterations Following an Early-Life Antibiotic Intervention. Front. Microbiol. 2017, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data: Figure 1. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef] [PubMed]
- Ohene-Adjei, S.; Chaves, A.V.; McAllister, T.A.; Benchaar, C.; Teather, R.M.; Forster, R.J. Evidence of increased diversity of methanogenic archaea with plant extract supplementation. Microb. Ecol. 2008, 56, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Auer, L.; Mariadassou, M.; O’Donohue, M.; Klopp, C.; Hernandez-Raquet, G. Analysis of large 16S rRNA Illumina data sets: Impact of singleton read filtering on microbial community description. Mol. Ecol. Resour. 2017, 17, e122–e132. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M.A.S.S. The vegan package. Commun. Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Fan, W.; et al. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef] [PubMed]
- Danzeisen, J.L.; Calvert, A.J.; Noll, S.L.; McComb, B.; Sherwood, J.S.; Logue, C.M.; Johnson, T.J. Succession of the turkey gastrointestinal bacterial microbiome related to weight gain. PeerJ 2013, 1, e237. [Google Scholar] [CrossRef] [PubMed]
- Danzeisen, J.L.; Clayton, J.B.; Huang, H.; Knights, D.; McComb, B.; Hayer, S.S.; Johnson, T.J. Temporal Relationships Exist Between Cecum, Ileum, and Litter Bacterial Microbiomes in a Commercial Turkey Flock, and Subtherapeutic Penicillin Treatment Impacts Ileum Bacterial Community Establishment. Front. Vet. Sci. 2015, 2, 56. [Google Scholar] [CrossRef]
- Liang, H.; Dai, Z.; Liu, N.; Ji, Y.; Chen, J.; Zhang, Y.; Yang, Y.; Li, J.; Wu, Z.; Wu, G. Dietary L-Tryptophan Modulates the Structural and Functional Composition of the Intestinal Microbiome in Weaned Piglets. Front. Microbiol. 2018, 9, 1736. [Google Scholar] [CrossRef]
- Wang, Q.; Cai, R.; Huang, A.; Wang, X.; Qu, W.; Shi, L.; Li, C.; Yan, H. Comparison of Oropharyngeal Microbiota in Healthy Piglets and Piglets With Respiratory Disease. Front. Microbiol. 2018, 9, 3218. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Chen, L.; Gerritsen, J.; Smidt, H.; Goldfine, H. The cellular lipids of Romboutsia. Biochim. Biophys. Acta 2016, 1861, 1076–1082. [Google Scholar] [CrossRef]
- Cheng, M.P.; Domingo, M.-C.; Lévesque, S.; Yansouni, C.P. A case report of a deep surgical site infection with Terrisporobacter glycolicus/T. Mayombei and review of the literature. BMC Infect. Dis. 2016, 16, 529. [Google Scholar] [CrossRef] [PubMed]
- González-Ortiz, G.; Pérez, J.F.; Hermes, R.G.; Molist, F.; Jiménez-Díaz, R.; Martín-Orúe, S.M. Screening the ability of natural feed ingredients to interfere with the adherence of enterotoxigenic Escherichia coli (ETEC) K88 to the porcine intestinal mucus. Br. J. Nutr. 2014, 111, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Alter, T.; Gaull, F.; Kasimir, S.; Gürtler, M.; Mielke, H.; Linnebur, M.; Fehlhaber, K. Prevalences and transmission routes of Campylobacter spp. strains within multiple pig farms. Vet. Microbiol. 2005, 108, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.J. The physiology and metabolism of Campylobacter jejuni and Helicobacter pylori. Symp. Ser. Soc. Appl. Microbiol. 2001, 90, 16S–24S. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Navarro, K.; Couch, R.D.; Hill, D.; Urban, J.F. Alterations in the porcine colon microbiota induced by the gastrointestinal nematode Trichuris suis. Infect. Immun. 2012, 80, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, R.W.; Li, W.; Beshah, E.; Dawson, H.D.; Urban, J.F. Worm burden-dependent disruption of the porcine colon microbiota by Trichuris suis infection. PLoS ONE 2012, 7, e35470. [Google Scholar] [CrossRef] [PubMed]
- Biswal, D. Helminth Infections and Gut Microbiota: The Futuristic Study of Pathogen Virulence and Gut Ecosystem. J. Mol. Biomark. Diagn. 2016, 7, 2. [Google Scholar] [CrossRef]
- Saraf, M.K.; Piccolo, B.D.; Bowlin, A.K.; Mercer, K.E.; LeRoith, T.; Chintapalli, S.V.; Shankar, K.; Badger, T.M.; Yeruva, L. Formula diet driven microbiota shifts tryptophan metabolism from serotonin to tryptamine in neonatal porcine colon. Microbiome 2017, 5, 77. [Google Scholar] [CrossRef]
- Chen, S.; He, S.; Wu, C.; Du, D. Characteristics of heterotrophic nitrification and aerobic denitrification bacterium Acinetobacter sp. T1 and its application for pig farm wastewater treatment. J. Biosci. Bioeng. 2019, 127, 201–205. [Google Scholar] [CrossRef]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea Are Interactive Components of Complex Microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef]
- Luo, Y.; Chen, H.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Tian, G.; Yu, J.; Huang, Z.; Luo, J.; et al. Dietary pea fiber increases diversity of colonic methanogens of pigs with a shift from Methanobrevibacter to Methanomassiliicoccus-like genus and change in numbers of three hydrogenotrophs. BMC Microbiol. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Federici, S.; Miragoli, F.; Pisacane, V.; Rebecchi, A.; Morelli, L.; Callegari, M.L. Archaeal microbiota population in piglet feces shifts in response to weaning: Methanobrevibacter smithii is replaced with Methanobrevibacter boviskoreani. FEMS Microbiol. Lett. 2015, 362, fnv064. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.-Y.; Yang, C.-F.; Zhu, W.-Y. Phylogenetic analysis of methanogens in the pig feces. Curr. Microbiol. 2011, 62, 1386–1389. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Su, Y.; Wright, A.-D.G.; Zhang, L.; Smidt, H.; Zhu, W. Lean breed Landrace pigs harbor fecal methanogens at higher diversity and density than obese breed Erhualian pigs. Archaea Vanc. BC 2012, 2012, 605289. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Bian, G.; Zhu, Z.; Smidt, H.; Zhu, W. Early methanogenic colonisation in the faeces of Meishan and Yorkshire piglets as determined by pyrosequencing analysis. Archaea Vanc. BC 2014, 2014, 547908. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Parisot, N.; Harris, H.M.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genom. 2014, 15, 679. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatani, M.; Inoue, R.; Tomonaga, S.; Fukuta, K.; Tsukahara, T. Production, Absorption, and Blood Flow Dynamics of Short-Chain Fatty Acids Produced by Fermentation in Piglet Hindgut during the Suckling–Weaning Period. Nutrients 2018, 10, 1220. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Upadhyaya, B.; McCormack, L.; Fardin-Kia, A.R.; Juenemann, R.; Nichenametla, S.; Clapper, J.; Specker, B.; Dey, M. Impact of dietary resistant starch type 4 on human gut microbiota and immunometabolic functions. Sci. Rep. 2016, 6, 28797. [Google Scholar] [CrossRef] [PubMed]
- Brugère, J.-F.; Ben Hania, W.; Arnal, M.-E.; Ribière, C.; Ballet, N.; Vandeckerkove, P.; Ollivier, B.; O’Toole, P.W. Archaea: Microbial Candidates in Next-generation Probiotics Development. J. Clin. Gastroenterol. 2018, 52, S71–S73. [Google Scholar] [CrossRef] [PubMed]
- Dou, S.; Gadonna-Widehem, P.; Rome, V.; Hamoudi, D.; Rhazi, L.; Lakhal, L.; Larcher, T.; Bahi-Jaber, N.; Pinon-Quintana, A.; Guyonvarch, A.; et al. Characterisation of Early-Life Fecal Microbiota in Susceptible and Healthy Pigs to Post-Weaning Diarrhoea. PLoS ONE 2017, 12, e0169851. [Google Scholar] [CrossRef] [PubMed]
- Eng, A.; Borenstein, E. Taxa-function robustness in microbial communities. Microbiome 2018, 6, 45. [Google Scholar] [CrossRef]
- Bian, G.; Deng, Z.; Liu, T. Strategies for terpenoid overproduction and new terpenoid discovery. Curr. Opin. Biotechnol. 2017, 48, 234–241. [Google Scholar] [CrossRef]
- Hill, P.; Heberlig, G.; Boddy, C. Sampling Terrestrial Environments for Bacterial Polyketides. Molecules 2017, 22, 707. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of Short-Chain Fatty Acid (mmol/L) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compartments | Acetate | Butyrate | Propionate | Valerate | Caproate | Iso Butyrate | Iso Valerate | Total SCFAs | ||||||||
| Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | Individual Values | Mean ± std | |
| Caecum | 67.41 | 60.87 ± 15.75 | 8.03 | 9.06 ± 2.55 | 13.28 | 17.67 ± 4.68 | 3.12 | 3.17 ± 0.85 | 0.40 | 0.37 ± 0.17 | 2.28 | 2.28 ± 0.63 | 2.57 | 2.84 ± 0.73 | 97.09 | 96.24 ± 22.15 |
| 54.52 | 8.68 | 17.61 | 2.93 | 0.06 | 2.53 | 3.58 | 89.92 | |||||||||
| 78.89 | 10.11 | 15.83 | 3.82 | 0.49 | 2.89 | 3.13 | 115.17 | |||||||||
| 33.90 | 4.72 | 13.18 | 1.63 | 0.34 | 1.09 | 1.50 | 56.36 | |||||||||
| 71.05 | 10.96 | 25.15 | 3.58 | 0.35 | 2.66 | 3.26 | 117.00 | |||||||||
| 59.43 | 11.84 | 20.95 | 3.92 | 0.55 | 2.23 | 2.99 | 101.91 | |||||||||
| Proximal colon | 33.18 | 39.00 ± 12.70 | 6.23 | 7.09 ± 2.37 | 12.88 | 14.03 ± 5.99 | 1.77 | 2.18 ± 0.83 | 0.08 | 0.23 ± 0.12 | 1.47 | 1.44 ± 0.55 | 2.59 | 2.23 ± 0.48 | 58.20 | 66.20 ± 21.59 |
| 47.73 | 7.10 | 10.78 | 2.31 | 0.37 | 1.87 | 2.40 | 72.55 | |||||||||
| 27.37 | 4.74 | 8.92 | 1.15 | 0.10 | 0.89 | 1.68 | 44.84 | |||||||||
| 46.17 | 7.34 | 20.87 | 1.99 | 0.33 | 1.33 | 2.26 | 80.29 | |||||||||
| 55.72 | 11.52 | 22.11 | 3.64 | 0.26 | 2.25 | 2.82 | 98.32 | |||||||||
| 23.84 | 5.63 | 8.60 | 2.19 | 0.23 | 0.85 | 1.63 | 42.98 | |||||||||
| Distal colon | 12.61 | 17.46 ± 6.65 | 1.17 | 3.11 ± 2.21 | 3.75 | 5.03 ± 3.34 | 0.23 | 0.89 ± 0.83 | 0.00 | 0.11 ± 0.12 | 0.44 | 0.46 ± 0.18 | 1.06 | 1.05 ± 0.44 | 19.28 | 28.11 ± 12.86 |
| 11.97 | 1.26 | 1.70 | 0.31 | 0.03 | 0.30 | 0.58 | 16.14 | |||||||||
| 13.06 | 1.43 | 2.34 | 0.23 | 0.00 | 0.20 | 0.54 | 17.80 | |||||||||
| 29.10 | 4.30 | 10.82 | 1.14 | 0.26 | 0.62 | 1.32 | 47.57 | |||||||||
| 17.01 | 3.84 | 5.10 | 1.09 | 0.13 | 0.50 | 1.16 | 28.83 | |||||||||
| 21.01 | 6.63 | 6.48 | 2.35 | 0.24 | 0.68 | 1.67 | 39.05 | |||||||||
| Rectum | 17.41 | 21.23 ± 7.47 | 4.51 | 3.51 ± 2.12 | 6.72 | 4.34 ± 2.60 | 0.56 | 1.03 ± 0.68 | 0.00 | 0.19 ± 0.21 | 1.25 | 0.90 ± 0.59 | 2.92 | 2.03 ± 1.18 | 33.37 | 33.22 ± 12.33 |
| 9.41 | 1.83 | 1.49 | 0.56 | 0.10 | 0.28 | 0.70 | 14.37 | |||||||||
| 29.08 | 1.13 | 5.27 | 0.64 | 0.00 | 0.52 | 1.26 | 37.90 | |||||||||
| 29.18 | 6.69 | 7.71 | 2.22 | 0.53 | 1.88 | 3.87 | 52.07 | |||||||||
| 21.50 | 4.64 | 2.88 | 1.49 | 0.17 | 0.89 | 2.04 | 33.60 | |||||||||
| 20.82 | 2.24 | 1.96 | 0.72 | 0.31 | 0.56 | 1.40 | 28.02 | |||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gresse, R.; Chaucheyras Durand, F.; Dunière, L.; Blanquet-Diot, S.; Forano, E. Microbiota Composition and Functional Profiling Throughout the Gastrointestinal Tract of Commercial Weaning Piglets. Microorganisms 2019, 7, 343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090343
Gresse R, Chaucheyras Durand F, Dunière L, Blanquet-Diot S, Forano E. Microbiota Composition and Functional Profiling Throughout the Gastrointestinal Tract of Commercial Weaning Piglets. Microorganisms. 2019; 7(9):343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090343
Chicago/Turabian StyleGresse, Raphaële, Frédérique Chaucheyras Durand, Lysiane Dunière, Stéphanie Blanquet-Diot, and Evelyne Forano. 2019. "Microbiota Composition and Functional Profiling Throughout the Gastrointestinal Tract of Commercial Weaning Piglets" Microorganisms 7, no. 9: 343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090343