Identification of Microbial Profiles in Heavy-Metal-Contaminated Soil from Full-Length 16S rRNA Reads Sequenced by a PacBio System


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Characterization
2.2. PCR Amplification of Bacterial 16S rRNA Gene and Sequencing by PacBio System
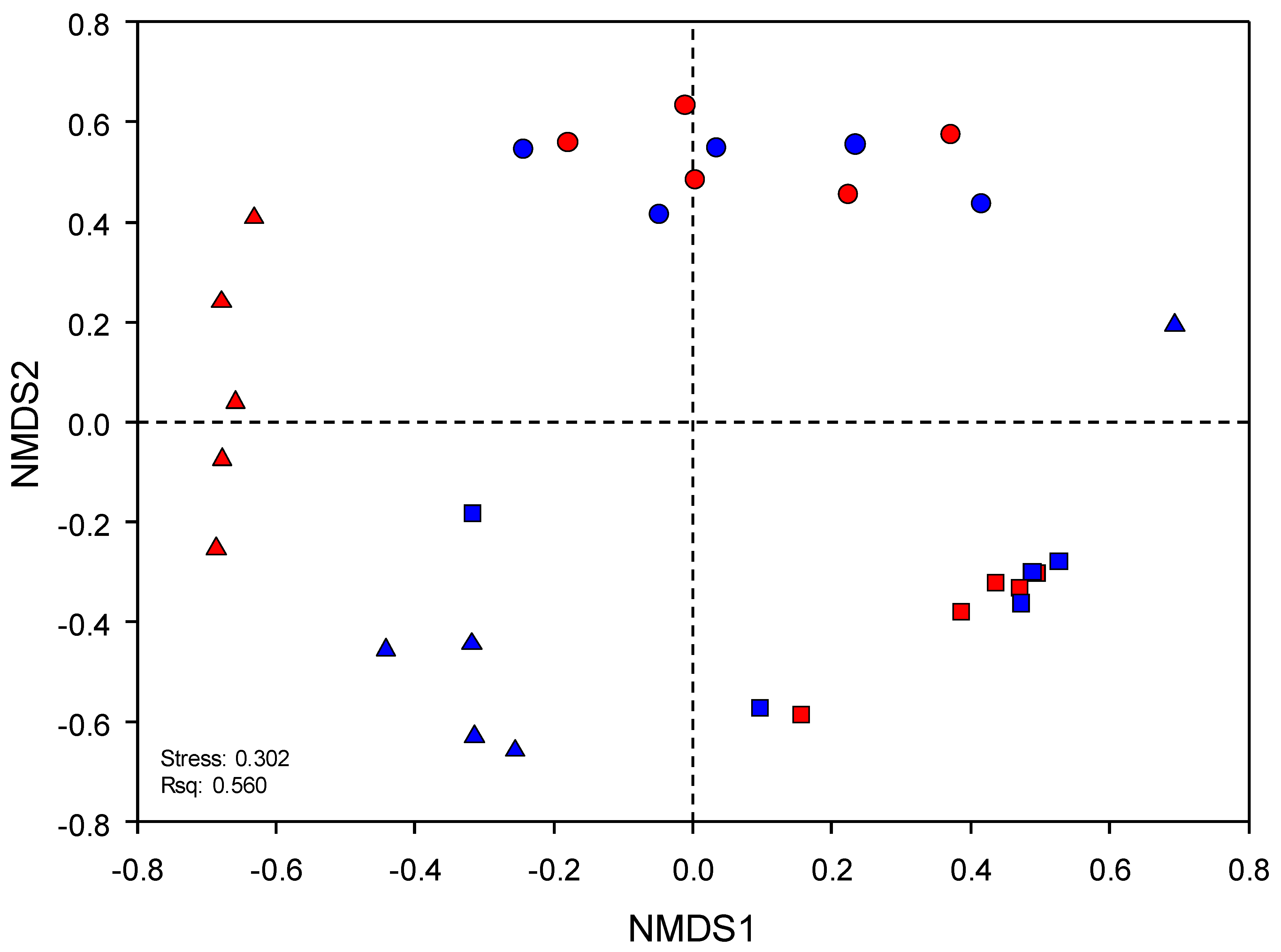
2.3. Sequence Analysis, Diversity Indices, and Statistics
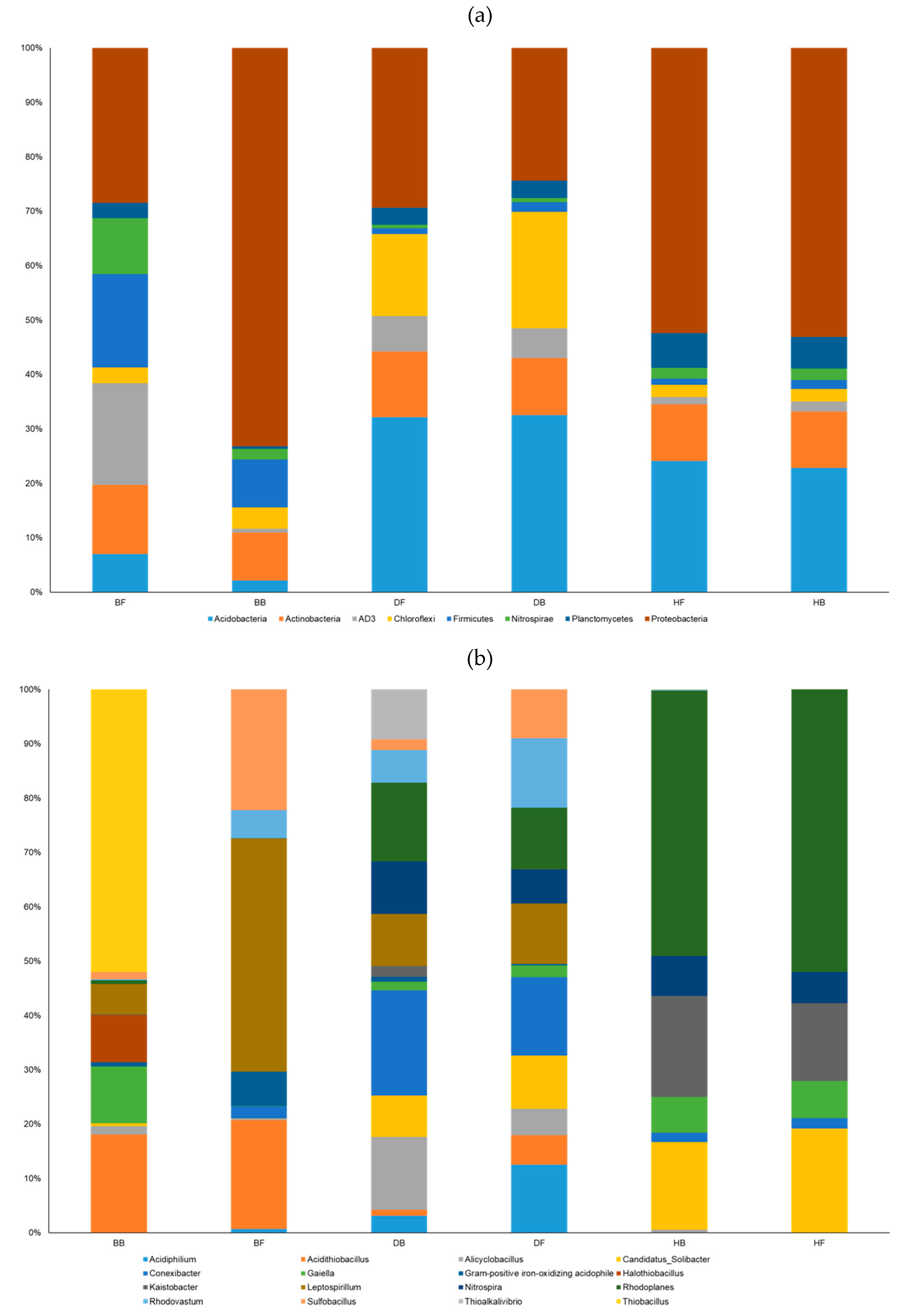
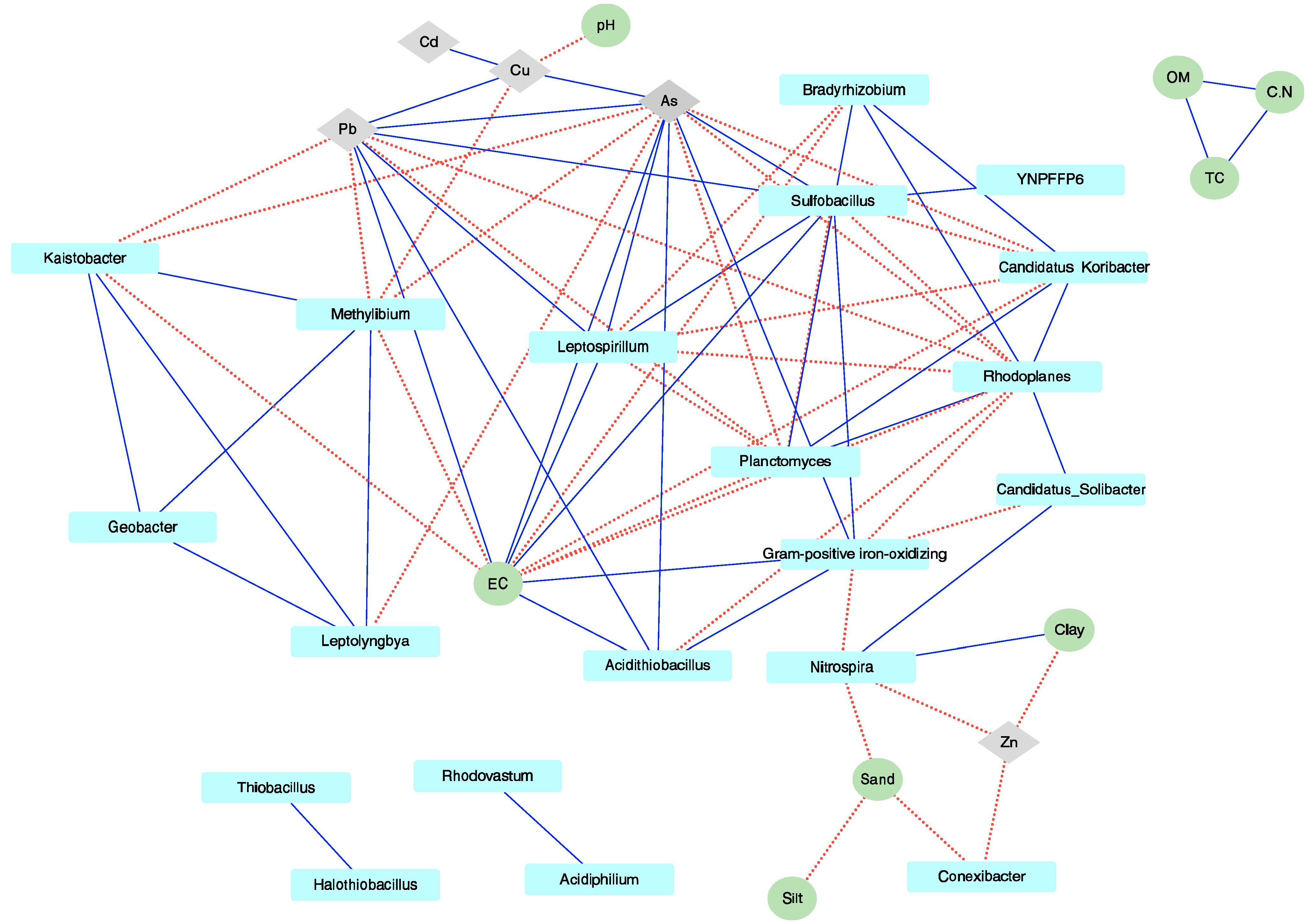
3. Results
3.1. Environmental Data
3.2. General Statistics for 16S rRNA Gene Amplification
3.3. Environmental Factors Significantly Affecting the Microbial Community
3.4. Predictive Functional Profiles from Microbial Communities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- MIRECO. 2016 Yearbook of MIRECO (Mine Reclamation Corp. in Korea) Statistics; Mine Reclamation Corp.: Wonju, Korea, 2017. [Google Scholar]
- NIER. Studies on the Assessment of Biological Exposure to Heavy Metals; National Institute of Environmental Research (NIER): Incheon, Korea, 1996. [Google Scholar]
- KMOE. Soil Environment Conservation Act in Korea; Ministry of Environment: Seoul, Korea, 2016. [Google Scholar]
- Gałązka, A.; Gałązka, R. Phytoremediation of polycyclic aromatic hydrocarbons in soils artificially polluted using plant-associated-endophytic bacteria and dactylis glomerata as the bioremediation plant. Pol. J. Microbiol. 2015, 64, 241–252. [Google Scholar] [CrossRef]
- Galazka, A.; Grzadziel, J.; Galazka, R.; Ukalska-Jaruga, A.; Strzelecka, J.; Smreczak, B. Genetic and functional diversity of bacterial microbiome in soils with long term impacts of petroleum hydrocarbons. Front. Microbiol. 2018, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.; Zhang, C.; Mathews, E.R.; Tang, C.; Franks, A.E. Microbial community dynamics in the rhizosphere of a cadmium hyper-accumulator. Sci. Rep. 2016, 6, 36067. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003, 44, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Edwards, K.J.; Bond, P.L.; Gihring, T.M.; Banfield, J.F. An archaeal iron-oxidizing extreme acidophile important in acid mine drainage. Science 2000, 287, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Tyson, G.W.; Webb, R.I.; Flanagan, J.; Hugenholtz, P.; Allen, E.E.; Banfield, J.F. Lineages of acidophilic archaea revealed by community genomic analysis. Science 2006, 314, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Comolli, L.R.; Dick, G.J.; Hauser, L.J.; Hyatt, D.; Dill, B.D.; Land, M.L.; Verberkmoes, N.C.; Hettich, R.L.; Banfield, J.F. Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8806–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salipante, S.J.; Kawashima, T.; Rosenthal, C.; Hoogestraat, D.R.; Cummings, L.A.; Sengupta, D.J.; Harkins, T.T.; Cookson, B.T.; Hoffman, N.G. Performance comparison of Illumina and ion torrent next-generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl. Env. Microbiol. 2014, 80, 7583–7591. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Morrison, H.G.; Benjamino, J.; Grim, S.L.; Graf, J. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS ONE 2014, 9, e94249. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Env. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Koh, H.W.; Kim, H.; Chae, J.C.; Park, S.J. Microbial community composition in the marine sediments of Jeju island: Next-generation sequencing surveys. J. Microbiol. Biotechnol. 2016, 26, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Currie, A.R.; Tait, K.; Parry, H.; de Francisco-Mora, B.; Hicks, N.; Osborn, A.M.; Widdicombe, S.; Stahl, H. Marine microbial gene abundance and community composition in response to ocean acidification and elevated temperature in two contrasting coastal marine sediments. Front. Microbiol. 2017, 8, 1599. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasken, R.S. Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 2012, 10, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; McLean, J.S. Recent advances in genomic DNA sequencing of microbial species from single cells. Nat. Rev. Genet. 2014, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Soergel, D.A.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Phillippy, A.M. One chromosome, one contig: Complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015, 23, 110–120. [Google Scholar] [CrossRef]
- Mosher, J.J.; Bowman, B.; Bernberg, E.L.; Shevchenko, O.; Kan, J.; Korlach, J.; Kaplan, L.A. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing. J. Microbiol. Methods 2014, 104, 59–60. [Google Scholar] [CrossRef]
- Myer, P.R.; Kim, M.; Freetly, H.C.; Smith, T.P. Metagenomic and near full-length 16S rRNA sequence data in support of the phylogenetic analysis of the rumen bacterial community in steers. Data Brief 2016, 8, 1048–1053. [Google Scholar] [CrossRef]
- Wagner, J.; Coupland, P.; Browne, H.P.; Lawley, T.D.; Francis, S.C.; Parkhill, J. Evaluation of PacBio sequencing for full-length bacterial 16S rRNA gene classification. BMC Microbiol. 2016, 16, 274. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Bowers, R.M.; Levy, A.; Gies, E.A.; Cheng, J.F.; Copeland, A.; Klenk, H.P.; et al. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Pootakham, W.; Mhuantong, W.; Yoocha, T.; Putchim, L.; Sonthirod, C.; Naktang, C.; Thongtham, N.; Tangphatsornruang, S. High resolution profiling of coral-associated bacterial communities using full-length 16S rRNA sequence data from PacBio SMRT sequencing system. Sci. Rep. 2017, 7, 2774. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Dueholm, M.S.; McIlroy, S.J.; Kirkegaard, R.H.; Nielsen, P.H.; Albertsen, M. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat. Biotechnol. 2018, 36, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Pedros-Alio, C. The rare bacterial biosphere. Ann. Rev. Mar. Sci. 2012, 4, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Webster, R. Replicate and randomize, or lie. Env. Microbiol. 2017, 19, 25–28. [Google Scholar] [CrossRef]
- MOE. Official Test Methods of Soil Contamination; Ministry of Environment, Korea: Seoul, Korea, 2017. [Google Scholar]
- Koh, H.W.; Hur, M.; Kang, M.S.; Ku, Y.B.; Ghai, R.; Park, S.J. Physiological and genomic insights into the lifestyle of arsenite-oxidizing Herminiimonas arsenitoxidans. Sci. Rep. 2017, 7, 15007. [Google Scholar] [CrossRef]
- Burt, R.; United, S.; Natural Resources Conservation, S.; National Soil Survey, C.; National Soil Survey, L. Soil Survey Laboratory Methods Manual; United States Dept. of Agriculture, Natural Resources Conservation Service: Washington, DC, USA, 2004.
- Jahn, R.; Blume, H.P.; Asio, V.B.; Spaargaren, O.; Schad, P. Guidelines for soil description, 4th ed; FAO: Rome, Italy, 2006. [Google Scholar]
- Myer, P.R.; Kim, M.; Freetly, H.C.; Smith, T.P. Evaluation of 16S rRNA amplicon sequencing using two next-generation sequencing technologies for phylogenetic analysis of the rumen bacterial community in steers. J. Microbiol. Methods 2016, 127, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, Z.; Yu, X.; Xue, Y.; Huang, B.; Yang, J. Invasion by cordgrass increases microbial diversity and alters community composition in a mangrove nature reserve. Front. Microbiol. 2017, 8, 2503. [Google Scholar] [CrossRef]
- Feinsinger, P.; Spears, E.E.; Poole, R.W. A simple measure of niche breadth. Ecology 1981, 62, 27–32. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package, R Package Version 2.4-5. 2017.
- Buttigieg, P.L.; Ramette, A. A guide to statistical analysis in microbial ecology: A community-focused, living review of multivariate data analyses. FEMS Microbiol. Ecol. 2014, 90, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Koh, H.W.; Rhee, S.K.; Fujitani, H.; Park, S.J. Detection and diversity of the nitrite oxidoreductase alpha subunit (nxrA) gene of Nitrospina in marine sediments. Microb. Ecol. 2017, 73, 111–122. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2010. [Google Scholar]
- Moberly, J.G.; Bernards, M.T.; Waynant, K.V. Key features and updates for origin 2018. J. Cheminform. 2018, 10, 5. [Google Scholar] [CrossRef]
- Harrell, F.E.J.; Dupont, C. Hmisc: Harrell Miscellaneous, R Package Version 4.1-1. 2018.
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bonett, D.G.; Wright, T.A. Sample size requirements for estimating pearson, kendall and spearman correlations. Psychometrika 2000, 65, 23–28. [Google Scholar] [CrossRef]
- González-Toril, E.; Gómez, F.; Malki, M.; Amils, R. The isolation and study of acidophilic microorganisms. In Methods in Microbiology; Academic Press: Cambridge, MA, USA, 2006; Volume 35, pp. 471–510. [Google Scholar]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome. Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Forney, L.J.; Zhou, X.; Brown, C.J. Molecular microbial ecology: Land of the one-eyed king. Curr. Opin. Microbiol. 2004, 7, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinje, H.; Liland, K.H.; Almoy, T.; Snipen, L. Comparing K-mer based methods for improved classification of 16S sequences. BMC Bioinform. 2015, 16, 205. [Google Scholar] [CrossRef] [PubMed]
- van Dorst, J.; Bissett, A.; Palmer, A.S.; Brown, M.; Snape, I.; Stark, J.S.; Raymond, B.; McKinlay, J.; Ji, M.; Winsley, T.; et al. Community fingerprinting in a sequencing world. FEMS Microbiol. Ecol. 2014, 89, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.I. Replicate or lie. Env. Microbiol. 2010, 12, 1806–1810. [Google Scholar] [CrossRef]
- Ferraz, M.C.M.A.; Lourençlo, J.C.N. The influence of organic matter content of contaminated soils on the leaching rate of heavy metals. Environ. Prog. 2000, 19, 53–58. [Google Scholar] [CrossRef]
- Uchimiya, M.; Bannon, D.I.; Wartelle, L.H. Retention of Heavy Metals by Carboxyl Functional Groups of Biochars in Small Arms Range Soil. J. Agric. Food Chem. 2012, 60, 1798–1809. [Google Scholar] [CrossRef]
- Adviento-Borbe, M.A.; Doran, J.W.; Drijber, R.A.; Dobermann, A. Soil electrical conductivity and water content affect nitrous oxide and carbon dioxide emissions in intensively managed soils. J. Env. Qual. 2006, 35, 1999–2010. [Google Scholar] [CrossRef]
- Krauss, S.; Schnitzler, W.H.; Grassmann, J.; Woitke, M. The influence of different electrical conductivity values in a simplified recirculating soilless system on inner and outer fruit quality characteristics of tomato. J. Agric. Food Chem. 2006, 54, 441–448. [Google Scholar] [CrossRef]
- Kim, J.M.; Roh, A.S.; Choi, S.C.; Kim, E.J.; Choi, M.T.; Ahn, B.K.; Kim, S.K.; Lee, Y.H.; Joa, J.H.; Kang, S.S.; et al. Soil pH and electrical conductivity are key edaphic factors shaping bacterial communities of greenhouse soils in Korea. J. Microbiol. 2016, 54, 838–845. [Google Scholar] [CrossRef]
- Atekwana, E.A.; Atekwana, E.A.; Werkema, D.D.; Allen, J.P.; Smart, L.A.; Duris, J.W.; Cassidy, D.P.; Sauck, W.A.; Rossbach, S. Evidence for microbial enhanced electrical conductivity in hydrocarbon-contaminated sediments. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Jordán, M.M.; Rincón-Mora, B.; Almendro-Candel, M.B. Heavy metal distribution and electrical conductivity measurements in biosolid pellets. J. Soils Sediments. 2016, 16, 1176–1182. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Env. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Zeng, Q.; Dong, Y.; An, S. Bacterial Community Responses to Soils along a Latitudinal and Vegetation Gradient on the Loess Plateau, China. PLoS ONE 2016, 11, e0152894. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Greening, C.; Carere, C.R.; Rushton-Green, R.; Harold, L.K.; Hards, K.; Taylor, M.C.; Morales, S.E.; Stott, M.B.; Cook, G.M. Persistence of the dominant soil phylum Acidobacteria by trace gas scavenging. Proc. Natl. Acad. Sci. USA 2015, 112, 10497–10502. [Google Scholar] [CrossRef]
- Goltsman, D.S.; Dasari, M.; Thomas, B.C.; Shah, M.B.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. New group in the Leptospirillum clade: Cultivation-independent community genomics, proteomics, and transcriptomics of the new species "Leptospirillum group IV UBA BS". Appl. Env. Microbiol 2013, 79, 5384–5393. [Google Scholar] [CrossRef]
- Diaby, N.; Dold, B.; Pfeifer, H.R.; Holliger, C.; Johnson, D.B.; Hallberg, K.B. Microbial communities in a porphyry copper tailings impoundment and their impact on the geochemical dynamics of the mine waste. Env. Microbiol. 2007, 9, 298–307. [Google Scholar] [CrossRef]
- Robertson, L.A.; Kuenen, J.G. The Genus Thiobacillus. In The Prokaryotes: Volume 5: Proteobacteria: Alpha and Beta Subclasses; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, 2006; pp. 812–827. [Google Scholar]
- Schippers, A.; Breuker, A.; Blazejak, A.; Bosecker, K.; Kock, D.; Wright, T.L. The biogeochemistry and microbiology of sulfidic mine waste and bioleaching dumps and heaps, and novel Fe(II)-oxidizing bacteria. Hydrometallurgy 2010, 104, 342–350. [Google Scholar] [CrossRef]
- Kelly, D.P.; Wood, A.P. Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2000, 50, 511–516. [Google Scholar] [CrossRef]
- Whaley-Martin, K.; Jessen, G.L.; Nelson, T.C.; Mori, J.F.; Apte, S.; Jarolimek, C.; Warren, L.A. The potential role of Halothiobacillus spp. in sulfur oxidation and acid generation in circum-neutral mine tailings reservoirs. Front. Microbiol. 2019, 10, 297. [Google Scholar] [CrossRef]
- Hiraishi, A.; Ueda, Y. Rhodoplanes gen. nov., a new genus of phototrophic bacteria including Rhodopseudomonas rosea as Rhodoplanes roseus comb. nov. and Rhodoplanes elegans sp. nov. Int. Syst. Bacteriol. 1994, 44, 665–673. [Google Scholar] [CrossRef]
- Gómez, P.; Paterson, S.; De Meester, L.; Liu, X.; Lenzi, L.; Sharma, M.D.; McElroy, K.; Buckling, A. Local adaptation of a bacterium is as important as its presence in structuring a natural microbial community. Nat. Commun. 2016, 7, 12453. [Google Scholar] [CrossRef]
- Brooks, A.N.; Turkarslan, S.; Beer, K.D.; Lo, F.Y.; Baliga, N.S. Adaptation of cells to new environments. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 544–561. [Google Scholar] [CrossRef]
- Seki, T.; Matsumoto, A.; Shimada, R.; Inahashi, Y.; Ōmura, S.; Takahashi, Y. Conexibacter arvalis sp. nov., isolated from a cultivated field soil sample. Int. J. Syst. Evol. Microbiol. 2012, 62, 2400–2404. [Google Scholar] [CrossRef]
- Monciardini, P.; Cavaletti, L.; Schumann, P.; Rohde, M.; Donadio, S. Conexibacter woesei gen. nov., sp. nov., a novel representative of a deep evolutionary line of descent within the class Actinobacteria. Int. J. Syst. Evol. Microbiol. 2003, 53, 569–576. [Google Scholar] [CrossRef]
- Deng, J.; Gu, Y.; Zhang, J.; Xue, K.; Qin, Y.; Yuan, M.; Yin, H.; He, Z.; Wu, L.; Schuur, E.A.G.; et al. Shifts of tundra bacterial and archaeal communities along a permafrost thaw gradient in Alaska. Mol. Ecol. 2015, 24, 222–234. [Google Scholar] [CrossRef]
- Krivushin, K.; Kondrashov, F.; Shmakova, L.; Tutukina, M.; Petrovskaya, L.; Rivkina, E. Two Metagenomes from Late Pleistocene Northeast Siberian Permafrost. Genome Announc. 2015, 3, e01380-14. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, S.; Jiang, Q.; Bai, Y.; Shen, G.; Li, S.; Ding, W. Using community analysis to explore bacterial indicators for disease suppression of tobacco bacterial wilt. Sci. Rep. 2016, 6, 36773. [Google Scholar] [CrossRef] [Green Version]
- Kerkhof, L.J.; Dillon, K.P.; Häggblom, M.M.; McGuinness, L.R. Profiling bacterial communities by MinION sequencing of ribosomal operons. Microbiome 2017, 5, 116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample* | Grouping name | No. of analyzed reads | OTU | Chao | Shannon | Simpson | Good’s coverage | Sampling site (GPS, altitude) |
|---|---|---|---|---|---|---|---|---|
| BB1 | BB | 2738 | 388 | 1629.03 | 3.79 | 8.62 | 0.89 | Bongwha (129°3’17.9"E, 36°51′45.6″N, 630 m) |
| BB2 | 2931 | 544 | 2092.42 | 4.65 | 24.99 | 0.87 | ||
| BB3 | 2240 | 355 | 1814.62 | 3.98 | 13.51 | 0.88 | ||
| BB4 | 2404 | 600 | 4411.03 | 4.14 | 5.74 | 0.78 | ||
| BB5 | 7769 | 1492 | 7657.55 | 4.66 | 9.77 | 0.84 | ||
| BF1 | BF | 5677 | 486 | 2820.18 | 2.73 | 3.67 | 0.93 | |
| BF2 | 4606 | 508 | 2393.16 | 4.01 | 12.06 | 0.92 | ||
| BF3 | 4420 | 547 | 2592.36 | 4.07 | 12.18 | 0.91 | ||
| BF4 | 3969 | 743 | 2787.00 | 5.25 | 53.47 | 0.87 | ||
| BF5 | 3784 | 601 | 3295.08 | 4.64 | 24.51 | 0.88 | ||
| DB1 | DB | 4107 | 792 | 2427.76 | 5.03 | 16.91 | 0.87 | Daegu (128°40’18.6"E, 35°46′52.5″N, 243 m) |
| DB2 | 2149 | 424 | 1513.36 | 4.30 | 8.74 | 0.86 | ||
| DB3 | 5250 | 1036 | 4241.84 | 5.09 | 13.70 | 0.86 | ||
| DB4 | 2762 | 551 | 1756.51 | 4.38 | 7.55 | 0.86 | ||
| DB5 | 8284 | 952 | 3784.81 | 4.99 | 40.10 | 0.92 | ||
| DF1 | DF | 5962 | 699 | 3354.82 | 4.17 | 13.56 | 0.91 | |
| DF2 | 5489 | 712 | 2880.12 | 4.15 | 8.73 | 0.91 | ||
| DF3 | 4019 | 711 | 3157.89 | 4.39 | 8.37 | 0.87 | ||
| DF4 | 3880 | 702 | 2803.69 | 4.13 | 5.05 | 0.87 | ||
| DF5 | 3521 | 478 | 1580.64 | 4.69 | 36.54 | 0.91 | ||
| HB1 | HB | 4712 | 2425 | 9038.20 | 7.57 | 627.07 | 0.61 | Hwaseong (126°55’47.6"E, 37°13′09.1″N, 105 m) |
| HB2 | 4634 | 2167 | 7723.74 | 7.30 | 355.46 | 0.65 | ||
| HB3 | 4489 | 2308 | 8079.12 | 7.52 | 557.24 | 0.61 | ||
| HB4 | 3825 | 2057 | 6954.94 | 7.50 | 521.60 | 0.59 | ||
| HB5 | 3360 | 1954 | 7019.94 | 7.60 | 726.93 | 0.55 | ||
| hf1 | HF | 3142 | 1673 | 5914.55 | 7.30 | 539.23 | 0.60 | |
| hf2 | 1662 | 1026 | 4310.03 | 7.09 | 348.38 | 0.51 | ||
| hf3 | 3720 | 2097 | 8645.22 | 7.57 | 607.90 | 0.56 | ||
| hf4 | 1490 | 1032 | 3987.65 | 7.42 | 861.26 | 0.45 | ||
| hf5 | 5707 | 2801 | 10771.69 | 7.62 | 661.09 | 0.63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hur, M.; Park, S.-J. Identification of Microbial Profiles in Heavy-Metal-Contaminated Soil from Full-Length 16S rRNA Reads Sequenced by a PacBio System. Microorganisms 2019, 7, 357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090357
Hur M, Park S-J. Identification of Microbial Profiles in Heavy-Metal-Contaminated Soil from Full-Length 16S rRNA Reads Sequenced by a PacBio System. Microorganisms. 2019; 7(9):357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090357
Chicago/Turabian StyleHur, Moonsuk, and Soo-Je Park. 2019. "Identification of Microbial Profiles in Heavy-Metal-Contaminated Soil from Full-Length 16S rRNA Reads Sequenced by a PacBio System" Microorganisms 7, no. 9: 357. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090357