Sample Preservation and Storage Significantly Impact Taxonomic and Functional Profiles in Metaproteomics Studies of the Human Gut Microbiome

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics
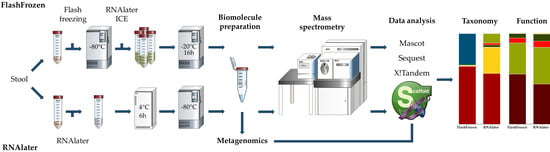
2.2. Sample Collection and Processing
2.3. Metagenomics and Metatranscriptomics
2.4. Prefractionation and Digestion
2.5. High-Pressure Liquid Chromatography and Mass Spectrometry
2.6. Database Searching
2.7. Criteria for Protein Identification
2.8. Further Analyses
3. Results
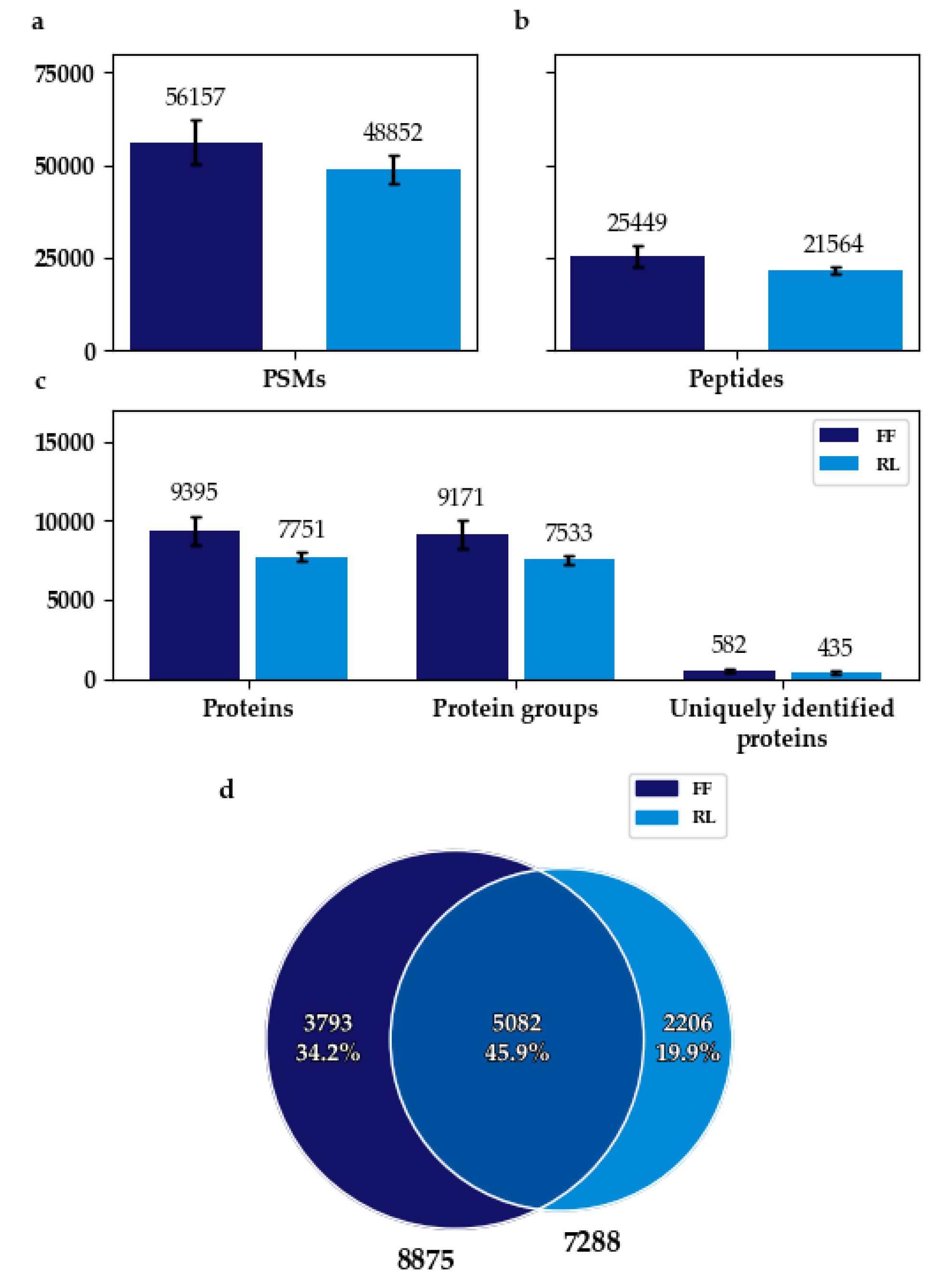
3.1. Flash Frozen Samples Achieved a Higher Protein Identification Rate
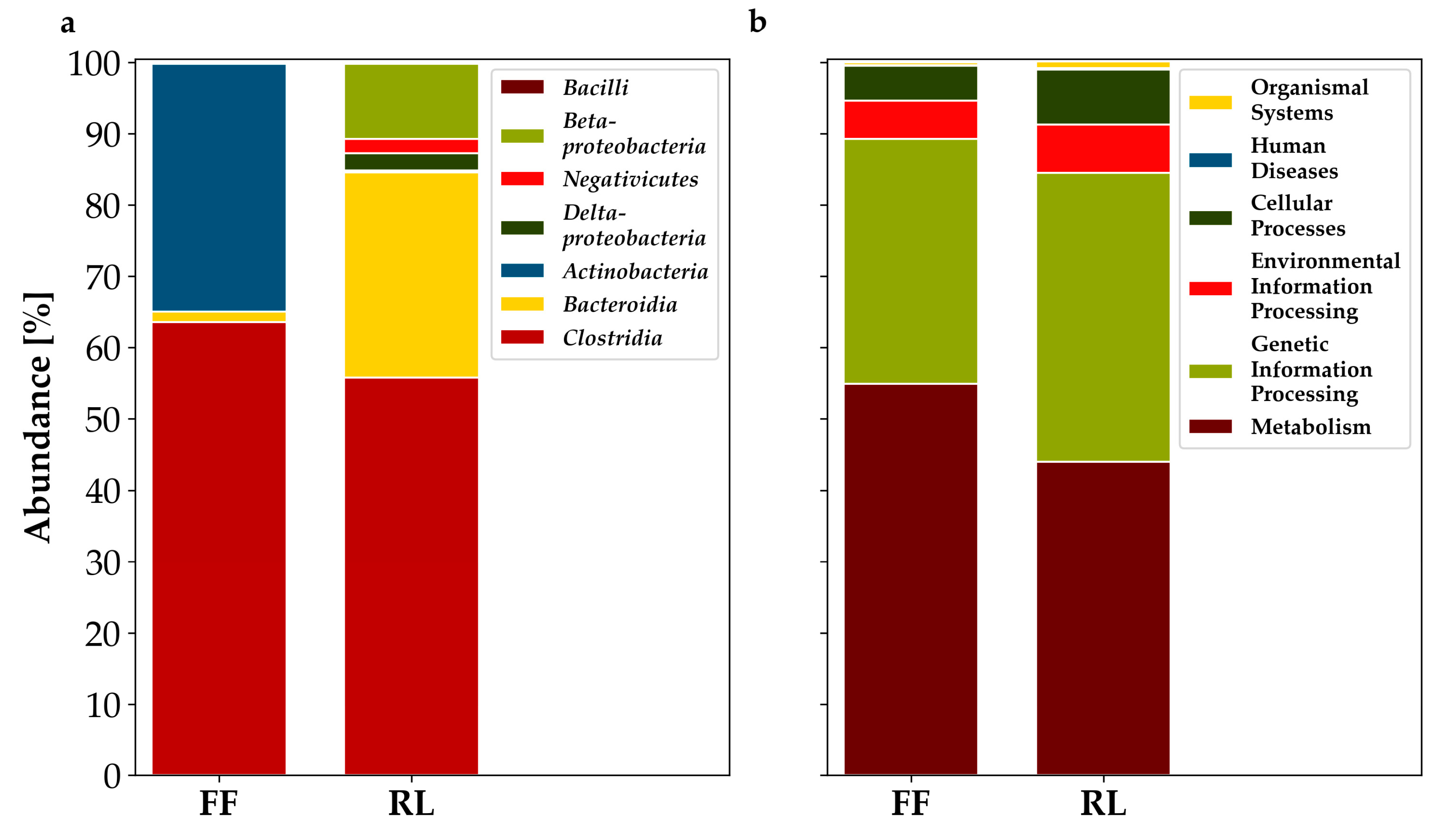
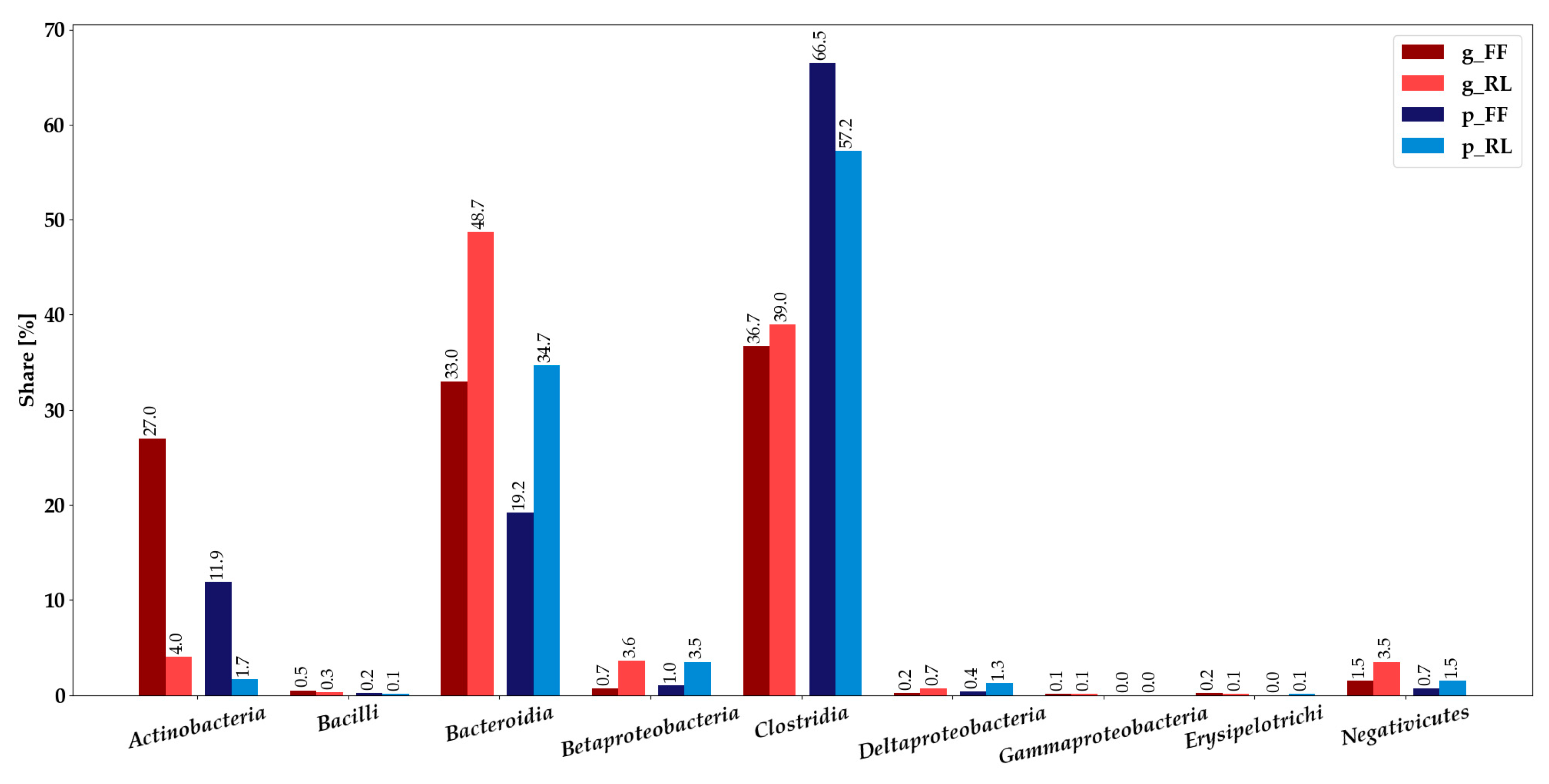
3.2. Metaproteomics-Based Taxonomic Profiles Differed Significantly between Storage Conditions
3.3. Integration of Metagenomics and Metaproteomics Data
3.4. Annotation of Identified Proteins Using Prophane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Bjork, J.R.; Easson, C.; Astudillo-Garcia, C.; Olson, J.B.; Erwin, P.M.; Lopez-Legentil, S.; Luter, H. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016, 7, 11870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.I.; Yatsunenko, T.; Manary, M.J.; Trehan, I.; Mkakosya, R.; Cheng, J.; Kau, A.L.; Rich, S.S.; Concannon, P.; Mychaleckyj, J.C.; et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 2013, 339, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Vazquez-Baeza, Y.; Callewaert, C.; Debelius, J.; Hyde, E.; Marotz, C.; Morton, J.T.; Swafford, A.; Vrbanac, A.; Dorrestein, P.C.; Knight, R. Impacts of the Human Gut Microbiome on Therapeutics. Annu. Rev. Pharm. Toxicol. 2018, 58, 253–270. [Google Scholar] [CrossRef]
- Drissi, F.; Merhej, V.; Angelakis, E.; El Kaoutari, A.; Carriere, F.; Henrissat, B.; Raoult, D. Comparative genomics analysis of Lactobacillus species associated with weight gain or weight protection. Nutr. Diabetes 2014, 4, e109. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L.; Smith, M.; Grange, C.; Watt, E.R.; El-Omar, E.M.; Mukhopadhya, I. Role of the gut microbiota in inflammatory bowel disease pathogenesis: What have we learnt in the past 10 years? World J. Gastroenterol. 2014, 20, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Versini, M.; Jeandel, P.Y.; Bashi, T.; Bizzaro, G.; Blank, M.; Shoenfeld, Y. Unraveling the Hygiene Hypothesis of helminthes and autoimmunity: Origins, pathophysiology, and clinical applications. BMC Med. 2015, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M.; et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Tillisch, K.; Labus, J.; Kilpatrick, L.; Jiang, Z.; Stains, J.; Ebrat, B.; Guyonnet, D.; Legrain-Raspaud, S.; Trotin, B.; Naliboff, B.; et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology 2013, 144, 1394–1401. [Google Scholar] [CrossRef]
- Bhargava, P.; Mowry, E.M. Gut microbiome and multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2014, 14, 492. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Suzuki, K.; Meek, B.; Doi, Y.; Muramatsu, M.; Chiba, T.; Honjo, T.; Fagarasan, S. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. USA 2004, 101, 1981–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanasamy, S.; Muller, E.E.; Sheik, A.R.; Wilmes, P. Integrated omics for the identification of key functionalities in biological wastewater treatment microbial communities. Microb. Biotechnol. 2015, 8, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Benndorf, D.; Kohrs, F.; De Vrieze, J.; Boon, N.; Hoffmann, M.; Rapp, E.; Schluter, A.; Sczyrba, A.; Reichl, U. Proteotyping of biogas plant microbiomes separates biogas plants according to process temperature and reactor type. Biotechnol. Biofuels 2016, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Hassa, J.; Maus, I.; Off, S.; Puhler, A.; Scherer, P.; Klocke, M.; Schluter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Beyond metagenomics, metatranscriptomics illuminates microbiome functionality in IBD. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 193. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Driggers, E.M. Integration of omics: more than the sum of its parts. Cancer Metab. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Bond, P.L. Microbial community proteomics: Elucidating the catalysts and metabolic mechanisms that drive the Earth’s biogeochemical cycles. Curr. Opin. Microbiol. 2009, 12, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, J.A.; Solis, N.; Cordwell, S.J. Beyond gene expression: The impact of protein post-translational modifications in bacteria. J. Proteom. 2014, 97, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Passow, C.N.; Kono, T.J.Y.; Stahl, B.A.; Jaggard, J.B.; Keene, A.C.; McGaugh, S.E. Nonrandom RNAseq gene expression associated with RNAlater and flash freezing storage methods. Mol. Ecol. Resour. 2019, 19, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Deane, J.; Rea, M.C.; O’Sullivan, Ó.; Ross, R.P.; O’Callaghan, G.; Plant, B.J.; Stanton, C. The Effects of Freezing on Faecal Microbiota as Determined Using MiSeq Sequencing and Culture-Based Investigations. PLoS ONE 2015, 10, e0119355. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Tito, R.Y.; Vanleeuwen, R.; Falony, G.; Raes, J. Practical considerations for large-scale gut microbiome studies. FEMS Microbiol. Rev. 2017, 41, S154–S167. [Google Scholar] [CrossRef] [PubMed]
- Menke, S.; Gillingham, M.A.F.; Wilhelm, K.; Sommer, S. Home-Made Cost Effective Preservation Buffer Is a Better Alternative to Commercial Preservation Methods for Microbiome Research. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Amir, A.; Metcalf, J.L.; Amato, K.R.; Xu, Z.Z.; Humphrey, G.; Knight, R. Preservation Methods Differ in Fecal Microbiome Stability, Affecting Suitability for Field Studies. MSystems 2016, 1, e00021-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tap, J.; Cools-Portier, S.; Pavan, S.; Druesne, A.; Öhman, L.; Törnblom, H.; Simren, M.; Derrien, M. Effects of the long-term storage of human fecal microbiota samples collected in RNAlater. Sci. Rep. 2019, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.Y.; Costea, P.I.; Kultima, J.R.; Li, S.S.; Zeller, G.; Sunagawa, S.; Bork, P. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.B.; Kastaniegaard, K.; Padurariu, S.; Gaihede, M.; Birkelund, S.; Andersen, V.; Stensballe, A. Proteome stability analysis of snap frozen, RNAlater preserved, and formalin-fixed paraffin-embedded human colon mucosal biopsies. Data Brief. 2016, 6, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Kim, S.-J.; Lee, S.-E.; Kwon, W.; Kim, H.; Han, Y.; Jang, J.-Y.; Kim, M.-S.; Lee, S.-W. Comprehensive proteome and phosphoproteome profiling shows negligible influence of RNAlater on protein abundance and phosphorylation. Clin. Proteom. 2019, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2016, 2, 16180. [Google Scholar] [CrossRef] [PubMed]
- Roume, H.; Heintz-Buschart, A.; Muller, E.E.L.; Wilmes, P. Chapter Eleven—Sequential Isolation of Metabolites, RNA, DNA, and Proteins from the Same Unique Sample. In Methods in Enzymology; DeLong, E.F., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 219–236. [Google Scholar]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.L.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.C.; Pinel, N.; May, P.; Wilmes, P. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016, 17, 260. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Mende, D.R.; Zeller, G.; Izquierdo-Carrasco, F.; Berger, S.A.; Kultima, J.R.; Coelho, L.P.; Arumugam, M.; Tap, J.; Nielsen, H.B.; et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 2013, 10, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Bonn, F.; Bartel, J.; Buttner, K.; Hecker, M.; Otto, A.; Becher, D. Picking vanished proteins from the void: How to collect and ship/share extremely dilute proteins in a reproducible and highly efficient manner. Anal. Chem 2014, 86, 7421–7427. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2016, 45, D1100–D1106. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. ELECTROPHORESIS 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. PROTEOMICS 2010, 10, 1265–1269. [Google Scholar] [CrossRef]
- Searle, B.C.; Turner, M.; Nesvizhskii, A.I. Improving Sensitivity by Probabilistically Combining Results from Multiple MS/MS Search Methodologies. J. Proteome Res. 2008, 7, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Keiblinger, K.M.; Schmid, E.; Sterflinger-Gleixner, K.; Ellersdorfer, G.; Roschitzki, B.; Richter, A.; Eberl, L.; Zechmeister-Boltenstern, S.; Riedel, K. Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. Isme J. 2012, 6, 1749–1762. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2015, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef]
- Dominianni, C.; Wu, J.; Hayes, R.B.; Ahn, J. Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiol. 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Hale, V.L.; Tan, C.L.; Knight, R.; Amato, K.R. Effect of preservation method on spider monkey (Ateles geoffroyi) fecal microbiota over 8 weeks. J. Microbiol. Methods 2015, 113, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Choo, J.M.; Leong, L.E.X.; Rogers, G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015, 5, 16350. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Yu, G.; Ma, B.; Ravel, J.; Goedert, J.J.; Sinha, R. Collection media and delayed freezing effects on microbial composition of human stool. Microbiome 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, S.-H.; Kuang, Y.-S.; He, J.-R.; Lu, J.-H.; Luo, B.-J.; Jiang, F.-J.; Liu, Y.-Z.; Papasian, C.J.; Xia, H.-M.; et al. Effect of short-term room temperature storage on the microbial community in infant fecal samples. Sci. Rep. 2016, 6, 26648. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Galperin, M.Y. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ. Microbiol. 2013, 15, 2631–2641. [Google Scholar] [CrossRef]
- Vogtmann, E.; Chen, J.; Amir, A.; Shi, J.; Abnet, C.C.; Nelson, H.; Knight, R.; Chia, N.; Sinha, R. Comparison of Collection Methods for Fecal Samples in Microbiome Studies. Am. J. Epidemiol. 2017, 185, 115–123. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hickl, O.; Heintz-Buschart, A.; Trautwein-Schult, A.; Hercog, R.; Bork, P.; Wilmes, P.; Becher, D. Sample Preservation and Storage Significantly Impact Taxonomic and Functional Profiles in Metaproteomics Studies of the Human Gut Microbiome. Microorganisms 2019, 7, 367. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090367
Hickl O, Heintz-Buschart A, Trautwein-Schult A, Hercog R, Bork P, Wilmes P, Becher D. Sample Preservation and Storage Significantly Impact Taxonomic and Functional Profiles in Metaproteomics Studies of the Human Gut Microbiome. Microorganisms. 2019; 7(9):367. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090367
Chicago/Turabian StyleHickl, Oskar, Anna Heintz-Buschart, Anke Trautwein-Schult, Rajna Hercog, Peer Bork, Paul Wilmes, and Dörte Becher. 2019. "Sample Preservation and Storage Significantly Impact Taxonomic and Functional Profiles in Metaproteomics Studies of the Human Gut Microbiome" Microorganisms 7, no. 9: 367. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms7090367