The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems

1
Barcelona Supercomputing Centre (BCS-CNS), Jordi Girona, 29., 08034 Barcelona, Spain
2
Institute for Research in Biomedicine (IRB), The Barcelona Institute of Science and Technology (BIST), 08034 Barcelona, Spain
3
Institució Catalana de Recerca i Estudis Avançats (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Microorganisms 2020, 8(2), 308; https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020308
Submission received: 5 February 2020
/
Revised: 14 February 2020
/
Accepted: 16 February 2020
/
Published: 23 February 2020
(This article belongs to the Special Issue Oral Microbiota in Health and Disease)
Abstract
:The human oral cavity is home to an abundant and diverse microbial community (i.e., the oral microbiome), whose composition and roles in health and disease have been the focus of intense research in recent years. Thanks to developments in sequencing-based approaches, such as 16S ribosomal RNA metabarcoding, whole metagenome shotgun sequencing, or meta-transcriptomics, we now can efficiently explore the diversity and roles of oral microbes, even if unculturable. Recent sequencing-based studies have charted oral ecosystems and how they change due to lifestyle or disease conditions. As studies progress, there is increasing evidence of an important role of the oral microbiome in diverse health conditions, which are not limited to diseases of the oral cavity. This, in turn, opens new avenues for microbiome-based diagnostics and therapeutics that benefit from the easy accessibility of the oral cavity for microbiome monitoring and manipulation. Yet, many challenges remain ahead. In this review, we survey the main sequencing-based methodologies that are currently used to explore the oral microbiome and highlight major findings enabled by these approaches. Finally, we discuss future prospects in the field.
1. Introduction
Much like the various terrestrial biomes that make up the Earth, the human microbiome is a series of distinct communities of bacteria, fungi, viruses, archaea, protists, and other microorganisms, whose compositions are dependent upon environmental conditions [1]. Different sites of the human body can be seen as unique biomes, with drastically different environments and nutrient availabilities, which in turn promote different communities. Yet even within a particular body site, the microbiome composition can be highly variable between individuals in different states of health, with distinct lifestyles, or due to a number of other factors [2]. The focus of this review will be the human oral microbiome, techniques to approaching its analysis, and outlining its typical composition as we currently know it, as well as its deviations under atypical conditions.
The oral cavity contains one of the most diverse and unique communities of microbes in the human body [3,4], yet this niche is relatively understudied as compared to the gut—at the time of writing this review, a PubMed search with “oral microbiome” resulted in 746 articles, as compared to 5605 with “gut microbiome”. A milliliter of saliva contains approximately 108 microbial cells [5], and an array of studies have detected up to 700 distinct prokaryotic taxa [6], with a typical healthy microbiome comprised of a range of about 100 to 200 distinct bacterial organisms [7]. The advent of next generation sequencing (NGS) techniques has opened new avenues for large-scale metagenomic studies in diverse populations, allowing for characterization of the microbiome structure and, in some cases, the functional roles and implications for health.
The mouth as a biome is home to multiple unique habitats, each of which has its own community of microorganisms. The microbiomes of the saliva, tongue, buccal mucosa, teeth surfaces, gums, palate, both subgingival and supragingival plaque, as well as the throat and tonsils, have all been characterized in multiple studies, showing overall similarities but with small-scale differences, such as higher levels of the genus Corynebacterium in both types of plaque [3,8] or higher levels of the phylum Firmicutes in both saliva and buccal mucosa as compared to plaque [8,9]. While some metagenomics studies look at these individual habitats separately, it is also not uncommon to use an oral rinse as a sample collection method, in order to obtain a representative sample of the overall oral microbiome [10,11,12].
Regardless of the particular biome or habitat being explored, the current trend in microbiome studies is largely in taking advantage of culture-independent NGS technologies, as they continue to decrease in both financial and computational cost, alongside the continuous expansions of databases of microbial genetic sequences. According to the expanded Human Oral Microbiome Database (HOMD) [13], only 57% of the oral bacterial species have been officially named, 13% have been cultivated yet remain unnamed, and 30% are uncultivated. Hence, not only do the NGS techniques make analyses relatively quick and easy, but they have also vastly expanded our awareness of unculturable and/or rare microbiota.
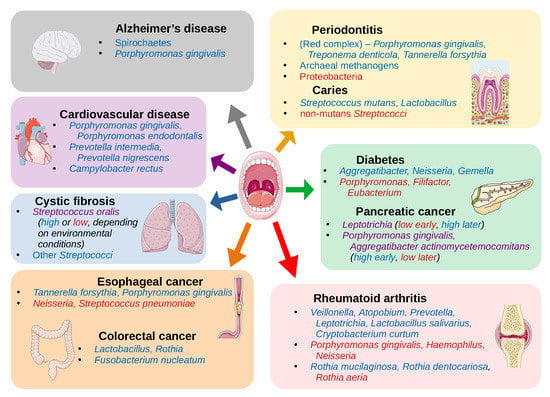
The mouth can be affected by several pathologies that have high prevalence among human populations, including periodontitis, gingivitis, and dental caries, all of which have been clearly related to alterations in the oral microbiome (see references in Table 1). However, the mouth constitutes an entry point to the respiratory and digestive systems, and it is highly vascularized, resulting in potential implications of the oral microbiome in other systemic diseases. Indeed, a growing number of studies have shown associations between other diseases and changes in the oral microbiome (Table 2). This suggests that oral microbiota may provide potential biomarkers in the diagnosis of some systemic diseases.
As we search for these deviations within populations, we must also consider the caveat that our knowledge of the human microbiome may be far from complete. Recent studies have collected data from previously unstudied populations and found not only differences in composition but even added many undiscovered species to public databases [44,45], highlighting strong disparities between different regions of the world. Most studies have been focused on European, North American, Chinese, or other so-called “WEIRD” populations. This refers to Western, educated, industrialized, rich, and democratic nations, an acronym that was proposed originally to denote a bias in psychology studies toward these societies, which at the time comprised about 13% of the world’s population, yet accounted for between 60% and 90% of subjects in psychology studies [46]. This early evidence of a similar bias in microbiome studies suggests that there remains the possibility of a much broader landscape of “healthy” microbiomes across different cultures. Furthermore, even within healthy sample sets of the same populations, distinct subgroups can be elucidated [10,47,48,49,50,51], Therefore, in addition to a focus on understanding the causes and effects of dysbiosis in the microbiome, there must also be a continued emphasis on fully characterizing the healthy microbiome to more reliably detect true deviations from the normal state.
Studies aiming to characterize the composition of the oral microbiome in diverse human populations are progressing rapidly, as are those looking for variation according to a growing number of parameters, particularly those related to health and disease. In this review, we provide a general overview of the state-of-the-art methodologies used to study the oral microbiome, and of the main results obtained during the last decade of intense research. Finally, we will discuss the current challenges and perspectives of this fast-moving field. Throughout the review, we will put a focus on the emerging roles of the oral microbiome in health and disease, and the new opportunities for therapeutics and diagnostics.
2. Technical Approaches to Study the Oral Microbiome
The costs of sequencing DNA have plummeted thanks to the introduction of NGS technologies, now allowing scientists to sequence several human genomes in a single day at a price of under $1000 per genome, a nearly one million-fold decrease from 20 years ago [52]. Similarly, there are a number of cost-efficient NGS techniques that can be used today when approaching microbiome studies, depending on what the researcher hopes to learn (Figure 1). The two most widely used approaches include whole metagenome shotgun sequencing (WMS) and 16S ribosomal RNA amplicon sequencing, both of which involve reading the DNA sequences of the microbes present in a sample and comparing them to a database of sequences to establish the relative quantities of the different organisms present in that sample. In WMS sequencing (Figure 1C), the DNA is randomly fragmented multiple times, allowing millions of short sequences to be read in parallel, and then they are reassembled into full (or partial) genomic sequences by connecting the overlapping ends [53]. However, 16S rRNA sequencing (Figure 1A), also known as 16S barcoding, has been used more frequently in metagenomic studies, since it is less expensive, both experimentally and computationally, permitting larger scale study designs. The 16S rRNA gene is common to all bacteria and archaea, and has highly conserved regions, which make it a useful marker gene for the use of universal primer sequences to isolate it for sequencing. Scattered amongst the conserved regions of the gene are nine hypervariable regions (named V1 through to V9), and it is these segments that allow the taxonomic identification of organisms when mapping reads to a database of known 16S rRNA sequences [54].
Of course, even with the use of ASVs for identification, 16S sequencing still lacks significant taxonomic resolution as compared to WMS sequencing, often only permitting distinction up to the genus level. Alternatives to 16S sequencing have also been proposed in order to improve resolution or to avoid bias due to the varying number of copies of the 16S gene in different species [60] (though there are methods to correct for it [61]). The rpoB gene, for instance, has the advantage of generally being single copy and having greater variation, which allows for deeper taxonomic resolution. However, the corresponding lack of conservation makes it less applicable as a universal marker [62]. A database of rpoB gene sequences is available from the FROGS (Find, Rapidly, Otus with Galaxy Solution) website [63]. Some have proposed that one or more housekeeping genes, like rpoB, should be sequenced along with the 16S gene, since they are ubiquitous and rapidly evolving, allowing for better taxonomic resolution than the 16S gene alone [64]. To distinguish closely related organisms, others have suggested a multilocus sequence analysis (MLSA) approach, wherein multiple housekeeping genes from distinct chromosomal loci are sequenced in parallel [65]. Nevertheless, the 16S rRNA gene remains the current standard for marker gene analyses of the microbiome.
All of the marker gene techniques mentioned are useful when asking the question, “What microorganisms are present in a sample?”, giving an overview of the microbial makeup across many samples. However, WMS sequencing can allow for the detection of species or even strains, in addition to functional annotations of microbiome samples [66], which can only be predicted based on known full genome sequences when performing 16S sequencing. So, WMS additionally gives insight into the functional potential of the microbiome, allowing researchers to ask the question “What can the microorganisms present actually do?”.
Metagenome studies can be further bolstered by the use of metatranscriptomics [67], metaproteomics [68], and metabolomics [69], though only the first of these utilizes NGS technologies. Metatranscriptomics (Figure 1D) answers the question, “what are the microorganisms doing?” Here, the idea is to profile the total microbial gene expression in a sample by capturing the total messenger RNA (mRNA) content, so this is particularly useful when exploring the functional activity of the microbiome in different conditions, like disease vs. health, different diets, or different times of the day. Metaproteomics (Figure 1E) is another approach to assessing the functional activity of a microbiome sample, but instead of sequencing genetic material, the idea is to catalog the abundances of the microbial proteins present in a sample. This is typically done by protein extraction and tandem mass spectrometry analysis (MS/MS) [68]. Metabolomics (Figure 1E), on the other hand, answers the question, “what are the microorganisms producing in a given sample?” The metabolome is the total set of small molecules produced by the microbiome (and the host) in a sample, and can be a strong indicator of the health or dysbiosis of a sample [70]. Metabolites are typically quantified by use of chromatography and detection techniques like mass spectrometry (MS) and nuclear magnetic resonance (NMR). Each of these techniques also has its own drawbacks, which prevent it from being as widely used as metagenomics. Metatranscriptomics can be hindered by the instability of mRNA and the excess of rRNA (though methods have been developed to counteract this [71]), and by the limited reference databases of transcriptomes [72]. Metaproteomics suffers from computational limitations when querying protein databases (which, nevertheless, remain incomplete), as well as a redundancy in annotations due to identical peptides in homologous proteins from different organisms, which may use them in different processes, thereby leaving the resulting taxonomic and functional quantifications ambiguous [68]. Recent tools, however, claim to combat both of these issues [73]. With metabolomics, the challenges lie in determining whether the metabolites are produced by the host or the microbiome, and associating them with the relevant genes and pathways, highlighting the need for the integration of this technique with other omics data [72].
All of these tools and techniques are frequently aimed at investigating the bacteriome, which makes up the most significant portion of the microbiome but not its entirety. To classify the composition of the mycobiome, the fungal component of the microbiome, researchers often use a marker region, much like the 16S rRNA gene, called the internal transcribed spacer region of the nuclear ribosomal RNA cistron, referred to as the ITS region (Figure 1A), which provides a similar taxonomic resolution to that of 16S sequencing for bacteria [74]. The virome, the viral component of the microbiome, can be difficult to approach since there are no conserved marker regions like in the 16S rRNA gene in bacteria or the ITS region in fungi. Thus, the full virome must be sampled and compared to known viral sequences. Two problems arise from this, as current viral databases lack the characterization of many viruses, and consequently, any new viral sequences that do not match closely to those in current databases would be difficult to classify [75]. Another challenge for virome studies is the relatively low proportion of viral nucleic acid content alongside that of other microbes. However, there have been enrichment procedures proposed to increase the content of viral nucleic acids [76]. More recently, a new approach called single virus genomics (SVG) has been proposed (Figure 1B), in which individual viruses are isolated via fluorescence-activated viral sorting (FAVS), and genomic material is amplified and sequenced [77].
Whatever the technique being employed, it is important that researchers come to a consensus on the exact procedure for collection and sequencing to ensure reproducibility. While some of these referenced studies have shown that the microbiome profile of a sample is not heavily influenced by the collection technique [78,79], these are focused on large-scale differences. However, as sequencing technologies become more efficient and microbiome-associated databases become more complete, researchers will continue to compare samples at finer scales, so minor technical variability stemming from different techniques of swabbing collection sites or using a different solution for oral rinse collections could potentially impact results. Some reviews of the current best practices have been published [80,81] and these should continue to be improved and built upon.
Depending on the investigator’s study goals, there is a wide variety of potential approaches to data analysis, and there are equally plentiful software packages available. The phyloseq [82] and microbiome [83] packages for R offer a means to organize the data from sequencing experiments alongside any metadata, and provide a collection of tools and tutorials for calculations and plotting in typical microbiome analyses. This includes functions for calculating the alpha diversity (the relative diversity of taxa present in a sample) and beta diversity (the relative distance between any two samples based on the overall composition, as well as plots for various ordination methods. From there, a bioinformatician can go in any number of directions, so here we will just mention some of the most prominent analyses. The vegan R package [84] offers options for multivariate tests like the anosim function (analysis of similarities) to determine differences in microbiome compositions between groups of samples, and the adonis function for permanova (permutational multivariate analysis of variance), which can apply linear models to determine sources of variation amongst samples. Linear mixed effects models can also be applied to determine the effects on various data points, like particular taxa, diversity levels, or other metadata variables. Standard or generalized linear models can be fitted using the lm or glm functions, respectively, from the core R package called stats [85]. Mixed effects models can also be used with the lmer and glmer functions from the lme4 package [86]. There are also software options available to predict the functional capability of a microbiome sample sequenced with a marker gene target like 16S rRNA. The tools PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states) [87] and Tax4Fun [88] use reference genome databases to attempt to reconstruct full metagenomes from each sample. Machine learning techniques have also been implemented to attempt to predict disease based on the microbiome composition [89], and some investigators have made their code publicly available [90,91]. The oral microbiome has even been used in a classifier for colorectal cancer with some success [92].
A recent point of contention in the field is that of the compositional nature of microbiome data and the implication for its analyses, something which some have begun to address from both an experimental [93] and a statistical [94] perspective. Since the reads produced in an NGS experiment are essentially random samples of the relative abundances of the organisms present, this cannot account for the implications of differences in the total abundances of organisms, which may be physiologically relevant. Vandeputte et al. described an experimental technique called quantitative microbiome profiling (QMP) [93] in which the total microbial load is determined by sampling an equal number of sequences per sample, and then correcting for the 16S copy number bias and the total number of cells from the sample. They were able to use this approach to reveal erroneous results from a standard experiment, suggesting a solution for some technical biases inherent in NGS studies. However, since most datasets to date have not been sequenced with any techniques like QMP, other solutions have been proposed to handle the typical asymmetrical datasets in microbiome studies. Rarefaction of read counts, in which a random sample of the same number of reads is extracted from each sample, was a common approach in earlier microbiome studies, but this practice has been discouraged as it omits biologically relevant information [95]. Gloor et al. instead suggested normalizing the data using a centered log-ratio transformation, which is minimally affected by the depth of reads for a sample [94]. They also provided a tutorial for the workflow that accompanies their publication on this topic.
3. The Oral Cavity and its Microbial Niches
Of all the habitats within the human body for which the microbiome is typically studied, the oral cavity warrants perhaps the most unique approach to study, in that it contains a number of highly distinct niches formed at the various surfaces within the mouth. Changes in the availability of oxygen, nutrients, and the pH-mediating effect of saliva [96] can promote the growth of different organisms, and conversely, these organisms can be involved in their own small niche construction [97] via biofilm formation and nutrient metabolism, which can produce effects both within the oral cavity (Table 1, Figure 2) and systemically (Table 2, Figure 2). Some researchers have chosen to study all these niches in parallel to compare them against each other [3,8,9], some have selected individual sites with particular focuses on localized dysbiosis in disease states [98,99,100,101,102,103,104,105,106], and others have used an oral rinse approach to capture an overall view of the oral cavity [10,11,12].
Studies focusing on specific oral niches typically aim to explore a disease relevant to that site. For instance, primary Sjögren’s syndrome (pSS) in the buccal mucosa was believed to be a potential reservoir for pathogens implicit in the disease, wherein the disease samples were shown to have higher Firmicutes/Proteobacteria ratios as compared to healthy controls, and higher abundances of 19 genera [98]. Various efforts have characterized the changes in subgingival and supragingival plaque related to periodontitis [99,100,101,102], as well as a study of subgingival plaque and buccal mucosa showing that both sites differed between periodontitis samples and healthy controls, with many of the same organisms affected in both sites, though they also displayed unique species colonization [103]. The tongue microbiome was explored in the elderly in Japan because of a potential connection between ingested microbes and pneumonia, which found that samples with worse dental health were enriched in pneumonia-associated bacteria [104]. The tongue was also targeted as a potential segment in diagnostic tools that would perhaps incorporate the microbiomes of the full gastrointestinal tract to detect pancreatic cancer [105]. The palatine tonsils were explored in HIV-infected patients to better understand the oral and systemic complications of the disease, and it was shown that the bacteriome was indeed significantly altered in infected individuals, but the mycobiome was not [106].
At a broad scale, the microbial composition throughout the regions of the oral cavity is fairly consistent, making it easily distinguishable from the microbiomes of other human body habitats [3,107,108,109,110]. However, while the niches in the oral cavity are largely composed of the same organisms, some may be present in different proportions. One study combining samples from 10 niches along the digestive tract in over 200 individuals from the United States placed these niches into four groups based on the similarity of overall composition [8]. One of the sites was the intestine, represented by stool samples, which were grouped alone, while the other nine were in the mouth and throat. One of the three other groups consisted of buccal mucosa, keratinized gingiva, and hard palate, another of tongue, saliva, palatine tonsils, and throat, while the last group contained subgingival and supragingival plaques. Though all of the non-stool niches were generally dominated by the phyla Firmicutes and Bacteroidetes, they based the groups more on small-scale differences. The first group was more unique than the other two non-stool groups, and was shown to have a considerably higher abundances of the genus Streptococcus and lower overall alpha diversity, which is a measure of the relative diversity of organisms present in a given sample. Group 3, containing the two types of gingival plaque, typically had higher alpha diversity. Comparisons between the compositions of these niches and their diversities have been corroborated in other studies [3,9,111]. The authors posit that the level of saliva flow in the mouth is a key factor determining the composition of the microbiome at each niche in the oral habitat because of its capacity to regulate pH and nutrient availability, but other major factors may include the type of surface and oxygen availability. The two plaques, for instance, form on the non-shedding surfaces of teeth where they produce biofilms, within which oxygen is limited, resulting in greater abundances of obligate anaerobic organisms in the subgingival plaque and of facultative anaerobic organisms in the supragingival plaque [8]. They suggest that one niche from each group could be used to represent all of the niches in that group, such as using the buccal mucosa microbiome as a proxy for both keratinized gingiva and hard palate microbiomes (group 1 niches). However, as sequencing techniques continue to improve and costs continue to decrease, we may find more and more subtle idiosyncrasies within each niche, due to combinations of the microenvironmental factors just mentioned, as well as any others. So, that decision will be up to the discretion of the researchers and the relevance to their studies.
Since these niches tend to have very similar overall microbiome compositions at all but the lowest taxonomic levels, many researchers choose to treat the oral cavity as an individual habitat and analyze global compositions and processes therein [10,12,112]. A few studies have shown that this is a viable and efficient method for sample collection to investigate the oral microbiome [78,79], and standardized procedures have been proposed [113,114]. Essentially, an individual would refrain from eating, drinking, brushing, or smoking (anything that might temporarily shift the typical microbial composition) for at least 30 min prior to collection. Then, they would swish with a buffer solution for about 30−60 s, and then spit the contents into a tube, which would later be centrifuged and sequenced. The practical benefits of using an oral rinse are the ease of collection, as it is a quick and non-invasive method to obtain oral microbial DNA from a study participant, as well as the ease of storage and transport, since these samples can be frozen and sequenced later without detriment to the quality of the samples [79,115]. This is advantageous for large-scale microbiome projects, as it allows for the collection of many samples, which may take weeks or months, that can later be sequenced together to minimize the potential technical bias inherent in sequencing projects, as mentioned in the previous section.
4. The Healthy Oral Microbiome and Definition of Stomatotypes
The field of microbiome research is arguably still in its infancy, as evidenced by the continuing efforts to expand the databases on known microbial genomes, and to combat the bias toward “WEIRD” populations, as mentioned in the introduction [44,45,116]. These factors alone make it difficult to effectively define what constitutes a “healthy” oral microbiome. However, on top of that, the accumulation of studies over the last decade or so have shown that, even within particular populations, there can potentially be multiple distinct trends of microbiome composition amongst individuals in relative general health [10,48,49,50,51]. As such, while we may be eager to investigate the microbiome’s relationship to disease, it is vital that we also continue to further define microbiome compositions in health among different populations, and explore the causes of shifts within or between these populations.
Projects based on oral rinse samples are ideal for observing how the microbiome of the oral habitat as a whole is affected by external factors. Of course, the phrase “external factors” could cover a wide spectrum of variables, but some with clear connections to the mouth are the water we drink and the food we eat. In a cohort of 1319 samples from healthy adolescents in Spain analyzed by 16S rRNA sequencing, it was shown that differences in the ionic composition of public drinking water was associated with shifts in the overall composition of the oral microbiome [10]. Samples from regions with greater alkalinity and greater levels of ions, such as sulfate (SO4) and sodium (Na), had higher abundances of genera, such as Porphyromonas and Flavobacterium, while regions with lower levels showed higher abundances of other genera, including Veillonella, Pseudomonas, and Ralstonia. Different diets have also been shown to contribute to variations in the microbiome composition, such as in the WMS-based study comparing the oral microbiomes from populations of hunter-gatherers (HGs) from the Philippines, traditional farmers (TFs) from the Philippines, and Western controls (WCs) from the Human Microbiome Project (samples from the United States) [116]. They showed that the HG samples had higher alpha-diversity while it was lower in WC samples, and TF samples fell in the middle. Likewise, there was a strong gradient in the abundances of the core oral genera Neisseria and Haemophilus, with high levels of Neisseria and low levels of Haemophilus in HG samples, the reverse in WC samples, and TF samples again falling in between. The HG samples, despite good oral health, also displayed higher abundances of a number of species typically considered to be oral pathogens associated with gingivitis and periodontitis by Western standards. Functional analyses revealed an increase in vitamin B5 biosynthesis pathways in HG samples and, to a lesser extent, in TF samples. Americans had been shown to consume greater quantities of foods with vitamin B5, so the authors posit that this lack in hunter-gatherer diets would select for organisms that synthesize it on their own. Conversely, they showed that WC samples, and to a lesser extent, TF samples were enriched in urease activity, particularly from Haemophilus spp. This urease counteracts the drops in pH that occur when bacteria degrade sugars into acidic compounds, so selecting for these organisms in WC samples, with their sugar- and starch-heavy diets makes sense. The authors thus suggest that organisms considered to be oral pathogens in Western populations could indeed be part of the healthy microbiomes of different populations like hunter-gatherer societies, and that pathogenic strains of these organisms would be selected based on the nutrient availability tied to diet.
Food and water are obvious influencing factors, but any number of other factors could also impact the microbiome. A common approach in the early stages of the analysis of the microbiome of any body habitat is to first look at the broad effects of such factors by clustering the samples based on the overall microbial composition. The notion of separating samples into clusters was discussed in an early NGS-based study on the gut microbiome, wherein the authors labeled the clusters “enterotypes”, implying the presence of different putative categories of gut microbial composition [47]. They found three distinct enterotypes, the separations of which were driven largely by differences in the abundances of particular organisms, namely the genera Bacteroides, Prevotella, and Ruminococcus, and corroborated these by finding very similar enterotypes from two separate sample sets. From this, they suggested that there may exist some limited number of equilibria of symbiotic states between a human host and its microbiome, which would arise due to different diets and lifestyles.
Many studies have since adopted this technique, and in studies of the oral cavity, similarly composed clusters of samples have also emerged, dubbed “stomatotypes” in one such study, as homage to the original term enterotype, but in reference to the mouth [10]. A summary of some of the genera of bacteria that have been found to co-occur in different stomatotypes across studies is found in Table 3. There have thus been shown at least two strongly corroborated stomatotypes, one including higher abundances of the Proteobacteria genera Neisseria and Haemophilus, and the other with higher abundances of the Bacteroidetes genus Prevotella and the Firmicutes genus Veillonella. Some studies have shown more than just these two stomatotypes, though the consensus of compositions is more varied. Some of the genera co-occur in different manners, depending on the study.
The explicit nature of stomatotypes may be very appealing as a means of differentiating samples, but these discrepancies highlight two major concerns. The first stems from the fragmentary nature of our current understanding of the microbiome due to the cultural biases and technical limitations already discussed. We do not yet have a complete picture of what may constitute the various potential equilibria of microbial abundances that lead to a particular stomatotype, because we have not explored the healthy microbiomes of many unique populations across the world, and many of the studies we already have lack the resolution to explain fine-scale distinctions between samples. As mentioned above, the hunter-gatherer populations from the Philippines that were sampled were enriched in Neisseria spp. while the Western controls were enriched in Haemophilus spp. [116]. This might suggest that samples from both populations would be clustered into stomatotype 1 from Table 3, since these two genera drive the equilibria of stomatotype 1, typically together but not in all studies [49]. Meanwhile, Bacteroidetes (the phylum containing Prevotella) and Veillonella, the drivers of stomatotype 2, fell more in the middle of the gradient between the HG and WC equilibria, further potential evidence that HG and WC may group together in stomatotype 1. How could one reconcile the strong differences displayed between these two populations and the evidence so far presented for the compositions within stomatotypes? This could likely be partially explained by the “WEIRD” bias in the studies presenting stomatotypes—perhaps these equilibria emerge in populations with westernized diets, medical treatments, and lifestyles. However, the question also ties into the second major concern, which is the statistical relevance of separating samples into discrete clusters. The notion of analyzing the gradients of microbial abundances was proposed as a response to the enterotype concept [117]. This has led to further discussions of the merits of enterotypes/stomatotypes, cautioning their use as predictive or diagnostic tools [118], and also suggesting improvements to their calculations while further emphasizing a focus on the gradients of abundance [107,119]. See Figure 3 for an example of the different gradients of the abundances of organisms in Table 3, which have been shown as stomatotype drivers, and how they associate with the stomatotypes found in a random subset of 500 samples from an oral microbiome dataset [10]. These, and other studies, suggest that stomatotypes are useful as a first step in exploring underlying variation among samples, which can then be further investigated through deeper analysis of shifts in particular organisms. For instance, in the study of adolescents in Spain, they drew connections to tap water composition by first observing maps of the distributions of samples in the two stomatotypes that they found, which were reminiscent of maps of water hardness values across Spain, and then later began to look at the effects on particular organisms within their samples [10].
5. Non-Bacterial Oral Microbes
Bacteria dominate both the research about the human oral microbiome, and the biomass within the oral habitat, with fungi estimated to comprise <0.1% [120]. Nevertheless, there is an appreciable diversity of fungal species present in the oral cavity, including species from the genera Candida, Aspergillus, Penicillium, Schizophyllum, Rhodotorula, and Gibberella [121]. Yet, two primary complications have limited the exploration of the mycobiome: (1) Difficulty in identifying many fungal species and (2) confusion in fungal nomenclature. Both of these issues have begun to be addressed in large part by the use of NGS technologies. Until recently, the diversity within the oral mycobiome was believed to be quite limited, dominated primarily by a few species of Candida [122]. This was largely because many fungi are difficult to cultivate in a laboratory, but advances in NGS technologies have revealed a wider array of fungal organisms than previously expected. One study found that the genus Malassezia was highly prevalent in the mouth [123] but had previously gone undetected in this body site because it has particular lipid requirements and needs specialized culture media to grow in a lab, and it was previously believed to be a pathogen on the skin [124]. However, even within metagenomic studies, there may be complications in categorizing the true fungal diversity. For instance, a study re-analyzing the samples in which Malassezia was detected was not able to find this genus in any of the samples [125], but it is possibly because this second study did not use the same DNA extraction protocol as the first, which included a step that used beads to help break cell walls and capsules. This highlights an inconsistency in the protocols for fungal metagenomic studies, and the need for standardization.
Independent of the technical concerns surrounding the collection and categorization of genetic material in fungal studies, there is also some ambiguity in the classifications of fungi. For instance, Malassezia are dimorphic, with both yeast and mycelial phases, and in the past had been placed in multiple genera [123]. The authors claim that, while the taxonomy for this particular genus has largely been resolved, older studies may miss this information, and this issue may also occur for other fungal organisms. Within the last decade, there was a push to end the system of dual nomenclature, as this approach came to be seen as archaic, and a single name classification has since begun to be adopted [126]. As fungal taxonomy continues to be expanded, NGS-based studies contribute greatly to the identification of new species, both with ITS-amplicon [74] and shotgun metagenomic techniques [127].
Complications in technical approaches and in classification have led to scarce investigations of the oral virome, but we can begin to draw a few conclusions from some of the recent work in this area. This is an important segment of microbiome research because not only can eukaryotic viruses affect the health of a host directly, but prokaryotic viruses can do so as well by altering the overall bacteriome composition and thus its function [75]. A study in Spain using single-virus genomics (SVG) and viral metagenomics in 15 saliva samples found 439 oral viruses, which they grouped into about 200 clusters that corresponded to genus-level classification [128]. They saw that most viruses were not consistently predominant, and it was difficult to define a core group of salivary viruses, and instead there were variable interpersonal compositions in the oral virome. However, 26 of their 200 viral clusters shared many genes, and most of these were Streptococcus phages, which is a reasonable finding since, as we have seen, Streptococcus is typically among the most abundant genera of oral bacteria, if not the most abundant, in Western oral microbiomes. Another study, also in Spain, of 72 healthy adult oral viromes had similar findings [129]. They found very few ubiquitous viruses while most were found only in individual samples, and once again Streptococcus phages were common. However, they did suggest a small core of oral viruses and pointed to the presence of viral cores in other body sites seen in other studies, including the lung in healthy samples [130], the gut even after fecal transplant [28], and the skin [29]. They also stress the specificity of this oral viral core to Western cultures, since virome research also suffers from the “WEIRD” bias mentioned above.
Protozoa and archaea are also components of the oral microbiome, though little has been said on either group. There do not appear to be any NGS-based explorations of oral protozoa, but instead they have been identified by microscopy techniques [30,131,132,133]. However, the presence of the 16S rRNA gene in archaea has led to the use of NGS techniques in some studies. All of the archaeal species thus far discovered in the oral cavity are methanogens (methane-producing organisms) of the phylum Euryarchaeota [134]. It has been shown that these archaea tend to be present at higher abundances in patients suffering from periodontitis [14,15]. However, it has been suggested that there may be more archaeal diversity that has as yet gone undetected, either because conventional methods have precluded the detection of other archaea, because they occur at low prevalence and abundance, or because of a lack of diversity in the populations sampled [135]. Each of these issues could be addressed with further explorations of NGS-based studies among diverse populations.
6. Oral Microbiome and Oral Diseases
The plant ecologist Robert Harding Whittaker, in defining terrestrial biomes in the 1970s, discussed gradients of environmental conditions ranging from favorable to extreme. He showed that both alpha and beta diversities decrease as biome conditions become more extreme. [136]. A parallel to this generalization has been seen with microbiome studies over the last decade if we consider that disease states equate to “extreme” environmental conditions within certain body sites. This frequently causes low alpha diversity (fewer distinct organisms), which leads to low beta diversity (uniqueness of an individual sample’s overall composition) as certain organisms become better equipped to dominate their habitat. Common diseases of the oral cavity, like periodontitis and dental caries, provide explicit examples of this phenomenon, wherein the microbiome composition is strongly tied to the disease state. However, at this stage in the development of the microbiome field, it is not always clear whether changes in microbial compositions lead to disease, or vice versa. Nevertheless, it is certainly worth discussing the associations that have been observed to begin to postulate microbiome-related mechanisms of disease origin or progression.
The species of the “red complex” (Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia) have historically been seen as the primary infective organisms implicated in periodontitis [137], but this was determined by culture-based studies, which thus missed much of the bacterial diversity present in samples. NGS techniques have since revealed other organisms that are also associated with periodontitis (Table 1, Figure 2), such as the classes Clostridia, Negativicutes, and Erysipelotrichia [16]; the genera Synergistes [17], Prevotella, and Fusobacterium [18]; and the species Filifactor alocis [16]; as well as the archaeal species Methanobrevibacter oralis, Methanobacterium curvum/congolense, and Methanosarcina mazeii [14,15]. Conversely, some organisms are associated with periodontal health, including the phylum Proteobacteria and the Firmicutes class Bacilli [16], and the genera Streptococcus, Actinomyces, and Granulicatella [19].
Clearly, there are many organisms associated in one way or another with periodontitis, but that raises the question of which may actually be causative agents, and which are merely impacted by environmental alterations in the disease state. One study that employed metatranscriptomics techniques compared the expression profiles of 160,000 genes and showed conserved differences in metabolism, despite variation in microbiome composition, suggesting that, in a disease state, the organisms present in a sample perform similar functions, even if species differ between samples [20]. This notion could be corroborated by another study, which proposed that methanogenic archaeal species develop syntrophic relationships by acting as “hydrogen sinks”, allowing for increased growth of pathogenic secondary fermenters. Members of the genus Treponema have a similar hydrogen-consuming activity, perhaps explaining their involvement in the “red complex”. Indeed, this study showed that abundances of Treponema and methanogenic archaea anti-correlated, suggesting that they may fill the same functional niche [15].
In dental caries, alpha diversity has been shown to diminish as the disease progresses and the species Streptococcus mutans has been found at high levels at early stages of caries development but not at later stages while other species of Streptococcus are associated with dental health [22]. It has been suggested that, while S. mutans is acidogenic and this may contribute to initial caries formation, other oral taxa are also acidogenic. The significant virulent factor in this situation is its ability to metabolize sucrose from a host’s diet into extracellular polysaccharides (EPS), which are necessary to produce cariogenic biofilms. Furthermore, adhesion between S. mutans and Candida albicans is promoted in this setting, with C. albicans providing additional acidogenesis [23].
Connections between the microbiome and cancer have also been explored recently. A number of species found in the oral cavity have been associated with oral cancer, including Capnocytophaga gingivalis, Prevotella melaninogenica, and Streptococcus mitis [24]. Table 1 lists a number of species that have been described as having associations with oral cancer, though there is the caveat that the samples in one of these studies were taken from tumor and non-tumor sites in the same patient, and thus the composition at both sites may in fact be affected by the disease, and it is unclear exactly what, if any, cancer-related treatment the patients have undergone [25]. Unfortunately, there do not appear to be many studies exploring this connection with modern techniques, nor is there much consensus among such studies on the microbiome composition in the presence of oral cancer, but there are a number of hypotheses on the potential cancer-promoting action of microbiota. It has been suggested that some of the normal oral bacteria, including Streptococcus salivarius, Streptococcus intermedius, and Streptococcus mitis, can convert ethanol to the carcinogen acetaldehyde [26,138] or upregulate cytokines and other proinflammatory molecules, leading to chronic inflammation that may be involved in carcinogenesis [139], and that bacterial toxins may also affect cell signaling pathways or damage DNA [140].
Esophageal cancer has also been explored, with associations seen with the periodontal pathogens Tannerella forsythia and Porphyromonas gingivalis (members of the “red complex”) [27]. The study showed that the genus Neisseria was linked to a lower risk of esophageal cancer, as was the carotenoid biosynthesis pathway, to which a number of Neisseria species can potentially contribute.
7. Oral Microbiome and Non-Oral Diseases
The microbiome of the oral cavity is by no means an isolated biome, but it is instead part of a highly interconnected series of microbiomes across the human body, forming a sort of micro-biosphere. As the entry point of nearly all ingested material, and due to its high vascularity, the oral cavity has ample opportunity to influence activity at other body sites. So, it is no surprise that, in addition to diseases of the oral cavity, the oral microbiome has been implicated in a number of systemic diseases.
The mouth is a direct route to both the lungs and the digestive system, so an association between oral taxa and disorders like cystic fibrosis (CF) [33] or colorectal cancer (CRC) [12,141,142,143] can perhaps be expected, given what we have already discussed. The pathogenicity of Pseudomonas aeruginosa, the primary agent in biofilm formation in the lungs of CF patients, can be inhibited by oral commensal streptococcal species, particularly Streptococcus oralis, through the production of hydrogen peroxide, which can disrupt biofilm production, but this was only observed if these streptococci were primary colonizers before the introduction of P. aeruginosa. However, in a typical CF lung environment, these streptococcal species actually stimulate the production of P. aeruginosa virulence factors, including elastase and pyocyanin [33].
Dysbiosis in the oral cavity resulting in periodontitis has been linked with oral, esophageal, gastric, lung, pancreatic, prostate, hematologic, and breast cancers [144,145], amongst others. Hypotheses for these connections include: Production of carcinogenic molecules like nitrosamines by nitrate-reducing taxa [146,147] or acetaldehyde by ethanol-metabolizing taxa [26,138], increased abundance of cancer-linked viruses like cytomegalovirus and Epstein–Barr virus [148,149], and, perhaps most prominently, increased proinflammatory markers stemming from immune reactions to periodontal disease like cytokines [139] and the receptor for advanced glycation end products (RAGE) [150].
Multiple studies have linked Fusobacterium nucleatum with CRC [141,142,143], as this is an oral commensal species that is highly invasive and adherent [151] and appears in adenoma samples of CRC patients. In adenoma cases, it was correlated with local inflammation, TNF-α, and IL-10 [141]. However, a recent NGS-based study found no association between Fusobacterium and CRC, but instead saw associations with the genera Lactobacillus and Rothia [12]. The authors suggest that the results of some other studies may actually be confounded by smoking, which has been shown to associate with F. nucleatum abundance in the mouth as well [152,153]. They also posit that, while Lactobacillus has been suggested as a probiotic when present in the gut microbiome [154,155], as seen in the previous section (see Table 1, Figure 2), this genus is associated with dental caries in the oral microbiome. Thus, Lactobacillus may not have a direct impact on colorectal carcinogenesis, but it could perhaps be an ancillary indicator of poor oral health, which we have seen is strongly linked to cancer.
Another study showed higher abundances of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in pancreatic cancer samples [31], both of which are keystone pathogens in periodontitis [18,21] As supporting evidence, they referenced a study that found that risk for pancreatic cancer was significantly increased in the presence of elevated serum antibodies to P. gingivalis [156], and another showing that both P. gingivalis and A. actinomycetemcomitans have the potential to initiate Toll-like receptor (TLR) pathways, which has been shown to be a driver of pancreatic carcinogenesis [157]. However, another study seems to contradict the first, finding a greater abundance of Leptotrichia in pancreatic cancer samples compared to healthy controls, and lower abundance of Porphyromonas, as well as lower abundance of Aggregatibacter (though the latter was not statistically significant) [32]. They suggest that a high ratio of Leptotrichia to Porphyromonas (LP ratio) is a biomarker for pancreatic cancer. In fact, they were able to reclassify one patient that was originally a healthy control but diagnosed with an unknown digestive disease. The individual’s high LP ratio prompted a re-evaluation that led to diagnosis of pancreatic cancer in that patient.
Combining the evidence presented in both studies may lead to a solution for the incongruous findings. Both reference the link between P. gingivalis antibodies and pancreatic cancer, so this may be due to the high abundance of this species prior to onset of the disease, leading to antibody production and eventual diminishing abundances. High initial P. gingivalis abundance would then potentially be linked to periodontal disease, which may then lead to various cancers as a result of systemic inflammation or any of the other mechanisms discussed here. Competition between Leptotrichia and Porphyromonas would explain their anti-correlation, so that as antibodies reduce the abundance of oral P. gingivalis, Leptotrichia is able to thrive. In fact, in the second study, some of the pancreatic cancer samples had low LP ratios, on par with non-pancreatic cancer LP ratios, so these may then have been early stage cases. This situation highlights a need for a greater focus on the temporal dynamics of the microbiome in these kinds of association studies to discover important factors across the onset, progression, and maintenance of disease states.
Some other systemic disorders are linked with periodontitis as well, like cardiovascular disease/atherosclerosis [34,35]. One study following over 3000 subjects for a 16-year period found that periodontitis with the loss of molars was linked with breast cancer (among other types), and premature death due to cancer and cardiovascular or gastrointestinal diseases [158]. As with the cancers that are linked to periodontitis, the resulting increase in systemic inflammation is a primary explanation for the link with cardiovascular disease [159]. This is largely due to the invasive nature of some of the associated taxa, like P. gingivalis, which also promotes invasiveness into host epithelial cells in species like Prevotella intermedia, an otherwise commensal oral species [160]. The proteins secreted by these organisms are implicated in their pathogenicity, such as gingipains from P. gingivalis, which aid in its biofilm formation during periodontitis [161], and subsequently activate cytokine production [162]. Studies linking periodontitis and atherosclerosis have relied on findings of oral bacteria colonizing atherosclerotic plaques [34,35] rather than on abundances of taxa in the oral cavity (Table 2, Figure 2). Although there appear to be fewer studies using NGS techniques of oral cavity samples to determine the associations between the oral microbiome composition and cardiovascular disease, the potential mechanisms of action by oral taxa make this another attractive area for investigation.
Rheumatoid arthritis (RA) has often been connected with periodontitis [36,163], once again implicating P. gingivalis, in this case for its production of gingipains and peptidylarginine deiminase, which enable protein citrullination, an important trigger for RA. However, recently it was shown that this direct connection may be erroneous [164]. In fact, studies of RA using 16S sequencing and whole metagenome shotgun sequencing, respectively, found that P. gingivalis was either not associated with RA [37] or actually more abundant in healthy controls compared to RA samples [38] while different organisms were associated with the disease (Table 2, Figure 2). This demonstrates how NGS studies can allow researchers to determine the veracity of previously held beliefs and potentially open new pathways for investigation.
Neurological disorders have also been associated with the oral microbiome. Perhaps the most complete study in this regard is the association of P. gingivalis with Alzheimer’s disease [40]. The authors of this study not only identified this bacterium in the brains of Alzheimer’s patients at levels that correlated with tau and ubiquitin aggregates (a hallmark of the disease), but also showed that P. gingivalis infection in mice resulted in brain colonization and increased the production of components of amyloid plaques. They went on to show that the gingipains proteases produced by P. gingivalis are neurotoxic and inhibit tau function. This suggests a direct connection between baring colonization of P. gingivalis and the origin or progression of Alzheimer’s disease and also suggests that gingipain inhibitors could be used to treat neurodegeneration in this disease. It has also been shown that typical oral species of the phylum Spirochaetes, including multiple species of the genus Treponema, often make up amyloid plaques, and that these organisms are capable of producing additional amyloid-β and amyloid-β protein precursor [41].
Dysbiosis of the oral microbiome is also implicated in disorders of the endocrine system. In this case, periodontitis appears to be a potential result of diabetes, as opposed to a potential cause as in most of the diseases discussed above. In one longitudinal study, the prevalence of periodontitis was 60% in subjects with diabetes and 36% in subjects without diabetes [165]. Some of the proposed mechanisms of the influence of diabetes on periodontal health include microangiopathy or alterations in the inflammatory response, collagen metabolism, or the glucose concentrations in gingival crevicular fluid [166]. Nevertheless, it has been proposed that periodontitis can also compound the effects of diabetes by upregulating the production of inflammatory factors like TNF-α, which can act as insulin antagonists [167]. Surprisingly, one NGS study showed that subjects with diabetes actually had lower abundances of the typical “red complex” species seen in periodontitis infections, Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola [43]. The authors note that potential confounders in their study include a higher mean plaque index and greater age in the subjects with diabetes. However, it is also possible that any of the proposed mechanisms of diabetic influence on periodontal health could result in a variation of the typical dysbiotic composition in periodontitis, or that the samples were collected at a stage in the progression of the disease in which the common pathogens are found at lower abundances. In either case, this presents another interesting opportunity for larger scale longitudinal studies, to discover potential alternative pathways to periodontal disease, and thus a greater comprehension of its mechanisms and potential treatments or preventative measures.
8. Clinical Potential of the Oral Microbiome/Manipulations and Perturbations of the Oral Microbiome
We have demonstrated here a few key examples of the wide-ranging action of the oral microbiome upon the human body. This opens vast possibilities for diagnosis and intervention, only some of which have begun to be explored, or even conceptualized. To fully leverage this potential, we will need to continue to probe the compositions and actions of oral microbiomes, both at small-scale interactions within individuals in different states of health, as well as at broad scales among different populations. The oral microbiome has already shown potential as a diagnostic tool. One machine learning-based study, which collected 2424 publicly available metagenomes from eight studies, showed that the performance of disease predictions was improved when using strain-level features (not feasible with 16S sequencing), and suggested that disease phenotypes are linked to “non-core” microbial genes/factors which may be found in variable genomic regions that are specific to strains/subspecies [90]. However, they caution the use of some potential biomarkers for diagnosing disease, such as the species Streptococcus anginosus, which actually associates with general dysbiosis rather than a particular disease. The authors still consider this work to be an early stage of modeling the healthy microbiome, so that it can be used to contrast states of dysbiosis associated with disease. Another attempt at using oral microbial abundances as biomarkers has shown some preliminary success in the early detection of colorectal cancer (CRC) lesions [92]. The fecal immune test (FIT) and the fecal occult blood test (FOBT) are typical non-invasive screening procedures used to detect CRC, but they suffer from poor sensitivity in detecting early lesions. The authors showed that a classification model based on oral microbiome samples had high specificity and greater sensitivity than standard tests in distinguishing CRC and polyp samples from healthy controls, with a further increase in sensitivity when combining with fecal microbiome samples. Thus, with further verification studies, the microbiome could be implemented to improve the rate of early detection of CRC.
In addition to a biomarker, the oral microbiome can be both a tool and a target for treating diseases. Commercially available probiotics, which contain live strains from bacterial genera, such as Bifidobacterium, Lactobacillus, and Streptococcus, have been shown to promote greater alpha diversity in the oral microbiome, though without making large-scale or permanent alterations to its composition [154]. As mentioned in the previous section, Lactobacillus in particular is a common and effective probiotic in the context of the gut microbiome, but it has been associated with dental caries, and indirectly with CRC. However, it is also possible that this effect is dependent upon the environmental context, as discussed above in the case of Streptococcus oralis during cystic fibrosis infection or Leptotrichia during pancreatic cancer. Thus, Lactobacillus without environmental conditions suitable for dental caries may instead help to promote greater alpha diversity and better oral health. This is another case of preliminary results with interesting potential that require deeper longitudinal study.
Mechanisms have been proposed for the beneficial impact of other potential probiotics as well, to counteract the progression of periodontal disease and caries, including strains of Streptococcus salivarius, which has been shown to downregulate inflammatory responses and to stimulate beneficial pathways like type I and II interferon responses [168]. Other potential probiotics for this use are Streptococcus dentisani [169] and Streptococcus A12 [170], which can buffer the acidic pH produced within cariogenic biofilms through arginine metabolism. The Proteobacteria species Bdellovibrio bacteriovorus has been suggested as a tool for potentially targeting periodontal pathogens [171]. It feeds on Gram-negative bacteria by invading them, eventually leading to lysis of its prey. Ex vivo experiments on saliva and subgingival plaque showed that B. bacteriovorus was able to attack two important oral pathogens, Fusobacterium nucleatum and Aggregatibacter actinomycetemcomitans, though not certain other desired targets, nor was it specific only to these targets.
Pathways and products of oral microbiota may also be treatment targets. The proinflammatory cytokine IL-17 was shown to be the most strongly upregulated in diabetic subjects and was associated with periodontitis while treatment with anti-IL-17 antibodies was able to mitigate this effect [172]. In Alzheimer’s disease, inhibitors of the gingipains proteases secreted by Porphyromonas gingivalis were able to reduce infection by this species in the brain as well as amyloid-β production and neuroinflammation [40].
There is still a lot to learn about the functionality of the oral microbiome, and its potential reaction to treatments. One instance stems from the “WEIRD” bias that has left many of the world’s populations out of this area of investigation. A study of “uncontacted” Amerindians in the Venezuelan Amazon, an isolated community with no known exposure to antibiotics, had bacteria carrying functional antibiotic resistance (AR) genes [45]. The authors suggest that “AR genes are likely poised for mobilization and enrichment upon exposure to pharmacological levels of antibiotics”. The authors emphasize the need for characterizing remote populations “before globalization of modern practices affects potentially beneficial bacteria harbored in the human body”. Thus, the context of microbiome studies must continue to be expanded before we can find the optimal approaches to treatment.
9. Conclusions and Future Outlook
Improvements in sequencing techniques and costs continue to propel the field of microbiome research forward, allowing for larger-scale approaches and wider understanding of its structure and function. However, over the last decade or so, much of the research has been exploratory and many investigators have taken their own approaches to perform experiments and analyses as the potentials and the limitations of these new techniques have been probed. Some amount of technical bias has undoubtedly permeated the results in this field as a result. As we begin to understand what NGS experiments show us, and as new approaches are developed, we must strive for standardized methods that will allow for reliable data comparison with minimal bias. The same is true for study designs and sample collection methods, as has been proposed [113]. The oral microbiome will benefit especially from concrete methodologies to ensure the proper context for a given study, because, depending on the goal of study, results may be dependent on particular niches within this habitat.
Current evidence allows for at least some tentative generalizations about the structure of the oral microbiome. Despite the arguments for the focus on gradients of abundances across samples of a population and against the reliance on stomatotypes (or other relevantly termed clusters of microbiome samples), stomatotypes allow for researchers to obtain a broad perspective and can help guide further analyses. The Neisseria/Haemophilus and Prevotella/Veillonella stomatotypes discussed above may represent relatively healthy compositions while the other stomatotypes that have been detected, which tend to be less consistent in terms of composition, may represent dysbiosis. The driver genera of the first two stomatotypes often appear to be associated with health while driver genera in other described stomatotypes are often associated with disease, like Porphyromonas, Rothia, and certain species of Streptococcus. Of course, there are exceptions, such as the associations that have been reported between periodontitis and Prevotella [18] or between dental caries and Neisseria [22], though to qualify that statement, as mentioned above, there may be alterations in the abundances of many taxa as a result of the disease state, not as a cause of the disease. So, as in the case of Prevotella in periodontitis or Neisseria in dental caries, these taxa may simply become opportunistic in the environment created by the disease (Neisseria may simply be taking advantage of the “hydrogen sink” created by archaeal methanogens and/or Treponema species, since its species tend to be acidogenic [173]), and therefore could still be considered to be associated with general oral health. Periodontitis and dental caries are heavily researched diseases, but in seeking associations between the microbiome and any disorder, it is important to thoroughly explore the mechanisms of disease progression before labelling any particular organism as a causative agent. Table 1 and Table 2 provided in this review, which list associated taxa, should be taken as just that: A list of potential associations to be further explored.
As has been emphasized throughout this review, our current conceptions of the oral microbiome are largely biased toward the lifestyles in “WEIRD” nations (Western, educated, industrialized, rich, and democratic) [46], as most of the investigators and studied samples come from these populations. The few studies referenced here that have incorporated non-Westernized samples highlight the holes in our current knowledge of the potential compositions and structures of the oral microbiome [44] and its functionality in particular contexts [45]. As researchers integrate more diverse populations into the field (as well as expanding the focus to include other domains of life present in the microbiome), they will be able to continually generate more extensive databases of human oral-associated taxonomy and functionality, allowing for more comprehensive studies.
Its position as the gateway to some of the most vital functions for human life tightly connects the oral cavity to the rest of the body and gives it a powerful influence on many of the body’s processes. Dysbiosis in the oral microbiome is clearly linked to a number of local and systemic diseases, though some of the particular associations and mechanisms of action remain conjectural pending further study. Some of the treatment examples discussed here appear straightforward, even despite the need for deeper verification of efficacy. However, the human microbiome is a multi-dimensional interconnected micro-biosphere, and perturbations may have as yet unforeseen effects, whether undetected in the short term or undeterminable in the long term without the appropriate study designs. We have already mentioned examples where unrelated organisms can fill some of the same functional niches (see the discussion above of Treponema and methanogenic archaeal species acting as hydrogen sinks in periodontitis) and their competition can be implicated in disease. Microbiome research is far from complete, and without a deep understanding of its nature, we should exercise caution and patience before widely exploiting its medical potential. This will require many more large-scale and longitudinal studies, greater focus on the functional component of the microbiome, and a stronger characterization of the varying structures of each microbiome of the body in different contexts, part of which will be to combat the WEIRD bias, which has thus far limited the scope of study, and the dearth of attention to the less prominent domains of the microbiome, like fungi, archaea, and viruses.
Funding
We acknowledge support from the Spanish Ministry of Economy, Industry, and Competitiveness (MEIC) for the EMBL partnership, and grant PGC2018-099921-B-I00 cofounded by European Regional Development Fund (ERDF); from the CERCA Programme/Generalitat de Catalunya; from the Catalan Research Agency (AGAUR) SGR423, and grants from the European Union’s Horizon 2020 research and innovation programme under the grant agreement ERC-2016-724173, and the Marie Sklodowska-Curie grant agreement H2020-MSCA-IF-2017-793699. The group also receives support from a INB Grant PT17/0009/0023 - ISCIII-SGEFI/ERDF.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [Green Version]
- Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Bihan, M.; Yooseph, S.; Methé, B.A. Analyses of the microbial diversity across the human microbiome. PLoS ONE 2012, 7, e32118. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D.; Do, T.; Beighton, D.; Devine, D.A. Influence of saliva on the oral microbiota. Periodontology 2000, 70, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Paster, B.J.; Olsen, I.; Aas, J.A.; Dewhirst, F.E. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontology 2000, 42, 80–87. [Google Scholar] [CrossRef]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; He, J.; Xue, J.; Wang, Y.; Li, K.; Zhang, K.; Guo, Q.; Liu, X.; Zhou, Y.; Cheng, L.; et al. Oral microbiome differs by age and location. Environ. Microbiol. 2015, 17, 699–710. [Google Scholar] [CrossRef]
- Willis, J.R.; González-Torres, P.; Pittis, A.A.; Bejarano, L.A.; Cozzuto, L.; Andreu-Somavilla, N.; Alloza-Trabado, M.; Valentín, A.; Ksiezopolska, E.; Company, C.; et al. Citizen science charts two major “stomatotypes” in the oral microbiome of adolescents and reveals links with habits and drinking water composition. Microbiome 2018, 6, 218. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Altemus, J.; Niazi, F.; Green, H.; Calhoun, B.C.; Sturgis, C.; Grobmyer, S.R.; Eng, C. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 2017, 8, 88122–88138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, I.; Vasquez, A.A.; Moyerbrailean, G.; Land, S.; Sun, J.; Lin, H.S.; Ram, J.L. Oral microbiome and history of smoking and colorectal cancer. J. Epidemiol. Res. 2016, 2, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): A resource for the microbiome of the human aerodigestive tract. MSystems 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarazzo, F.; Ribeiro, A.C.; Feres, M.; Faveri, M.; Mayer, M.P.A. Diversity and quantitative analysis of Archaea in aggressive periodontitis and periodontally healthy subjects. J. Clin. Periodontol. 2011, 38, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Lepp, P.W.; Brinig, M.M.; Ouverney, C.C.; Palm, K.; Armitage, G.C.; Relman, D.A. Methanogenic Archaea and human periodontal disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6176–6181. [Google Scholar] [CrossRef] [Green Version]
- Griffen, A.L.; Beall, C.J.; Campbell, J.H.; Firestone, N.D.; Kumar, P.S.; Yang, Z.K.; Podar, M.; Leys, E.J. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012, 6, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Diversity and morphology of members of the phylum “synergistetes” in periodontal health and disease. Appl. Environ. Microbiol. 2009, 75, 3777–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costalonga, M.; Herzberg, M.C. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol. Lett. 2014, 162, 22–38. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.C.; Li, S.; et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS ONE 2012, 7, e37919. [Google Scholar] [CrossRef] [Green Version]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the human oral microbiome during health and disease. MBio 2014, 5, e01012–e01014. [Google Scholar] [CrossRef] [Green Version]
- HAUBEK, D. The highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans: Evolutionary aspects, epidemiology and etiological role in aggressive periodontitis. APMIS 2010, 118. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.L.; Leys, E.J.; Gasparovich, S.R.; Firestone, N.D.; Schwartzbaum, J.A.; Janies, D.A.; Asnani, K.; Griffen, A.L. Bacterial 16S sequence analysis of severe caries in young permanent teeth. J. Clin. Microbiol. 2010, 48, 4121–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.; Bowen, W.H. Candida albicans and Streptococcus mutans: A potential synergistic alliance to cause virulent tooth decay in children. Future Microbiol. 2014, 9, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.L.; Haffajee, A.D.; Devlin, P.M.; Norris, C.M.; Posner, M.R.; Goodson, J.M. The salivary microbiota as a diagnostic indicator of oral cancer: A descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 2005, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Pushalkar, S.; Ji, X.; Li, Y.; Estilo, C.; Yegnanarayana, R.; Singh, B.; Li, X.; Saxena, D. Comparison of oral microbiota in tumor and non-tumor tissues of patients with oral squamous cell carcinoma. BMC Microbiol. 2012, 12, 144. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ganly, I. The oral microbiome and oral cancer. Clin. Lab. Med. 2014, 34, 711–719. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Pei, Z.; Yang, L.; Purdue, M.P.; Freedman, N.D.; Jacobs, E.J.; Gapstur, S.M.; Hayes, R.B.; Ahn, J. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017, 77, 6777–6787. [Google Scholar] [CrossRef] [Green Version]
- Broecker, F.; Russo, G.; Klumpp, J.; Moelling, K. Stable core virome despite variable microbiome after fecal transfer. Gut Microbes 2017, 8, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. Temporal stability of the human skin microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [Green Version]
- Wantland, W.W.; Wantland, E.M.; Remo, J.W.; Winquist, D.L. Studies on Human Mouth Protozoa. J. Dent. Res. 1958, 37, 949–950. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, P.J.; Fletcher, E.M.; Gibbons, S.M.; Bouvet, M.; Doran, K.S.; Kelley, S.T. Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ 2015, 3, e1373. [Google Scholar] [CrossRef]
- Whiley, R.A.; Fleming, E.V.; Makhija, R.; Waite, R.D. Environment and colonisation sequence are key parameters driving cooperation and competition between Pseudomonas aeruginosa cystic fibrosis strains and oral commensal streptococci. PLoS ONE 2015, 10, e0115513. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.; Wang, C.Y. Mechanisms involved in the association between periodontal diseases and cardiovascular disease. Oral Dis. 2011, 17, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhibber-Goel, J.; Singhal, V.; Bhowmik, D.; Vivek, R.; Parakh, N.; Bhargava, B.; Sharma, A. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes 2016, 2. [Google Scholar] [CrossRef]
- Roszyk, E.; Puszczewicz, M. Role of human microbiome and selected bacterial infections in the pathogenesis of rheumatoid arthritis. Reumatologia 2017, 55, 242–250. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.H.; Weissmann, G.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Miklossy, J. Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J. Alzheimers Dis. 2016, 53, 1459–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo, S.; Schuh, C.M.A.P.; Vicente, B.; Aguayo, L.G. Association between alzheimer’s disease and oral and gut microbiota: Are pore forming proteins the missing link? J. Alzheimers Dis. 2018, 65, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Casarin, R.C.V.; Barbagallo, A.; Meulman, T.; Santos, V.R.; Sallum, E.A.; Nociti, F.H.; Duarte, P.M.; Casati, M.Z.; Gonçalves, R.B. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J. Periodontal Res. 2013, 48, 30–36. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, J.; Heine, S.; Norenzayan, A. The weirdest people in the world? Behav. Brain Sci. 2010, 33, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Zaura, E.; Brandt, B.W.; Prodan, A.; Teixeira de Mattos, M.J.; Imangaliyev, S.; Kool, J.; Buijs, M.J.; Jagers, F.L.; Hennequin-Hoenderdos, N.L.; Slot, D.E.; et al. On the ecosystemic network of saliva in healthy young adults. ISME J. 2017, 11, 1218–1231. [Google Scholar] [CrossRef]
- De Filippis, F.; Vannini, L.; La Storia, A.; Laghi, L.; Piombino, P.; Stellato, G.; Serrazanetti, D.I.; Gozzi, G.; Turroni, S.; Ferrocino, I.; et al. The same microbiota and a potentially discriminant metabolome in the saliva of omnivore, ovo-lacto-vegetarian and Vegan individuals. PLoS ONE 2014, 9, e112373. [Google Scholar] [CrossRef] [Green Version]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef]
- Takeshita, T.; Kageyama, S.; Furuta, M.; Tsuboi, H.; Takeuchi, K.; Shibata, Y.; Shimazaki, Y.; Akifusa, S.; Ninomiya, T.; Kiyohara, Y.; et al. Bacterial diversity in saliva and oral health-related conditions: The Hisayama Study. Sci. Rep. 2016, 6, 22164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malla, M.A.; Dubey, A.; Kumar, A.; Yadav, S.; Hashem, A.; Allah, E.F. Exploring the Human Microbiome: The Potential Future Role of Next-Generation Sequencing in Disease Diagnosis and Treatment. Front. Immunol. 2019, 9, 2868. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [Green Version]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech, X.Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur rapidly resolves single-nucleotide community sequence patterns. MSystems 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Kembel, S.W.; Wu, M.; Eisen, J.A.; Green, J.L. Incorporating 16S gene copy number information improves estimates of microbial diversity and abundance. PLoS Comput. Biol. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Quince, C.; Pijl, A.S.; de Hollander, M.; Kowalchuk, G.A. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS ONE 2012, 7, e30600. [Google Scholar] [CrossRef] [PubMed]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, rapidly, OTUs with galaxy solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Ogier, J.C.; Pagès, S.; Galan, M.; Barret, M.; Gaudriault, S. rpoB, a promising marker for analyzing the diversity of bacterial communities by amplicon sequencing. BMC Microbiol. 2019, 19, 171. [Google Scholar] [CrossRef] [Green Version]
- Martens, M.; Dawyndt, P.; Coopman, R.; Gillis, M.; De Vos, P.; Willems, A. Advantages of multilocus sequence analysis for taxonomic studies: A case study using 10 housekeeping genes in the genus Ensifer (including former Sinorhizobium). Int. J. Syst. Evol. Microbiol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.A. Metatranscriptomics: Eavesdropping on Complex Microbial Communities. Microbe 2009, 4, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nepi, S.; Saccenti, E.; Schäfer, H.; Schütz, B.; Spraul, M.; Tenori, L. Individual human phenotypes in metabolic space and time. J. Proteome Res. 2009, 8, 4264–4271. [Google Scholar] [CrossRef]
- Peano, C.; Pietrelli, A.; Consolandi, C.; Rossi, E.; Petiti, L.; Tagliabue, L.; De Bellis, G.; Landini, P. An efficient rRNA removal method for RNA sequencing in GC-rich bacteria. Microb. Inform. Exp. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol. Bioinform. Online 2016, 12, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easterly, C.W.; Sajulga, R.; Mehta, S.; Johnson, J.; Kumar, P.; Hubler, S.; Mesuere, B.; Rudney, J.; Griffin, T.J.; Jagtap, P.D. metaQuantome: An integrated, quantitative metaproteomics approach reveals connections between taxonomy and protein function in complex microbiomes. Mol. Cell. Proteom. 2019, 18, S82–S91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Emerging view of the human virome. Transl. Res. 2012, 160, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Thurber, R.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef]
- Allen, L.Z.; Ishoey, T.; Novotny, M.A.; McLean, J.S.; Lasken, R.S.; Williamson, S.J. Single virus genomics: A new tool for virus discovery. PLoS ONE 2011, 6, e17722. [Google Scholar] [CrossRef]
- Lim, Y.; Totsika, M.; Morrison, M.; Punyadeera, C. The saliva microbiome profiles are minimally affected by collection method or DNA extraction protocols. Sci. Rep. 2017, 7, 8523. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Peters, B.A.; Min, D.; Ahn, J.; Hayes, R.B. Comparison of the oral microbiome in mouthwash and whole saliva samples. PLoS ONE 2018, 13, e0194729. [Google Scholar] [CrossRef] [Green Version]
- Mallick, H.; Ma, S.; Franzosa, E.A.; Vatanen, T.; Morgan, X.C.; Huttenhower, C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017, 18, 228. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Susan Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Leo, L.; Shetty, S. Microbiome R Package. Available online: http://microbiome.github.io (accessed on 18 February 2020).
- Jari, O.F.; Guillaume, B.; Michael, F.; Roeland, K.; Pierre, L.; Dan, M.; Peter, R.M.; Minchin, P.R.; O’Hara, R.B.; Gavin, L.S.; et al. Vegan: Community Ecology Package; R Package Version, 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 18 February 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 18 February 2020).
- Douglas, B.; Martin, M.; Ben, B.; Steve, W. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front. Genet. 2019, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvallet, C.; Gibbons, S.M.; Gurry, T.; Irizarry, R.A.; Alm, E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017, 8, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M. Bacteriology of Humans an Ecological Perspective; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Laubichler, M.D.; Renn, J. Extended evolution: A conceptual framework for integrating regulatory networks and niche construction. J. Exp. Zool. B Mol. Dev. Evol. 2015, 324, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Meulen, T.A.; Harmsen, H.J.M.; Bootsma, H.; Liefers, S.C.; Vich, V.A.; Zhernakova, A.; Fu, J.; Wijmenga, C.; Spijkervet, F.K.L.; Kroese, F.G.M.; et al. Dysbiosis of the buccal mucosa microbiome in primary Sjögren’s syndrome patients. Rheumatology 2018, 57, 2225–2234. [Google Scholar] [CrossRef]
- Ganesan, S.M.; Joshi, V.; Fellows, M.; Dabdoub, S.M.; Nagaraja, H.N.; O’Donnell, B.; Deshpande, N.R.; Kumar, P.S. A tale of two risks: Smoking, diabetes and the subgingival microbiome. ISME J. 2017, 11, 2075–2089. [Google Scholar] [CrossRef] [Green Version]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Moutsopoulos, N.M.; Konkel, J.E. Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol. 2018, 39, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Mark, W.J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Shi, M.; Zhen, M.; Wang, C.; Hu, W.; Nie, Y.; Wu, X. Comparison of subgingival and buccal mucosa microbiome in chronic and aggressive periodontitis: A pilot study. Front. Cell. Infect. Microbiol. 2019, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Asakawa, M.; Takeshita, T.; Furuta, M.; Kageyama, S.; Takeuchi, K.; Hata, J.; Ninomiya, T.; Yamashita, Y. Tongue Microbiota and Oral Health Status in Community-Dwelling Elderly Adults. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Ren, Z.; Li, A.; Li, J.; Xu, S.; Zhang, H.; Jiang, J.; Yang, J.; Luo, Q.; Zhou, K.; et al. Tongue coating microbiome data distinguish patients with pancreatic head cancer from healthy controls. J. Oral Microbiol. 2019, 11, 1563409. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Aoki, K.; Ishii, Y.; Tateda, K. The palatine tonsil bacteriome, but not the mycobiome, is altered in HIV infection. BMC Microbiol. 2018, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 2013, 9, e1002863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debelius, J.; Song, S.J.; Vazquez-Baeza, Y.; Xu, Z.Z.; Gonzalez, A.; Knight, R. Tiny microbes, enormous impacts: What matters in gut microbiome studies? Genome Biol. 2016, 17, 217. [Google Scholar] [CrossRef] [Green Version]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial community variation in human body habitats across space and time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the dynamic microbiome to life with animations. Cell Host Microbe 2017, 21, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Eren, A.M.; Borisy, G.G.; Huse, S.M.; Mark Welch, J.L. Oligotyping analysis of the human oral microbiome. Proc. Natl. Acad. Sci. USA 2014, 111, E2875–E2884. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Fukuma, N.; Totsika, M.; Kenny, L.; Morrison, M.; Punyadeera, C. The performance of an oral microbiome biomarker panel in predicting oral cavity and oropharyngeal cancers. Front. Cell. Infect. Microbiol. 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Lu, D.Y. Procurement, transportation, and storage of saliva, buccal swab, and oral wash specimens. In Methods in Molecular Biology; Yong, W., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1897. [Google Scholar]
- XIT Genomic DNA from Buccal Cells for Extraction of Genomic DNA from Buccal/Cheek Cells. G-Biosciences. Available online: https://www.gbiosciences.com/image/pdfs/protocol/786-341_protocol.pdf (accessed on 18 February 2020).
- Pramanik, R.; Thompson, H.; Kistler, J.O.; Wade, W.G.; Galloway, J.; Peakman, T.; Proctor, G.B. Effects of the UK Biobank collection protocol on potential biomarkers in saliva. Int. J. Epidemiol. 2012, 41, 1786–1797. [Google Scholar] [CrossRef] [Green Version]
- Lassalle, F.; Spagnoletti, M.; Fumagalli, M.; Shaw, L.; Dyble, M.; Walker, C.; Thomas, M.G.; Bamberg Migliano, A.; Balloux, F. Oral microbiomes from hunter-gatherers and traditional farmers reveal shifts in commensal balance and pathogen load linked to diet. Mol. Ecol. 2018, 27, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, I.; Claesson, M.; O’Toole, P.; Shanahan, F. Categorization of the gut microbiota: Enterotypes or gradients? Nat. Rev. Microbiol. 2012, 10, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Ward, T.L.; McKinlay, C.E.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Rethinking “enterotypes”. Cell Host Microbe 2014, 16, 433–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.L.; Bor, B.; Agnello, M.; Shi, W.; He, X. Ecology of the oral microbiome: Beyond bacteria. Trends Microbiol. 2017, 25, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Peters, B.A.; Wu, J.; Hayes, R.B.; Ahn, J. The oral fungal mycobiome: Characteristics and relation to periodontitis in a pilot study. BMC Microbiol. 2017, 17, 157. [Google Scholar] [CrossRef]
- Bandara, H.M.H.N.; Panduwawala, C.P.; Samaranayake, L.P. Biodiversity of the human oral mycobiome in health and disease. Oral Dis. 2019, 25, 363–371. [Google Scholar] [CrossRef]
- Dupuy, A.K.; David, M.S.; Li, L.; Heider, T.N.; Peterson, J.D.; Montano, E.A.; Dongari-Bagtzoglou, A.; Diaz, P.I.; Strausbaugh, L.D. Redefining the human oral mycobiome with improved practices in amplicon-based taxonomy: Discovery of Malassezia as a prominent commensal. PLoS ONE 2014, 9, e90899. [Google Scholar] [CrossRef] [Green Version]
- Saunders, C.W.; Scheynius, A.; Heitman, J. Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS Pathog. 2012, 8, e1002701. [Google Scholar] [CrossRef]
- Wu, G.; Zhao, H.; Li, C.; Rajapakse, M.P.; Wong, W.C.; Xu, J.; Saunders, C.W.; Reeder, N.L.; Reilman, R.A.; Scheynius, A.; et al. Genus-wide comparative genomics of malassezia delineates its phylogeny, physiology, and niche adaptation on human skin. PLoS Genet. 2015, 11, e1005614. [Google Scholar] [CrossRef]
- Hibbett, D.; Taylor, J. Fungal systematics: Is a new age of enlightenment at hand? Nat. Rev. Microbiol. 2013, 11, 129–133. [Google Scholar] [CrossRef]
- Donovan, P.D.; Gonzalez, G.; Higgins, D.G.; Butler, G.; Ito, K. Identification of fungi in shotgun metagenomics datasets. PLoS ONE 2018, 13, e0192898. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz Peña, M.J.; Martinez-Hernandez, F.; Garcia-Heredia, I.; Lluesma Gomez, M.; Fornas, Ò.; Martinez-Garcia, M. Deciphering the human virome with single-virus genomics and metagenomics. Viruses 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Brocal, V.; Moya, A. The analysis of the oral DNA virome reveals which viruses are widespread and rare among healthy young adults in Valencia (Spain). PLoS ONE 2018, 13, e0191867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willner, D.; Furlan, M.; Haynes, M.; Schmieder, R.; Angly, F.E.; Silva, J.; Tammadoni, S.; Nosrat, B.; Conrad, D.; Rohwer, F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE 2009, 4, e7370. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Molet, B.; Haag, R.; Kremer, M. [Protozoa of the human oral cavity (epidemiological correlations and pathogenic possibilities]. J. Biol. Buccale 1981, 9, 155–161. [Google Scholar] [PubMed]
- Chomicz, L.; Piekarczyk, J.; Starościak, B.; Fiedor, P.; Piekarczyk, B.; Szubińska, D.; Zawadzki, P.J.; Walski, M. Comparative studies on the occurrence of protozoans, bacteria and fungi in the oral cavity of patients with systemic disorders. Acta Parasitol. 2002, 47, 147–153. [Google Scholar]
- Cielecka, D.; Chomicz, L.; Piekarczyk, J.; Walski, M.; Zawadzki, P.J.; Bednarczyk, A.; Szubińska, D. Oral cavity condition and the occurrence of parasitic protozoans in patients with genetic diseases. Acta Parasitol. 2000, 45, 107–112. [Google Scholar]
- Wade, W.G. The oral microbiome in health and disease. Pharmacol. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef]
- Horz, H.P. Archaeal lineages within the human microbiome: Absent, rare or elusive? Life 2015, 5, 1333–1345. [Google Scholar] [CrossRef]
- Whittaker, R.H. Communities and Ecosystems; MacMillan Publishing Company, Inc.: New York, NY, USA, 1975. [Google Scholar]
- Socransky, S.; Haffajee, A.; Cugini, M.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Kurkivuori, J.; Salaspuro, V.; Kaihovaara, P.; Kari, K.; Rautemaa, R.; Grönroos, L.; Meurman, J.H.; Salaspuro, M. Acetaldehyde production from ethanol by oral streptococci. Oral Oncol. 2007, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Meurman, J.H. Oral microbiota and cancer. J. Oral Microbiol. 2010, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lax, A. Bacterial toxins and cancer—A case to answer? Nat. Rev. Microbiol. 2005, 3, 343–349. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.N.; Araújo-Pérez, F.; Azcárate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, S.G.; Katz, J. The association between periodontal disease and cancer: A review of the literature. J. Dent. 2010, 38, 83–95. [Google Scholar] [CrossRef]
- Michaud, D.S.; Fu, Z.; Shi, J.; Chung, M. Periodontal disease, tooth loss, and cancer risk. Epidemiol. Rev. 2017, 39, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Abnet, C.C.; Qiao, Y.L.; Dawsey, S.M.; Dong, Z.W.; Taylor, P.R.; Mark, S.D. Tooth loss is associated with increased risk of total death and death from upper gastrointestinal cancer, heart disease, and stroke in a Chinese population-based cohort. Int. J. Epidemiol. 2005, 34, 467–474. [Google Scholar] [CrossRef]
- Mirvish, S.S. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 1995, 93, 17–48. [Google Scholar] [CrossRef]
- Chalabi, M.; Moghim, S.; Mogharehabed, A.; Najafi, F.; Rezaie, F. EBV and CMV in chronic periodontitis: A prevalence study. Arch. Virol. 2008, 153, 1917. [Google Scholar] [CrossRef] [PubMed]
- Slots, J.; Sugar, C.; Kamma, J.J. Cytomegalovirus periodontal presence is associated with subgingival Dialister pneumosintes and alveolar bone loss. Oral Microbiol. Immunol. 2002, 17, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Tateno, T.; Ueno, S.; Hiwatashi, K.; Matsumoto, M.; Okumura, H.; Setoyama, T.; Uchikado, Y.; Sakoda, M.; Kubo, F.; Ishigami, S.; et al. Expression of receptor for advanced glycation end products (RAGE) is related to prognosis in patients with esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009, 16, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.W.; Shi, W.; Huang, G.T.; Kinder Haake, S.; Park, N.H.; Kuramitsu, H.; Genco, R.J. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect. Immun. 2000, 68, 3140–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzarro, S.; Loos, B.G.; Laine, M.L.; Crielaard, W.; Zaura, E. Subgingival microbiome in smokers and non-smokers in periodontitis: An exploratory study using traditional targeted techniques and a next-generation sequencing. J. Clin. Periodontol. 2013, 40, 483–492. [Google Scholar] [CrossRef]
- Moon, J.H.; Lee, J.H.; Lee, J.Y. Subgingival microbiome in smokers and non-smokers in Korean chronic periodontitis patients. Mol. Oral Microbiol. 2015, 30, 227–241. [Google Scholar] [CrossRef]
- Dassi, E.; Ferretti, P.; Covello, G.; Speccher, A.; Migazzi, A.; Bosco, B.; Rajashekar, B.; Zarbo, C.; Ballabio, C.; Rossetto, D.; et al. The short-term impact of probiotic consumption on the oral cavity microbiome. Sci. Rep. 2018, 8, 10476. [Google Scholar] [CrossRef] [Green Version]
- Saxelin, M. Probiotic formulations and applications, the current probiotics market, and changes in the marketplace: A european perspective. Clin. Infect. Dis. 2008, 46, S76–S79. [Google Scholar] [CrossRef] [Green Version]
- Michaud, D.S.; Izard, J.; Wilhelm-Benartzi, C.S.; You, D.H.; Grote, V.A.; Tjønneland, A.; Dahm, C.C.; Overvad, K.; Jenab, M.; Fedirko, V.; et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013, 62, 1764–1770. [Google Scholar] [CrossRef]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Söder, B.; Jin, L.J.; Klinge, B.; Söder, P.Ö. Periodontitis and premature death: A 16-year longitudinal study in a Swedish urban population. J. Periodontal Res. 2007, 42, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Pramod, K.; Tahir, M.A.; Ansari, S.H. Autoimmune responses in periodontal diseases. Autoimmun. Rev. 2011, 10, 426–431. [Google Scholar] [CrossRef]
- Rangé, H.; Labreuche, J.; Louedec, L.; Rondeau, P.; Planesse, C.; Sebbag, U.; Bourdon, E.; Michel, J.B.; Bouchard, P.; Meilhac, O. Periodontal bacteria in human carotid atherothrombosis as a potential trigger for neutrophil activation. Atherosclerosis 2014, 236, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, A.; Miura, M.; Fujise, O.; Hamachi, T.; Nishimura, F. Porphyromonas gingivalis gingipain is involved in the detachment and aggregation of Aggregatibacter actinomycetemcomitans biofilm. Mol. Oral Microbiol. 2014, 29, 131–143. [Google Scholar] [CrossRef]
- Jayaprakash, K.; Khalaf, H.; Bengtsson, T. Gingipains from Porphyromonas gingivalis play a significant role in induction and regulation of CXCL8 in THP-1 cells. BMC Microbiol. 2014, 14, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissick, A.; Redman, R.S.; Jones, M.; Rangan, B.V.; Reimold, A.; Griffiths, G.R.; Mikuls, T.R.; Amdur, R.L.; Richards, J.S.; Kerr, G.S. Association of periodontitis with rheumatoid arthritis: A pilot study. J. Periodontol. 2010, 81, 223–230. [Google Scholar] [CrossRef]
- Konig, M.F.; Paracha, A.S.; Moni, M.; Bingham, C.O., III; Andrade, F. Defining the role of Porphyromonas gingivalis peptidylarginine deiminase (PPAD) in rheumatoid arthritis through the study of PPAD biology. Ann. Rheum. Dis. 2015, 74, 2054–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mealey, B.L.; Oates, T.W. Diabetes mellitus and periodontal diseases. J. Periodontol. 2006, 77, 1289–1303. [Google Scholar] [CrossRef]
- Taylor, G.W. Bidirectional Interrelationships Between Diabetes and Periodontal Diseases: An Epidemiologic Perspective. Ann. Periodontol. 2001, 6, 99–112. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Dasanayake, A.P.; Chhun, N. Does periodontal therapy reduce the risk for systemic diseases? Dent. Clin. N. Am. 2010, 54, 163–181. [Google Scholar] [CrossRef]
- Devine, D.A.; Marsh, P.D.; Meade, J. Modulation of host responses by oral commensal bacteria. J. Oral Microbiol. 2015, 7, 26941. [Google Scholar] [CrossRef] [PubMed]
- López-López, A.; Camelo-Castillo, A.; Ferrer, M.D.; Simon-Soro, Á.; Mira, A. Health-associated niche inhabitants as oral probiotics: The case of Streptococcus dentisani. Front. Microbiol. 2017, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Palmer, S.R.; Ahn, S.J.; Richards, V.P.; Williams, M.L.; Nascimento, M.M.; Burne, R.A. A highly arginolytic Streptococcus species that potently antagonizes Streptococcus mutans. Appl. Environ. Microbiol. 2016, 82, 2187–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loozen, G.; Boon, N.; Pauwels, M.; Slomka, V.; Rodrigues Herrero, E.; Quirynen, M.; Teughels, W. Effect of Bdellovibrio bacteriovorus HD100 on multispecies oral communities. Anaerobe 2015, 35, 45–53. [Google Scholar] [CrossRef]
- Xiao, E.; Mattos, M.; Vieira, G.H.A.; Chen, S.; Corrêa, J.D.; Wu, Y.; Albiero, M.L.; Bittinger, K.; Graves, D.T. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe 2017, 22, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Knapp, J.S. Historical perspectives and identification of Neisseria and related species. Clin. Microbiol. Rev. 1988, 1, 415–431. [Google Scholar] [CrossRef]
Figure 1.
Schematics of standard techniques used in microbiome studies. (A) Marker gene sequencing techniques can use primers to target certain conserved regions of a genome to capture intermittent variable regions, which can then be used to identify organisms in a sample rapidly and inexpensively. The 16S rRNA gene is the most commonly used marker gene in bacteria and archaea, and in the figure, primers are used to capture the V3 and V4 variable regions together, a common approach for 16S sequencing. The internal transcribed spacer (ITS) region of the nuclear rRNA cistron in fungi is made of two segments, which can be captured with primers targeting the 18S, 5.8S, and 28S rRNA sections that surround them. (B–D) Instead of targeting one small segment of the genome, these techniques capture the entirety of the genetic material from an organism. (B) Single virus genomics (SVG) uses a fluorescent stain to isolate individual virus particles in a sample by fluorescence-activated virus sorting (FAVS), wherein they are embedded in an agarose bead before undergoing whole genome amplification and sequencing. (C) Whole metagenome shotgun sequencing (WMS) involves the fragmentation of all DNA in a sample, sequencing of the fragments, and assembly of the sequences, which can then be mapped to reference genomes, or de novo assembly can be performed. (D) Metatranscriptomics also involves a shotgun sequencing approach, but it is performed after mRNA extraction. The outputs then allow for differential gene expression analysis. (E) Metabolomics and metaproteomics allow for quantification of the metabolites and proteins produced by the microbiome in a sample, respectively. Mass spectrometry is a common approach to quantification. Mock metabolite shapes in Figure 1 were generated using the JSME Molecular Editor by Peter Ertl and Bruno Bienfait licensed under CC-BY-NC-SA 3.0. Images of body sites and organs in Figure 1 and Figure 2 were obtained from Servier Medical Art by Servier licensed under CC-BY 3.0.Traditionally, 16S sequences were clustered into groups with at least 97% identity, called operational taxonomic units (OTUs), which have been used as proxies for species-level or, more commonly, genus-level taxonomic identification. A number of software tools are available, which convert reads to sample-by-OTU feature tables, such as QIIME [55] and mothur [56]. However, newer approaches are better able to control for amplicon sequencing errors, and thereby obviate the use of arbitrary identity thresholds, allowing for single-nucleotide resolution with amplicon sequence variants (ASVs) [57]. Software options for ASV methods include DADA2 [58] and Deblur [59].
Figure 1.
Schematics of standard techniques used in microbiome studies. (A) Marker gene sequencing techniques can use primers to target certain conserved regions of a genome to capture intermittent variable regions, which can then be used to identify organisms in a sample rapidly and inexpensively. The 16S rRNA gene is the most commonly used marker gene in bacteria and archaea, and in the figure, primers are used to capture the V3 and V4 variable regions together, a common approach for 16S sequencing. The internal transcribed spacer (ITS) region of the nuclear rRNA cistron in fungi is made of two segments, which can be captured with primers targeting the 18S, 5.8S, and 28S rRNA sections that surround them. (B–D) Instead of targeting one small segment of the genome, these techniques capture the entirety of the genetic material from an organism. (B) Single virus genomics (SVG) uses a fluorescent stain to isolate individual virus particles in a sample by fluorescence-activated virus sorting (FAVS), wherein they are embedded in an agarose bead before undergoing whole genome amplification and sequencing. (C) Whole metagenome shotgun sequencing (WMS) involves the fragmentation of all DNA in a sample, sequencing of the fragments, and assembly of the sequences, which can then be mapped to reference genomes, or de novo assembly can be performed. (D) Metatranscriptomics also involves a shotgun sequencing approach, but it is performed after mRNA extraction. The outputs then allow for differential gene expression analysis. (E) Metabolomics and metaproteomics allow for quantification of the metabolites and proteins produced by the microbiome in a sample, respectively. Mass spectrometry is a common approach to quantification. Mock metabolite shapes in Figure 1 were generated using the JSME Molecular Editor by Peter Ertl and Bruno Bienfait licensed under CC-BY-NC-SA 3.0. Images of body sites and organs in Figure 1 and Figure 2 were obtained from Servier Medical Art by Servier licensed under CC-BY 3.0.Traditionally, 16S sequences were clustered into groups with at least 97% identity, called operational taxonomic units (OTUs), which have been used as proxies for species-level or, more commonly, genus-level taxonomic identification. A number of software tools are available, which convert reads to sample-by-OTU feature tables, such as QIIME [55] and mothur [56]. However, newer approaches are better able to control for amplicon sequencing errors, and thereby obviate the use of arbitrary identity thresholds, allowing for single-nucleotide resolution with amplicon sequence variants (ASVs) [57]. Software options for ASV methods include DADA2 [58] and Deblur [59].

Figure 2.
Oral and systemic diseases associated with the oral microbiome. A representation of the associations found between diseases with increases or decreases of the abundances of organisms in the oral cavity (listed in Table 1 and Table 2). Organisms listed in blue have been shown to be increased in abundance in the oral cavity in individuals presenting with the noted disease, and organisms listed in red have been shown to be decreased. Those in purple may be either increased or decreased depending on the conditions or progression of the disease. Images of body sites and organs in Figure 1 and Figure 2 were obtained from Servier Medical Art by Servier licensed under CC-BY 3.0.
Figure 2.
Oral and systemic diseases associated with the oral microbiome. A representation of the associations found between diseases with increases or decreases of the abundances of organisms in the oral cavity (listed in Table 1 and Table 2). Organisms listed in blue have been shown to be increased in abundance in the oral cavity in individuals presenting with the noted disease, and organisms listed in red have been shown to be decreased. Those in purple may be either increased or decreased depending on the conditions or progression of the disease. Images of body sites and organs in Figure 1 and Figure 2 were obtained from Servier Medical Art by Servier licensed under CC-BY 3.0.

Figure 3.
Gradients of abundances of consensus stomatotype-driving genera. Using a random subset of 500 samples from an oral microbiome dataset [10], samples were clustered into two stomatotypes using the weighted Unifrac distance measure. Type 1 samples are represented by circles and type 2 samples by squares. In each box, samples are colored by the total relative abundance of the indicated organisms. Overlaid are arrows indicating the tendency of the abundances of each organism noted in Table 3. In this subset of samples, Neisseria and Haemophilus strongly associate with stomatotype 1 samples, Prevotella strongly associates with stomatotype 2 samples while Veillonella does so weakly. The “variable stomatotype” drivers are indeed variable in their associations in this instance. Streptococcus shows a clear gradient but does not conform to either stomatotype. Gemella and Rothia, which have been shown to co-occur with Streptococcus in stomatotypes in the literature, do the same here, with Rothia more associated with stomatotype 1. However, Porphyromonas, which has been shown to co-occur with Streptococcus, Gemella, or Neisseria previously, associates with none of these here, and instead is strongly associated with stomatotype 2.
Figure 3.
Gradients of abundances of consensus stomatotype-driving genera. Using a random subset of 500 samples from an oral microbiome dataset [10], samples were clustered into two stomatotypes using the weighted Unifrac distance measure. Type 1 samples are represented by circles and type 2 samples by squares. In each box, samples are colored by the total relative abundance of the indicated organisms. Overlaid are arrows indicating the tendency of the abundances of each organism noted in Table 3. In this subset of samples, Neisseria and Haemophilus strongly associate with stomatotype 1 samples, Prevotella strongly associates with stomatotype 2 samples while Veillonella does so weakly. The “variable stomatotype” drivers are indeed variable in their associations in this instance. Streptococcus shows a clear gradient but does not conform to either stomatotype. Gemella and Rothia, which have been shown to co-occur with Streptococcus in stomatotypes in the literature, do the same here, with Rothia more associated with stomatotype 1. However, Porphyromonas, which has been shown to co-occur with Streptococcus, Gemella, or Neisseria previously, associates with none of these here, and instead is strongly associated with stomatotype 2.


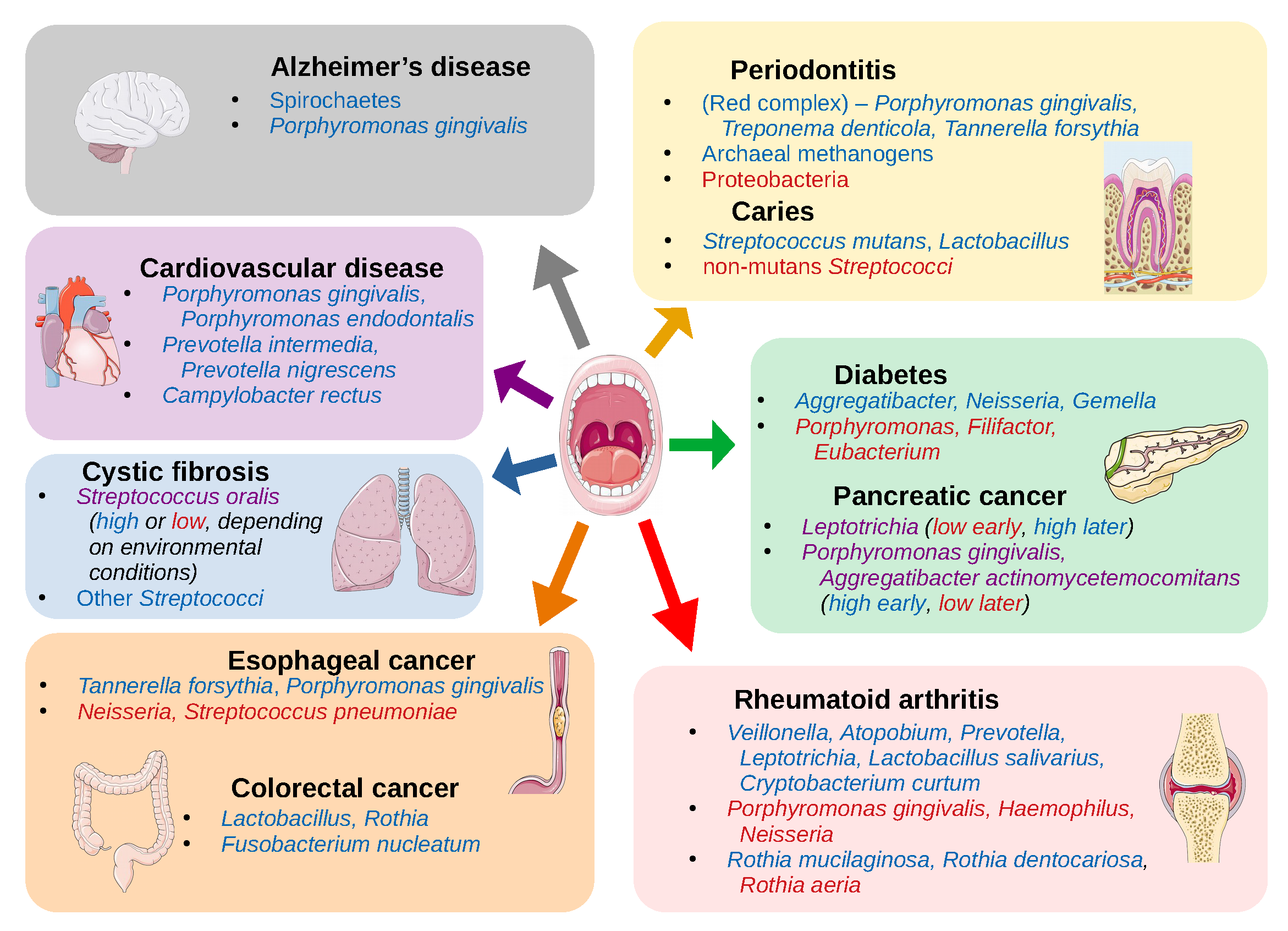{kind=link}
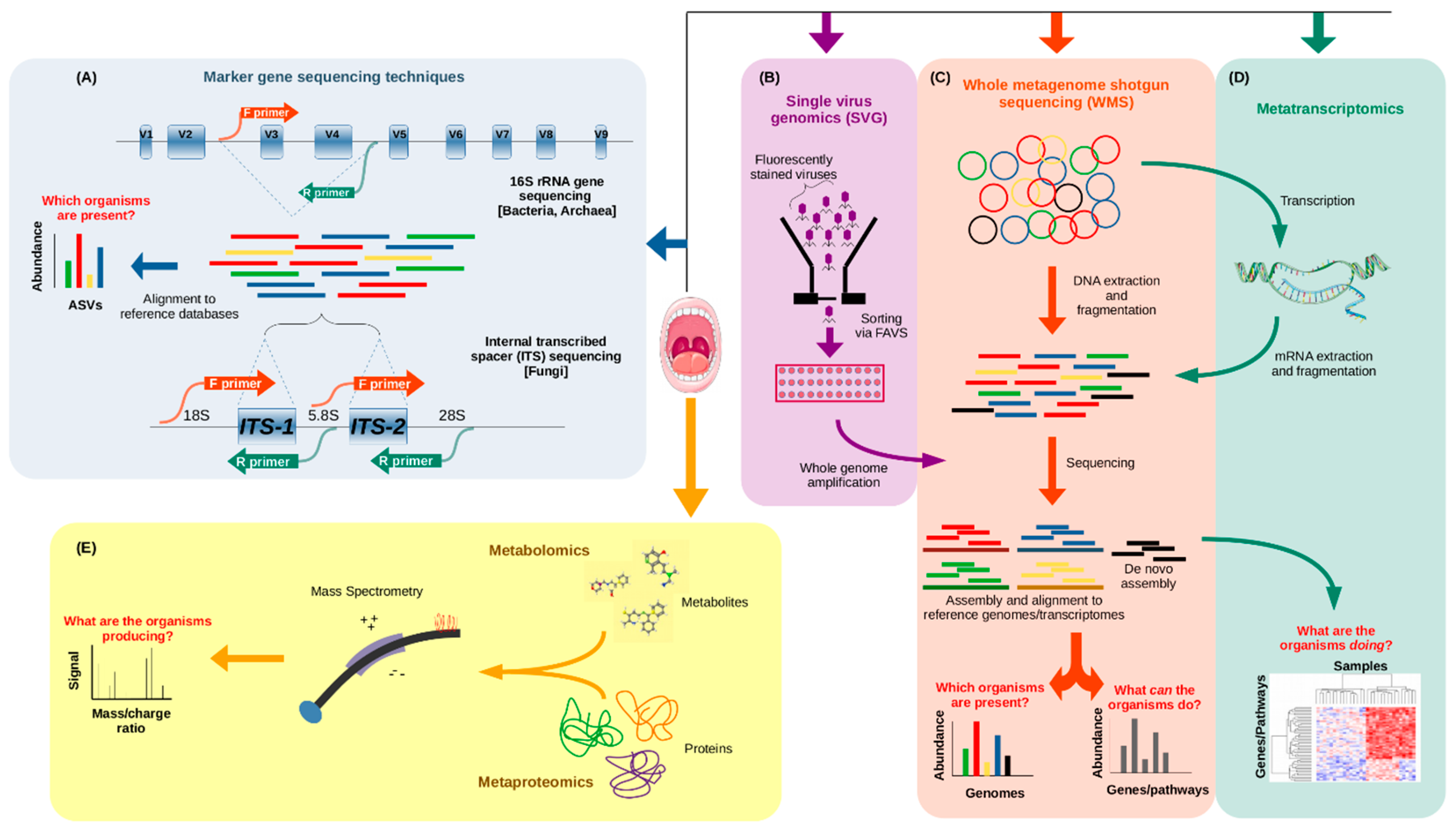{kind=link}
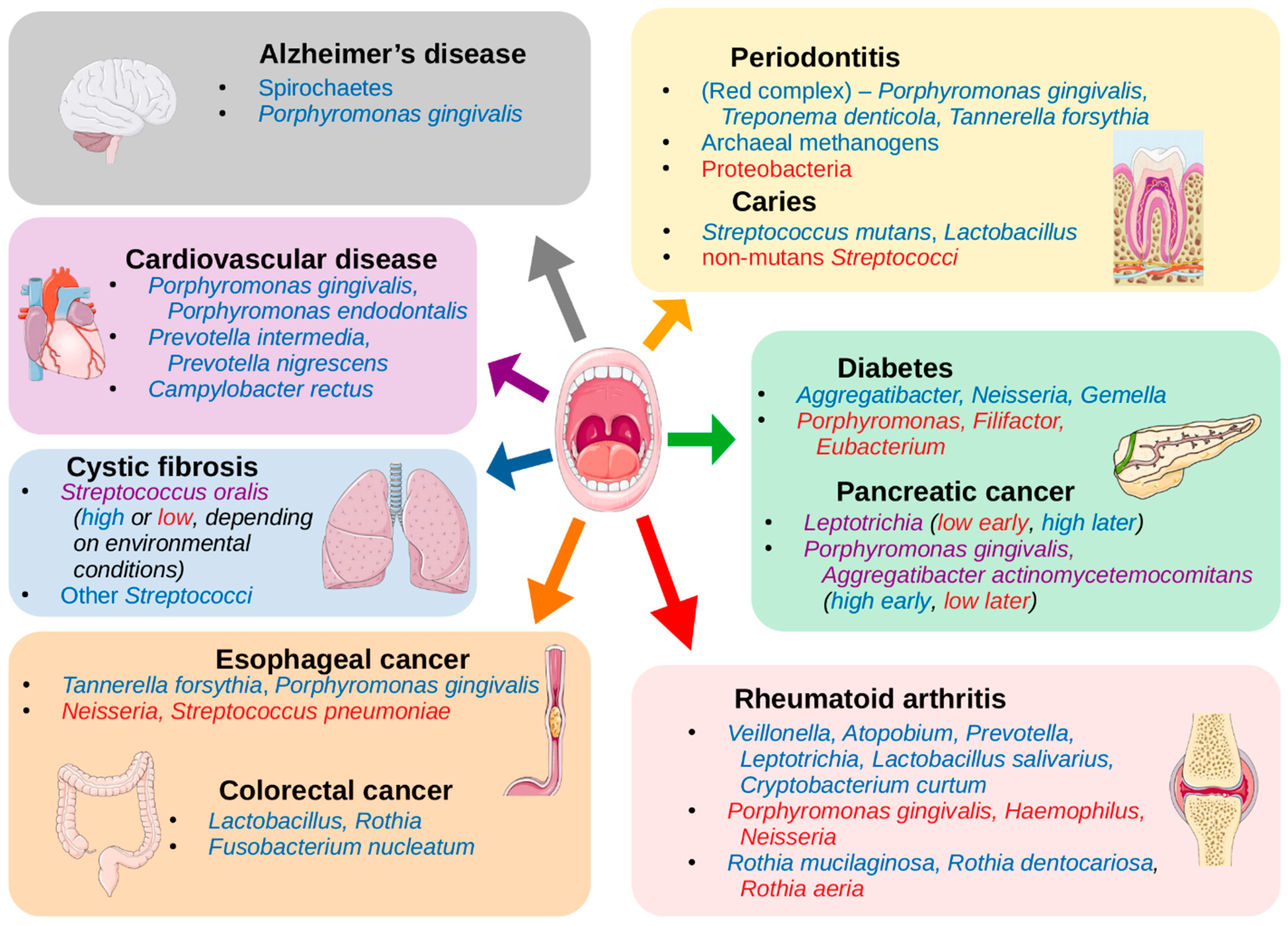{kind=link}
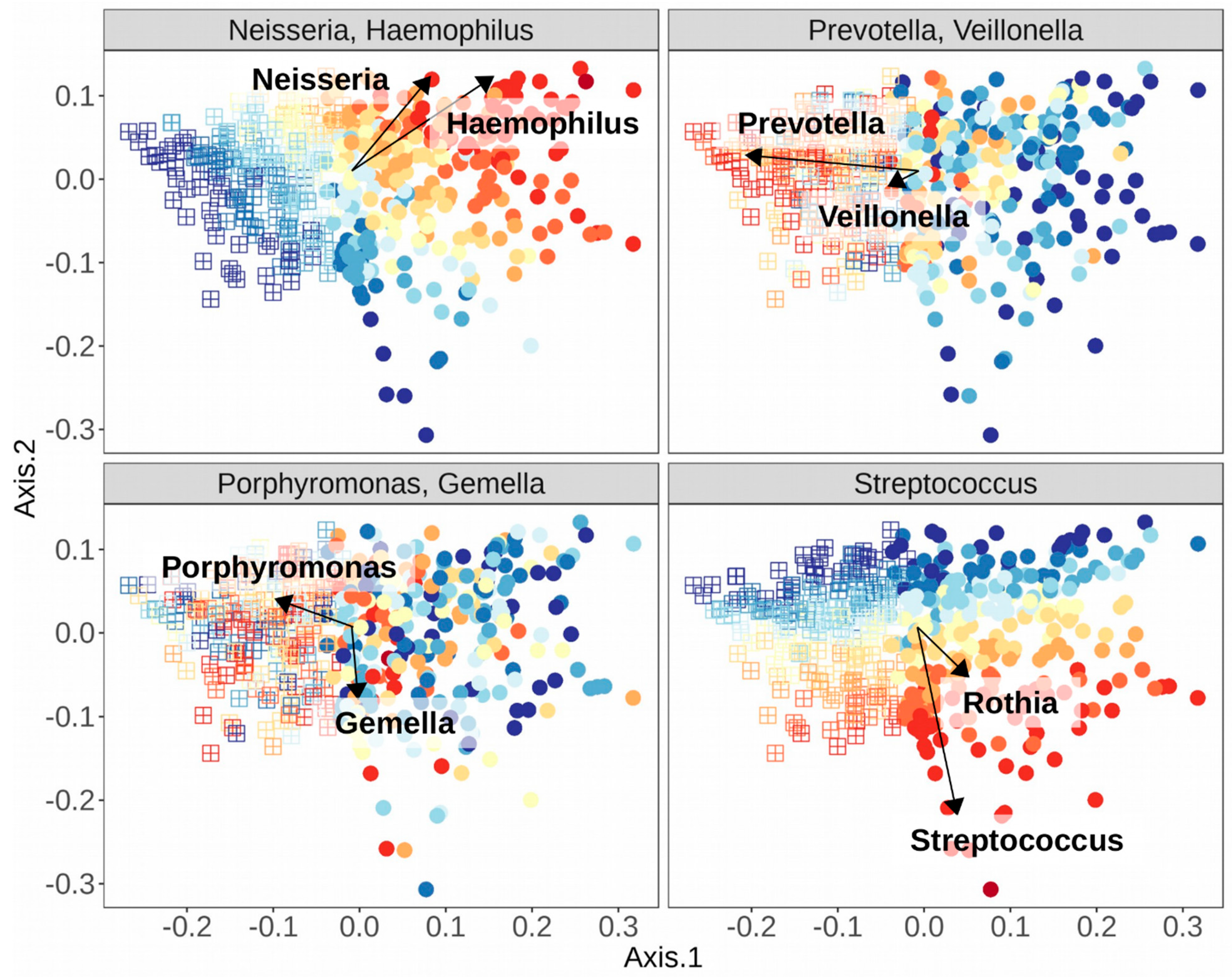{kind=link}
Table 1.
Examples of metagenomic studies of associations between the oral microbiome and oral diseases. The first column indicates a disease, the second indicates organisms that have been found at higher abundances in individuals presenting with the disease, the third indicates organisms at lower abundances, and the fourth contains the references to the literature, which displays these findings. (*) indicates taxa associated with oral cancer from a study in which samples were from tumor and non-tumor sites in the same patients and disease treatment is not specified.
Table 1.
Examples of metagenomic studies of associations between the oral microbiome and oral diseases. The first column indicates a disease, the second indicates organisms that have been found at higher abundances in individuals presenting with the disease, the third indicates organisms at lower abundances, and the fourth contains the references to the literature, which displays these findings. (*) indicates taxa associated with oral cancer from a study in which samples were from tumor and non-tumor sites in the same patients and disease treatment is not specified.
| Disease | Associated Organisms | Inhibited Organisms | Reference |
|---|---|---|---|
| Periodontitis | Phyla: Spirochaetes, Synergistetes and Bacteroidetes Classes: Clostridia, Negativicutes and Erysipelotrichia Genera: Prevotella, Fusobacterium Species: Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia, Filifactor alocis, Parvimonas micra, Aggregatibacter actinomycetemcomitans Archaea: Methanobrevibacter oralis, Methanobacterium curvum/congolense, and Methanosarcina mazeii | Phyla: Proteobacteria Classes: Bacilli Genera: Streptococcus, Actinomyces, Granulicatella | [14,15,16,17,18,19,20,21] |
| Dental caries | Genera: Neisseria, Selenomonas, Propionibacterium Species: Streptococcus mutans, Lactobacillus spp. Fungi: Candida albicans | Species: non-mutans Streptococci, Corynebacterium matruchotii, Capnocytophaga gingivalis, Eubacterium IR009, Campylobacter rectus, Lachnospiraceae sp. C1 | [22,23] |
| Oral cancer | Species: Capnocytophaga gingivalis, Prevotella melaninogenica and Streptococcus mitis, Peptostreptococcus stomatis*, Streptococcus salivarius*, Streptococcus gordonii*, Gemella haemolysans*, Gemella morbillorum*, Johnsonella ignava* and Streptococcus parasanguinis I* | Species: Granulicatella adiacens* | [24,25,26] |
| Esophageal cancer | Species: Tannerella forsythia, Porphyromonas gingivalis | Genera: Neisseria Species: Streptococcus pneumoniae | [27] |
Table 2.
Examples of metagenomic studies of associations between the oral microbiome and systemic diseases. The first column indicates a disease, the second indicates organisms that have been found at higher abundances in individuals presenting with the disease, the third indicates organisms at lower abundances, and the fourth contains the references to literature which displays these findings.
Table 2.
Examples of metagenomic studies of associations between the oral microbiome and systemic diseases. The first column indicates a disease, the second indicates organisms that have been found at higher abundances in individuals presenting with the disease, the third indicates organisms at lower abundances, and the fourth contains the references to literature which displays these findings.
| Disease | Associated Organisms | Inhibited Organisms | Reference |
|---|---|---|---|
| Colorectal cancer | Genera: Lactobacillus, Rothia Species: Fusobacterium nucleatum | [12,28,29,30] | |
| Pancreatic cancer | Genera: Leptotrichia (later in progression of disease) Species: Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans (at onset of disease) | Genera: Leptotrichia (at onset of disease) Species: Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans (later in progression of disease) | [31,32] |
| Cystic fibrosis | Species: Streptococcus oralis (depends on environmental conditions), S. mitis, S. gordonii and S. sanguinis | Species: Streptococcus oralis (depends on environmental conditions) | [33] |
| Cardiovascular disease | Species: Campylobacter rectus, Porphyromonas gingivalis, Porphyromonas endodontalis, Prevotella intermedia, Prevotella nigrescens, (oral commensals that were found on athersclerotic plaques - not necessarily at high abundance in oral cavity) | [34,35] | |
| Rheumatoid arthritis | Genera: Veillonella, Atopobium, Prevotella, Leptotrichia Species: Rothia mucilaginosa, Rothia dentocariosa, Lactobacillus salivarius, Cryptobacterium curtum | Genera: Haemophilus, Neisseria Species: Porphyromonas gingivalis, Rothia aeria | [36,37,38,39] |
| Alzheimer’s disease | Phyla: Spirochaetes Species: Porphyromonas gingivalis | [40,41,42] | |
| Diabetes | Genera: Aggregatibacter, Neisseria, Gemella, Eikenella, Selenomonas, Actinomyces, Capnocytophaga, Fusobacterium, Veillonella, Streptococcus | Genera: Porphyromonas, Filifactor, Eubacterium, Synergistetes, Tannerella, Treponema | [43] |
Table 3.
Consensus stomatotype-driving genera. Genera that have been shown in the literature to strongly drive the distinction between samples of oral microbiome datasets by differences in their abundances. The stomatotype numbers are arbitrarily assigned. The genera are listed in the second column, along with notes on associations between the organisms where relevant. The third column contains the references to the literature, which shows these stomatotype associations.
Table 3.
Consensus stomatotype-driving genera. Genera that have been shown in the literature to strongly drive the distinction between samples of oral microbiome datasets by differences in their abundances. The stomatotype numbers are arbitrarily assigned. The genera are listed in the second column, along with notes on associations between the organisms where relevant. The third column contains the references to the literature, which shows these stomatotype associations.
| Genus | References | |
|---|---|---|
| Stomatotype 1 | ||
| Neisseria | [10,48,49,51] | |
| Haemophilus | [10,48,51] | |
| Stomatotype 2 | ||
| Prevotella | [10,48,49,51] | |
| Veillonella | [10,48,51] | |
| Variable Stomatotypes | ||
| Streptococcus—varies depending on study and species | [10,48,49,51] | |
| Gemella—co-occurs with Streptococcus and Porphyromonas | [49,51] | |
| Porphyromonas—may co-occur with Streptococcus, Gemella, or Neisseria | [48,51] | |
| Rothia—co-occurs with varying species of Streptococcus, depending on study | [49,51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Willis, J.R.; Gabaldón, T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms 2020, 8, 308. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020308
AMA Style
Willis JR, Gabaldón T. The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems. Microorganisms. 2020; 8(2):308. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020308
Chicago/Turabian StyleWillis, Jesse R., and Toni Gabaldón. 2020. "The Human Oral Microbiome in Health and Disease: From Sequences to Ecosystems" Microorganisms 8, no. 2: 308. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8020308
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.