Whole Genome Sequencing and Comparative Genomic Analyses of Lysinibacillus pakistanensis LZH-9, a Halotolerant Strain with Excellent COD Removal Capability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Sampling, and Media
2.2. Enrichment, Isolation and Identification of Halotolerant Bacteria
2.3. COD Removal Efficiency Experiments
2.4. DNA Extraction, Genome Sequencing, Assembly and Annotation
2.5. Average Nucleotide Identity (ANI) and Whole Genome Alignments
2.6. Pan-Genome Analyses and Models Extrapolation of Lysinibacillus
2.7. Phylogenic Analyses
2.8. Genome Annotation
2.9. Prediction of Mobile Genetic Elements
2.10. Genome-wide Detection of Positively Selected Genes and Codon Adaption Index (CAI) Calculation
3. Results and Discussion
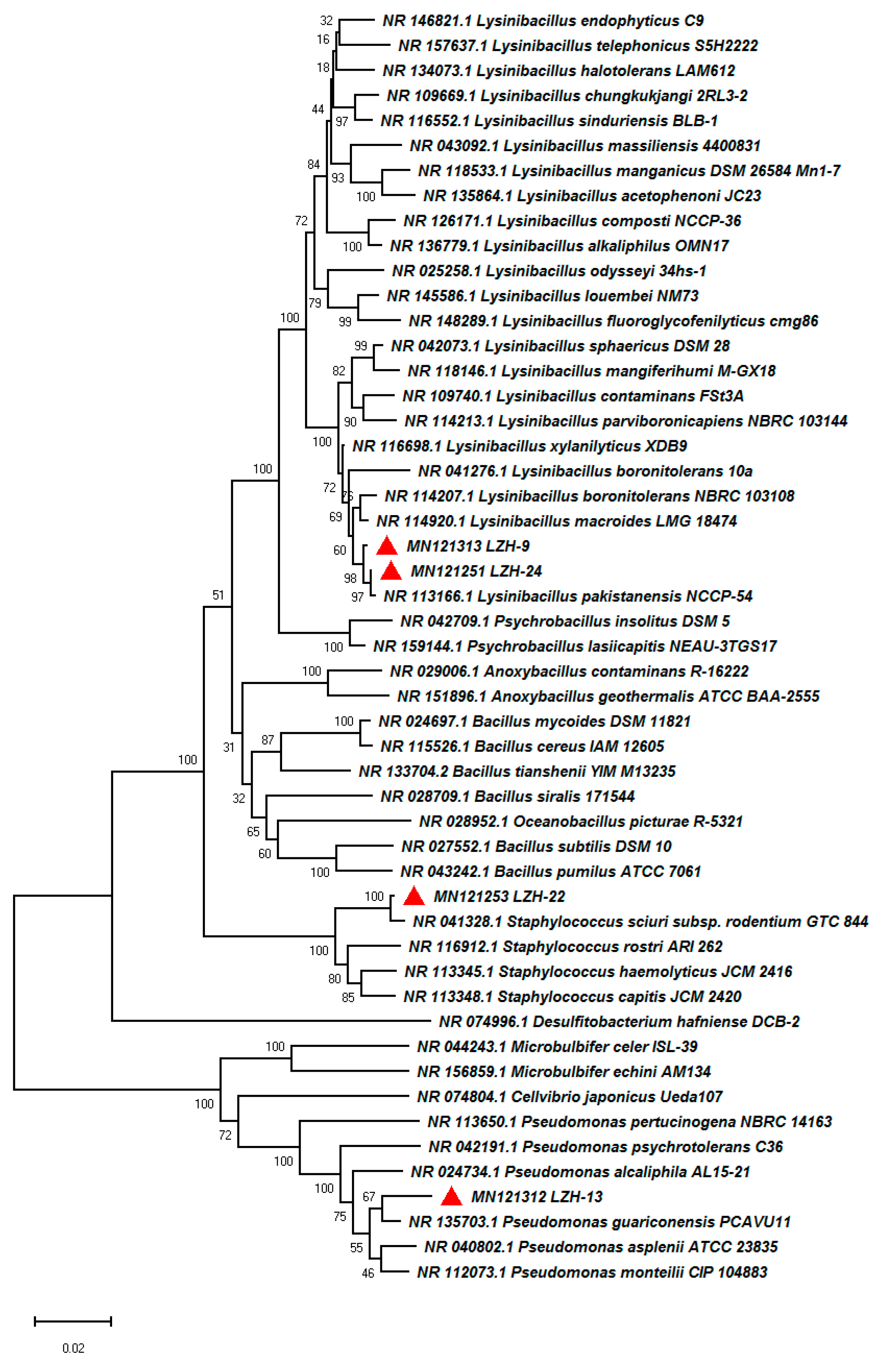
3.1. Isolation and Identification of Halotolerant Bacteria
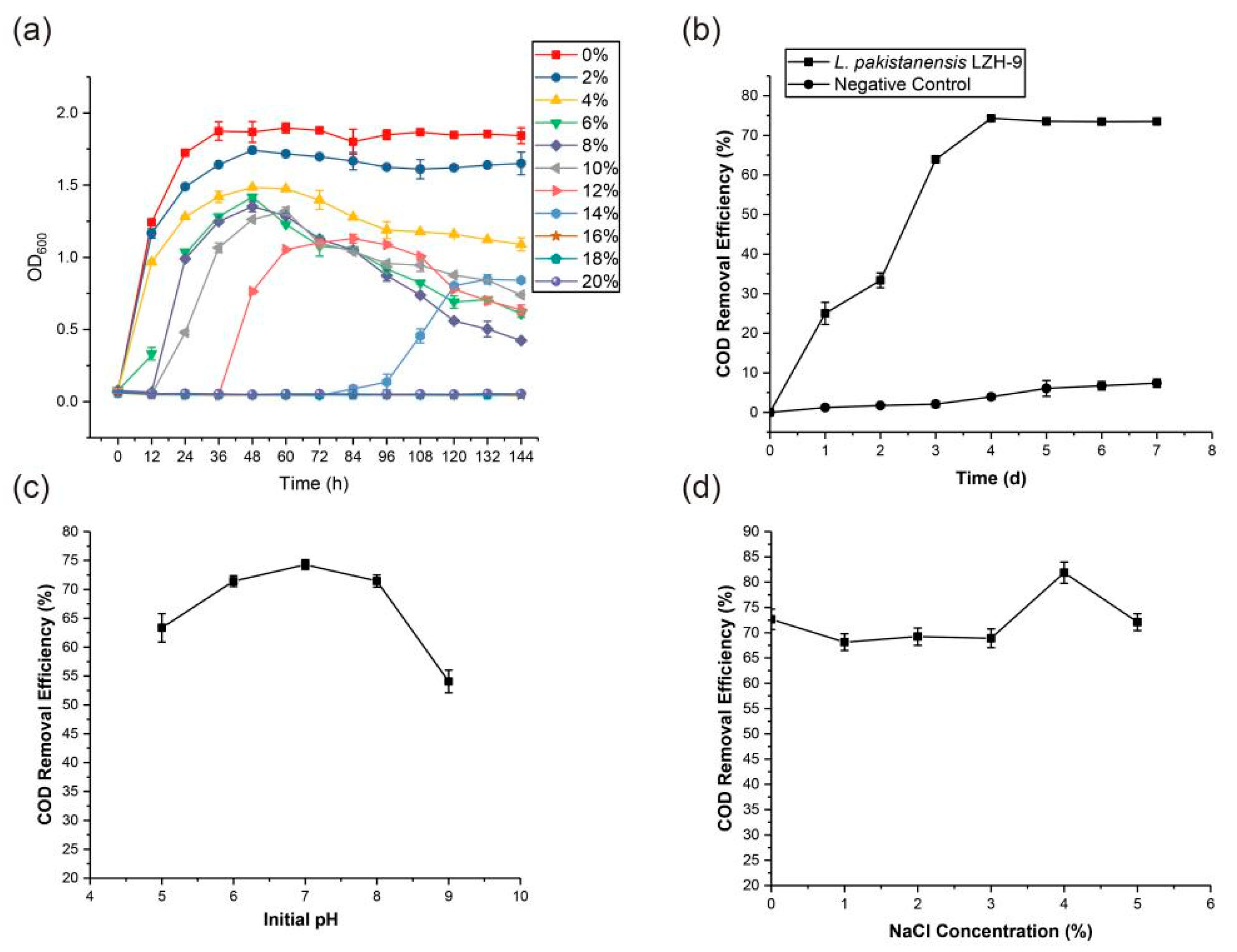
3.2. Optimization of COD Removal Efficiency with Strain LZH-9
3.3. Genomic Features
3.4. Core and Pan-genome of Lysinibacillus
3.5. Functional Potential and Phylogenetic Analyses
3.5.1. Carbon Metabolism
3.5.2. Nitrogen and Sulfur Metabolism
3.5.3. Energy Conservation and Transduction
3.6. Environmental Adaption
3.6.1. Resistance to Antibiotics and Toxic Metals
3.6.2. Capsular and Extracellular Polysacchrides
3.6.3. Halotolerance and Resistance to Osmotic Stress
3.6.4. Xenobiotics Biodegradation and Metabolism
3.6.5. Mobile Genetic Elements and CRISPR-Cas Systems
3.7. Positive Selection Analyses
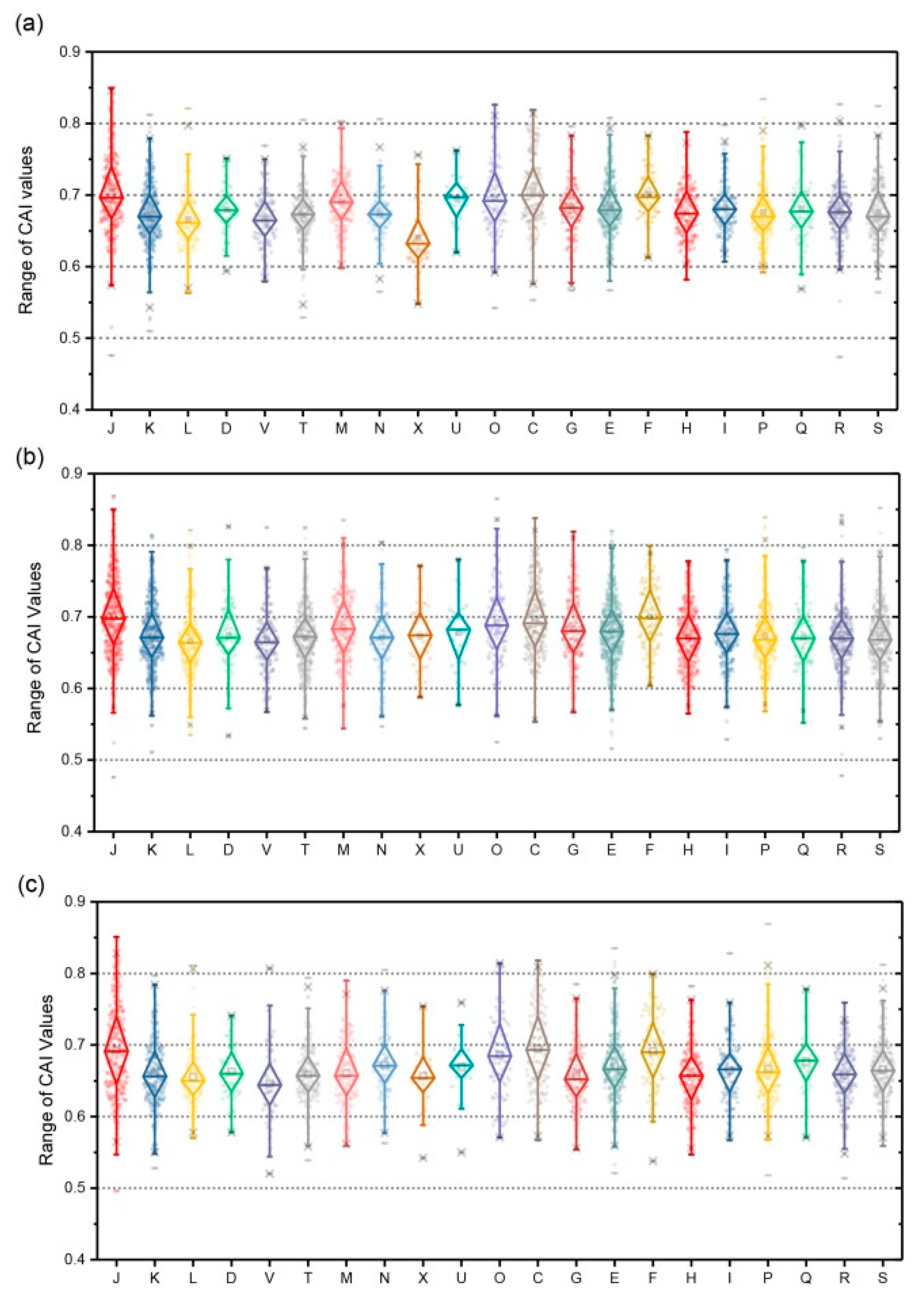
3.8. Expression Assessment with Codon Adaption Index (CAI)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiang, Y.; Wei, L.; Zhang, H.; Yang, K.; Wang, H. Removal performance and microbial communities in a sequencing batch reactor treating hypersaline phenol-laden wastewater. Bioresour. Technol. 2016, 218, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Jorfi, S.; Kujlu, R.; Ghafari, S.; Soltani, R.D.C.; Haghighifard, N.J. A novel salt-tolerant bacterial consortium for biodegradation of saline and recalcitrant petrochemical wastewater. J. Environ. Manag. 2017, 191, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Hiroe, J.; Murakami, M.; Fukami, H.; Tachiki, T. Treament of hypersaline-containing wastewater with salt- tolerant microorganisms. J. Biosci. Bioeng. 2001, 91, 222–224. [Google Scholar] [CrossRef]
- Soltani, R.D.C.; Jorfi, S.; Ramezani, H.; Purfadakari, S. Ultrasonically induced ZnO–biosilica nanocomposite for degradation of a textile dye in aqueous phase. Ultrason. Sonochem. 2016, 28, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Soltani, R.D.C.; Jorfi, S.; Safari, M.; Rajaei, M.S. Enhanced sonocatalysis of textile wastewater using bentonite-supported ZnO nanoparticles: Response surface methodological approach. J. Environ. Manag. 2016, 179, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, O.; Moletta, R. Treatment of organic pollution in industrial saline wastewater: A literature review. Water Res. 2006, 40, 3671–3682. [Google Scholar] [CrossRef]
- Castillo-Carvajal, L.C.; Sanz-Martín, J.L.; Barragán-Huerta, B.E. Biodegradation of organic pollutants in saline wastewater by halophilic microorganisms: A review. Environ. Sci. Pollut. Res. 2014, 21, 9578–9588. [Google Scholar] [CrossRef]
- Chung, T.-P.; Tseng, H.-Y.; Juang, R.-S. Mass transfer effect and intermediate detection for phenol degradation in immobilized Pseudomonas putida systems. Process. Biochem. 2003, 38, 1497–1507. [Google Scholar] [CrossRef]
- Lee, C.S.; Jung, Y.-T.; Park, S.; Oh, T.-K.; Yoon, J.-H. Lysinibacillus xylanilyticus sp. nov., a xylan-degrading bacterium isolated from forest humus. Int. J. Syst. Evol. Microbiol. 2010, 60, 281–286. [Google Scholar] [CrossRef]
- Kämpfer, P.; Martin, K.; Glaeser, S.P. Lysinibacillus contaminans sp. nov., isolated from surface water. Int. J. Syst. Evol. Microbiol. 2013, 63, 3148–3153. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Q.; Xu, L.; Wu, X.-L. Lysinibacillus alkalisoli sp. nov., isolated from saline–alkaline soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Li, G.; An, T.; Guo, B.; Sun, L.; Zu, L.; Ren, A. Biodegradation of ethanethiol in aqueous medium by a new Lysinibacillus sphaericus strain RG-1 isolated from activated sludge. Biogeochemistry 2010, 21, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Hu, Z.H.; Zhang, L.L.; Yu, X.; Chen, J. A novel dichloromethane-degrading Lysinibacillus sphaericus strain wh22 and its degradative plasmid. Appl. Microbiol. Biotechnol. 2009, 82, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ren, Y.; Deng, X.; Wei, C. Dual substrates biodegradation kinetics of m-cresol and pyridine by Lysinibacillus cresolivorans. J. Hazard. Mater. 2011, 186, 1136–1140. [Google Scholar] [CrossRef]
- Liang, B.; Lu, P.; Li, H.; Li, R.; Li, S.; Huang, X. Biodegradation of fomesafen by strain Lysinibacillus sp. ZB-1 isolated from soil. Chemosphere 2009, 77, 1614–1619. [Google Scholar] [CrossRef]
- Esmaeili, A.; Pourbabaee, A.A.; Alikhani, H.A.; Shabani, F.; Esmaeili, E. Biodegradation of Low-Density Polyethylene (LDPE) by Mixed Culture of Lysinibacillus xylanilyticus and Aspergillus niger in Soil. PLoS ONE 2013, 8, e71720. [Google Scholar] [CrossRef] [Green Version]
- Saratale, R.G.; Saratale, G.D.; Govindwar, S.; Kim, D.S. Exploiting the efficacy ofLysinibacillussp. RGS for decolorization and detoxification of industrial dyes, textile effluent and bioreactor studies. J. Environ. Sci. Heal. Part A 2015, 50, 176–192. [Google Scholar] [CrossRef]
- Kong, D.; Wang, Y.; Li, Y.; Song, J.; Zhai, Y.; Zhang, C.; Wang, H.; Chen, X.; Zhao, B.; Ruan, Z. Lysinibacillus halotolerans sp. nov., isolated from saline-alkaline soil. Int. J. Syst. Evol. Microbiol. 2014, 64, 2593–2598. [Google Scholar] [CrossRef]
- He, M.; Li, X.; Liu, H.; Miller, S.J.; Wang, G.; Rensing, C. Characterization and genomic analysis of a highly chromate resistant and reducing bacterial strain Lysinibacillus fusiformis ZC1. J. Hazard. Mater. 2011, 185, 682–688. [Google Scholar] [CrossRef]
- Chaudhari, A.U.; Tapase, S.; Markad, V.L.; Kodam, K. Simultaneous decolorization of reactive Orange M2R dye and reduction of chromate by Lysinibacillus sp. KMK-A. J. Hazard. Mater. 2013, 262, 580–588. [Google Scholar] [CrossRef]
- Bahuguna, A.; Lily, M.K.; Munjal, A.; Singh, R.N.; Dangwal, K.; Lily, M.K. Desulfurization of dibenzothiophene (DBT) by a novel strain Lysinibacillus sphaericus DMT-7 isolated from diesel contaminated soil. J. Environ. Sci. 2011, 23, 975–982. [Google Scholar] [CrossRef]
- Rahman, A.; Nahar, N.; Nawani, N.; Jass, J.; Desale, P.; Kapadnis, B.; Hossain, K.; Saha, A.K.; Ghosh, S.; Olsson, B.; et al. Isolation and characterization of a Lysinibacillus strain B1-CDA showing potential for bioremediation of arsenics from contaminated water. J. Environ. Sci. Heal. Part A 2014, 49, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Saratale, R.G.; Gandhi, S.S.; Purankar, M.V.; Kurade, M.B.; Govindwar, S.; Oh, S.E.; Saratale, G.D. Decolorization and detoxification of sulfonated azo dye C.I. Remazol Red and textile effluent by isolated Lysinibacillus sp. RGS. J. Biosci. Bioeng. 2013, 115, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Zhou, D.; Liu, Y.; Yu, R.; Qiu, G.; Zeng, W. Effects of pH value on the expression of key iron/sulfur oxidation genes during bioleaching of chalcopyrite on thermophilic condition. Ann. Microbiol. 2019, 69, 627–635. [Google Scholar] [CrossRef]
- Ai, C.; Yan, Z.; Chai, H.; Gu, T.; Wang, J.; Chai, L.; Qiu, G.; Zeng, W. Increased chalcopyrite bioleaching capabilities of extremely thermoacidophilic Metallosphaera sedula inocula by mixotrophic propagation. J. Ind. Microbiol. Biotechnol. 2019, 46, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, X.; Shen, L.; Li, J.; Yu, R.; Liu, Y.; Qiu, G.; Zeng, W. Whole Genome Sequencing and Comparative Genomics Analyses of Pandoraea sp. XY-2, a New Species Capable of Biodegrade Tetracycline. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Duan, J.; Fang, H.; Su, B.; Chen, J.; Lin, J. Characterization of a halophilic heterotrophic nitrification–aerobic denitrification bacterium and its application on treatment of saline wastewater. Bioresour. Technol. 2015, 179, 421–428. [Google Scholar] [CrossRef]
- Ai, C.; Yan, Z.; Zhou, H.; Hou, S.; Chai, L.; Qiu, G.; Zeng, W. Metagenomic insights into the effects of seasonal temperature variation on functional potentials of activated sludge. Microorganisms 2019, 7, 713. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.; McCombie, W.R. Library Quantification: Fluorometric Quantitation of Double-Stranded or Single-Stranded DNA Samples Using the Qubit System. Cold Spring Harb. Protoc. 2016, 2017. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.; Bergman, N.H.; Phillippy, A. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; De Silva, N.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Boil. 2015, 16, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Coleman-Derr, D.; Chen, G.; Gu, Y.Q. OrthoVenn: A web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015, 43, W78–W84. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Boil. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.-F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. Adv. Struct. Saf. Stud. 2009, 537, 113–137. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Boil. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.Y.; Glass, E.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2016, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. dbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2017, 46, D516–D521. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.; Choi, J.; Ahn, K.; Park, B.; Park, J.; Kang, S.; Lee, Y.-H. Fungal cytochrome P450 database. BMC Genom. 2008, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Finn, R.D. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2015, 44, D343–D350. [Google Scholar] [CrossRef] [Green Version]
- Gerlt, J.A. Genomic Enzymology: Web Tools for Leveraging Protein Family Sequence–Function Space and Genome Context to Discover Novel Functions. Biochemistry 2017, 56, 4293–4308. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F. Simon Fraser University Research Computing Group IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michálik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahm, A.; Bens, M.; Platzer, M.; Szafranski, K. PosiGene: Automated and easy-to-use pipeline for genome-wide detection of positively selected genes. Nucleic Acids Res. 2017, 45, e100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigbo, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Boil. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Kargi, F.; Dincer, A.R. Biological Treatment of Saline Wastewater by Fed-Batch Operation. J. Chem. Technol. Biotechnol. 1997, 69, 167–172. [Google Scholar] [CrossRef]
- Dinçer, A.; Kargı, F. Performance of rotating biological disc system treating saline wastewater. Process. Biochem. 2001, 36, 901–906. [Google Scholar] [CrossRef]
- Bomfim, N.; Nassar, N. Development of cassava periclinal chimera may boost production. Genet. Mol. Res. 2014, 13, 819–830. [Google Scholar] [CrossRef]
- Crane, B.R.; Sudhamsu, J.; Patel, B.A. Bacterial Nitric Oxide Synthases. Annu. Rev. Biochem. 2010, 79, 445–470. [Google Scholar] [CrossRef]
- Kers, J.A.; Wach, M.J.; Krasnoff, S.B.; Widom, J.; Cameron, K.D.; Bukhalid, R.A.; Gibson, D.M.; Crane, B.R.; Loria, R. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature 2004, 429, 79–82. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Y.; Yuan, M.; Du, G.; Chen, J.; Kang, Z. A Bacillus paralicheniformis Iron-Containing Urease Reduces Urea Concentrations in Rice Wine. Appl. Environ. Microbiol. 2017, 83, e01258-17. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Le Brun, N.E. Studies of the Cytochrome Subunits of Menaquinone:CytochromecReductase (bcComplex) ofBacillus subtilis. J. Boil. Chem. 1998, 273, 8860–8866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzschke, A.; Bettenbrock, K. All three quinone species play distinct roles in ensuring optimal growth under aerobic and fermentative conditions in E. coli K12. PLoS ONE 2018, 13, e0194699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecsi, I.; Hards, K.; Ekanayaka, N.; Berney, M.; Hartman, T.E.; Jacobs, W.R.; Cook, G.M. Essentiality of Succinate Dehydrogenase in Mycobacterium smegmatis and Its Role in the Generation of the Membrane Potential Under Hypoxia. mBio 2014, 5, e01093-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carsten, S.; Serdar, T.; Dong-Woo, L.; Fevzi, D. Cytochrome c biogenesis: The Ccm system. Trends Microbiol. 2010, 18, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Hutchings, M.I.; Hong, H.-J.; Buttner, M.J. The vancomycin resistance VanRS two-component signal transduction system of Streptomyces coelicolor. Mol. Microbiol. 2006, 59, 923–935. [Google Scholar] [CrossRef]
- Bisicchia, P.; Bui, N.K.; Aldridge, C.; Vollmer, W.; Devine, K. Acquisition of VanB-type vancomycin resistance by Bacillus subtilis: The impact on gene expression, cell wall composition and morphology. Mol. Microbiol. 2011, 81, 157–178. [Google Scholar] [CrossRef]
- Thompson, M.K.; Keithly, M.E.; Goodman, M.C.; Hammer, N.D.; Cook, P.D.; Jagessar, K.L.; Harp, J.; Skaar, E.P.; Armstrong, R.N. Structure and Function of the Genomically Encoded Fosfomycin Resistance Enzyme, FosB, from Staphylococcus aureus. Biochemistry 2014, 53, 755–765. [Google Scholar] [CrossRef]
- Li, W.; Atkinson, G.C.; Thakor, N.V.; Allas, Ü.; Lu, C.-C.; Chan, K.-Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V.; et al. Mechanism of Tetracycline Resistance by Ribosomal Protection Protein Tet(O). Nat. Commun. 2013, 4, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aires, J.; Doucet-Populaire, F.; Butel, M.J. Tetracycline Resistance Mediated by tet(W), tet(M), and tet(O) Genes of Bifidobacterium Isolates from Humans. Appl. Environ. Microbiol. 2007, 73, 2751–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, A.L.; Van Der Verren, S.; Näsvall, J.; Gutiérrez-De-Terán, H.; Selmer, M. Structural mechanism of AadA, a dual-specificity aminoglycoside adenylyltransferase from Salmonella enterica. J. Boil. Chem. 2018, 293, 11481–11490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burckhardt, R.M.; Escalante-Semerena, J.C. In Bacillus subtilis, the SatA (Formerly YyaR) Acetyltransferase Detoxifies Streptothricin via Lysine Acetylation. Appl. Environ. Microbiol. 2017, 83, e01590-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burckhardt, R.M.; Escalante-Semerena, J.C. Insights into the Function of the N-Acetyltransferase SatA That Detoxifies Streptothricin in Bacillus subtilis and Bacillus anthracis. Appl. Environ. Microbiol. 2019, 85, 85. [Google Scholar] [CrossRef] [Green Version]
- Bernard, R.; Guiseppi, A.; Chippaux, M.; Foglino, M.; Denizot, F. Resistance to Bacitracin in Bacillus subtilis: Unexpected Requirement of the BceAB ABC Transporter in the Control of Expression of Its Own Structural Genes. J. Bacteriol. 2007, 189, 8636–8642. [Google Scholar] [CrossRef] [Green Version]
- Stéphanie, C.F.; Céline, B.A.; Annick, G.; Fran Ois, D.; Maryline, F. A new highly conserved antibiotic sensing/resistance pathway in firmicutes involves an ABC transporter interplaying with a signal transduction system. PLoS ONE 2011, 6, e15951. [Google Scholar] [CrossRef]
- Furuta, Y.; Harima, H.; Ito, E.; Maruyama, F.; Ohnishi, N.; Osaki, K.; Ogawa, H.; Squarre, D.; Hang’Ombe, B.M.; Higashi, H. Loss of Bacitracin Resistance Due to a Large Genomic Deletion among Bacillus anthracis Strains. mSystems 2018, 3, e00182-18. [Google Scholar] [CrossRef] [Green Version]
- Cariss, S.J.L.; Constantinidou, C.; Patel, M.D.; Takebayashi, Y.; Hobman, J.; Penn, C.W.; Avison, M.B. YieJ (CbrC) Mediates CreBC-Dependent Colicin E2 Tolerance in Escherichia coli. J. Bacteriol. 2010, 192, 3329–3336. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, M.; Audurier, A.; Cossart, P. Plasmid-borne cadmium resistance genes in Listeria monocytogenes are similar to cadA and cadC of Staphylococcus aureus and are induced by cadmium. J. Bacteriol. 1994, 176, 3040–3048. [Google Scholar] [CrossRef] [Green Version]
- Anton, A.; Große, C.; Reißmann, J.; Pribyl, T.; Nies, D.H. CzcD Is a Heavy Metal Ion Transporter Involved in Regulation of Heavy Metal Resistance in Ralstonia sp. Strain CH34. J. Bacteriol. 1999, 181, 6876–6881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Genet. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Maseda, H.; Lu, Z.; Utsumi, M.; Zhang, Z.; Sugiura, N. Investigations into the biodegradation of microcystin-LR mediated by the biofilm in wintertime from a biological treatment facility in a drinking-water treatment plant. Bioresour. Technol. 2012, 106, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Szanto, M.; Pavlov, V. Biofilm development of the polyethylene-degrading bacterium Rhodococcus ruber. Appl. Microbiol. Biotechnol. 2006, 72, 346–352. [Google Scholar] [CrossRef]
- Radchenkova, N.; Boyadzhieva, I.; Atanasova, N.; Poli, A.; Finore, I.; Di Donato, P.; Nicolaus, B.; Panchev, I.; Kuncheva, M.; Kambourova, M. Extracellular polymer substance synthesized by a halophilic bacterium Chromohalobacter canadensis 28. Appl. Microbiol. Biotechnol. 2018, 102, 4937–4949. [Google Scholar] [CrossRef]
- Zivkovic, M.; Miljkovic, M.; Rúas-Madiedo, P.; Strahinic, I.; Tolinacki, M.; Golic, N.; Kojic, M. Exopolysaccharide Production and Ropy Phenotype Are Determined by Two Gene Clusters in Putative Probiotic Strain Lactobacillus paraplantarum BGCG11. Appl. Environ. Microbiol. 2015, 81, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Boels, I.C.; Beerthuyzen, M.M.; Kosters, M.H.W.; Van Kaauwen, M.P.W.; Kleerebezem, M.; De Vos, W.M. Identification and Functional Characterization of the Lactococcus lactis rfb Operon, Required for dTDP-Rhamnose Biosynthesis. J. Bacteriol. 2004, 186, 1239–1248. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Schell, M.A. Molecular characterization of the eps gene cluster of Pseudomonas solanacearum and its transcriptional regulation at a single promoter. Mol. Microbiol. 1995, 16, 977–989. [Google Scholar] [CrossRef]
- Minic, Z.; Marie, C.; Delorme, C.; Faurie, J.-M.; Mercier, G.; Ehrlich, D.; Renault, P. Control of EpsE, the Phosphoglycosyltransferase Initiating Exopolysaccharide Synthesis in Streptococcus thermophilus, by EpsD Tyrosine Kinase. J. Bacteriol. 2006, 189, 1351–1357. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, M.; Rahayu, J.; Sterling, G. A General Framework Based on Dynamic Constraints for the Enrichment of a Topological Theory of Spatial Simulation. Comput. Vis. 2005, 3684, 366–373. [Google Scholar] [CrossRef]
- Benachour, A.; Ladjouzi, R.; Le Jeune, A.; Hébert, L.; Thorpe, S.; Courtin, P.; Chapot-Chartier, M.-P.; Prajsnar, T.K.; Foster, S.J.; Mesnage, S. The Lysozyme-Induced Peptidoglycan N-Acetylglucosamine Deacetylase PgdA (EF1843) Is Required for Enterococcus faecalis Virulence. J. Bacteriol. 2012, 194, 6066–6073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Sudiarta, I.P.; Kodama, T.; Fukushima, T.; Ara, K.; Ozaki, K.; Sekiguchi, J. Identification and Characterization of a Novel Polysaccharide Deacetylase C (PdaC) from Bacillus subtilis. J. Boil. Chem. 2012, 287, 9765–9776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, D.; Urch, J.E.; Cate, J.M.T.; Rao, V.A.; Van Aalten, D.M.F.; Crielaard, W. Streptococcus mutans SMU.623c Codes for a Functional, Metal-Dependent Polysaccharide Deacetylase That Modulates Interactions with Salivary Agglutinin. J. Bacteriol. 2008, 191, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, E.G.; Nishida, A.; Weston, A.; Bañuelos, M.S.; Potter, K.; Conery, J.; Guillemin, K. Agent-Based Modeling Demonstrates How Local Chemotactic Behavior Can Shape Biofilm Architecture. mSphere 2019, 4, e00285-19. [Google Scholar] [CrossRef] [Green Version]
- Xavier, K.B.; Bassler, B.L. Interference with AI-2-mediated bacterial cell–cell communication. Nature 2005, 437, 750–753. [Google Scholar] [CrossRef] [Green Version]
- Laganenka, L.; Sourjik, V. Autoinducer 2-Dependent Escherichia coli Biofilm Formation Is Enhanced in a Dual-Species Coculture. Appl. Environ. Microbiol. 2018, 84, e02638-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ku, X.; Zhang, X.; Zhang, Y.; Chen, G.; Chen, F.; Zeng, W.; Li, J.; Zhu, L.; He, Q. The AI-2/luxS Quorum Sensing System Affects the Growth Characteristics, Biofilm Formation, and Virulence of Haemophilus parasuis. Front. Microbiol. 2019, 9, 62. [Google Scholar] [CrossRef]
- Sewald, X.; Saum, S.H.; Palm, P.; Pfeiffer, F.; Oesterhelt, D.; Müller, V. Autoinducer-2-Producing Protein LuxS, a Novel Salt- and Chloride-Induced Protein in the Moderately Halophilic Bacterium Halobacillus halophilus. Appl. Environ. Microbiol. 2006, 73, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-H.; Lu, C.-W.; Shyu, Y.-T.; Lin, S.-S. Revealing the Saline Adaptation Strategies of the Halophilic Bacterium Halomonas beimenensis through High-throughput Omics and Transposon Mutagenesis Approaches. Sci. Rep. 2017, 7, 13037. [Google Scholar] [CrossRef]
- Price-Whelan, A.; Poon, C.K.; Benson, M.A.; Eidem, T.T.; Roux, C.M.; Boyd, J.M.; Dunman, P.M.; Torres, V.J.; Krulwich, T.A. Transcriptional Profiling of Staphylococcus aureus During Growth in 2 M NaCl Leads to Clarification of Physiological Roles for Kdp and Ktr K+ Uptake Systems. mBio 2013, 4, e00407-13. [Google Scholar] [CrossRef] [Green Version]
- Lang, E.; Guyot, S.; Alvarez-Martin, P.; Perrier-Cornet, J.-M.; Gervais, P. Caco-2 Invasion by Cronobacter sakazakii and Salmonella enterica Exposed to Drying and Heat Treatments in Dried State in Milk Powder. Front. Microbiol. 2017, 8, 1893. [Google Scholar] [CrossRef] [PubMed]
- Domiínguez-Ferreras, A.; Muñoz, S.; Olivares, J.; Soto, M.J.; Sanjuán, J. Role of Potassium Uptake Systems in Sinorhizobium meliloti Osmoadaptation and Symbiotic Performance. J. Bacteriol. 2009, 191, 2133–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laimins, L.A.; Rhoads, D.B.; Epstein, W. Osmotic control of kdp operon expression in Escherichia coli. Proc. Natl. Acad. Sci. USA 1981, 78, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levina, N.; Totemeyer, S.; Stokes, N.R.; Louis, P.; Jones, M.A.; Booth, I.R. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: Identification of genes required for MscS activity. EMBO J. 1999, 18, 1730–1737. [Google Scholar] [CrossRef]
- McLaggan, D.; Jones, M.A.; Gouesbet, G.; Levina, N.; Lindey, S.; Epstein, W.; Booth, I.R. Analysis of the kefA2 mutation suggests that KefA is a cation-specific channel involved in osmotic adaptation in Escherichia coli. Mol. Microbiol. 2002, 43, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Vieira-Pires, R.; Szollosi, A.; Cabral, J.H.M. The structure of the KtrAB potassium transporter. Nature 2013, 496, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Szollosi, A.; Vieira-Pires, R.; Teixeira-Duarte, C.M.; Rocha, R.; Cabral, J.H.M. Dissecting the Molecular Mechanism of Nucleotide-Dependent Activation of the KtrAB K+ Transporter. PLoS Boil. 2016, 14, e1002356. [Google Scholar] [CrossRef]
- Tamara, H.; Erhard, B. Protection of Bacillus subtilis against cold stress via compatible-solute acquisition. J. Bacteriol. 2011, 193, 1552–1562. [Google Scholar] [CrossRef] [Green Version]
- Holtmann, G.J.; Bremer, E. Thermoprotection of Bacillus subtilis by Exogenously Provided Glycine Betaine and Structurally Related Compatible Solutes: Involvement of Opu Transporters. J. Bacteriol. 2004, 186, 1683–1693. [Google Scholar] [CrossRef] [Green Version]
- Bashir, A.; Hoffmann, T.; Smits, S.H.J.; Bremer, E. Dimethylglycine Provides Salt and Temperature Stress Protection to Bacillus subtilis. Appl. Environ. Microbiol. 2014, 80, 2773–2785. [Google Scholar] [CrossRef] [Green Version]
- Scholz, A.; Stahl, J.; Berardinis, V.; Müller, V.; Averhoff, B. Osmotic stress response in Acinetobacter baylyi: Identification of a glycine-betaine biosynthesis pathway and regulation of osmoadaptive choline uptake and glycine-betaine synthesis through a choline-responsive BetI repressor. Environ. Microbiol. Rep. 2016, 8, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Bremer, E. Guardians in a stressful world: The Opu family of compatible solute transporters from Bacillus subtilis. Boil. Chem. 2017, 398, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Wensing, A.; Brosius, M.; Steil, L.; Völker, U.; Bremer, E. Osmotic Control of opuA Expression in Bacillus subtilis and Its Modulation in Response to Intracellular Glycine Betaine and Proline Pools. J. Bacteriol. 2012, 195, 510–522. [Google Scholar] [CrossRef] [PubMed]
- André, S.; Jason, B.; Linda, B.S.; Susanne, K.; Jutta, G.; Gudrun, H.; Kay, D.; Wolfram, W.; Erhard, B. Cation-pi interactions as determinants for binding of the compatible solutes glycine betaine and proline betaine by the periplasmic ligand-binding protein ProX from Escherichia coli. J. Biol. Chem. 2004, 279, 5588–5596. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukkarasu, A.; Kumar, V. Role of proP and proU in betaine uptake by Yersinia enterocolitica under cold and osmotic stress conditions. Appl. Environ. Microbiol. 2009, 75, 1471–1477. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, A.M.; Uratsu, S.L. A single base pair change in proline biosynthesis genes causes osmotic stress tolerance. J. Bacteriol. 1988, 170, 5943–5945. [Google Scholar] [CrossRef] [Green Version]
- Sleator, R.D.; Gahan, C.G.; Hill, C. Mutations in the Listerial proB Gene Leading to Proline Overproduction: Effects on Salt Tolerance and Murine Infection. Appl. Environ. Microbiol. 2001, 67, 4560–4565. [Google Scholar] [CrossRef] [Green Version]
- Diehl, R.C.; Guinn, E.J.; Capp, M.W.; Tsodikov, O.V.; Record, M.T. Quantifying Additive Interactions of the Osmolyte Proline with Individual Functional Groups of Proteins: Comparisons with Urea and Glycine Betaine, Interpretation of m-Values. Biochemistry 2013, 52, 5997–6010. [Google Scholar] [CrossRef] [Green Version]
- Harayama, S.; Rekik, M.; Ngai, K.L.; Ornston, L.N. Physically associated enzymes produce and metabolize 2-hydroxy-2,4-dienoate, a chemically unstable intermediate formed in catechol metabolism via meta cleavage in Pseudomonas putida. J. Bacteriol. 1989, 171, 6251–6258. [Google Scholar] [CrossRef] [Green Version]
- Springael, D.; Top, E.M. Horizontal gene transfer and microbial adaptation to xenobiotics: New types of mobile genetic elements and lessons from ecological studies. Trends Microbiol. 2004, 12, 53–58. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the Immune System of Bacteria and Archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankapalli, K.; Saladi, S.; Awadia, S.S.; Goswami, A.V.; Samaddar, M.; D’Silva, P. Robust Glyoxalase activity of Hsp31, a ThiJ/DJ-1/PfpI Family Member Protein, Is Critical for Oxidative Stress Resistance inSaccharomyces cerevisiae. J. Boil. Chem. 2015, 290, 26491–26507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, K.P.; Choi, D.; Kim, I.; Min, B.; Park, C. Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 2011, 81, 926–936. [Google Scholar] [CrossRef]
- Mujacic, M.; Bader, M.W.; Baneyx, F.; Baneyx, F. Escherichia coli Hsp31 functions as a holding chaperone that cooperates with the DnaK-DnaJ-GrpE system in the management of protein misfolding under severe stress conditions. Mol. Microbiol. 2003, 51, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Mirna, M.; Fran Ois, B. Chaperone Hsp31 contributes to acid resistance in stationary-phase Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 1014–1018. [Google Scholar] [CrossRef] [Green Version]
- Behrens-Kneip, S. The role of SurA factor in outer membrane protein transport and virulence. Int. J. Med Microbiol. 2010, 300, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Soltes, G.R.; Schwalm, J.; Ricci, D.P.; Silhavy, T.J. The Activity of Escherichia coli Chaperone SurA Is Regulated by Conformational Changes Involving a Parvulin Domain. J. Bacteriol. 2016, 198, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Ricci, D.P.; Schwalm, J.; Gonzales-Cope, M.; Silhavy, T.J. The Activity and Specificity of the Outer Membrane Protein Chaperone SurA Are Modulated by a Proline Isomerase Domain. mBio 2013, 4, e00540-13. [Google Scholar] [CrossRef] [Green Version]
- Hennecke, G.; Nolte, J.; Volkmer-Engert, R.; Schneider-Mergener, J.; Behrens, S. The Periplasmic Chaperone SurA Exploits Two Features Characteristic of Integral Outer Membrane Proteins for Selective Substrate Recognition. J. Boil. Chem. 2005, 280, 23540–23548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Li, M.; Fu, H.; Meng, Q.; Gao, H. Shewanella oneidensis FabB: A β-ketoacyl-ACP Synthase That Works with C16:1-ACP. Front. Microbiol. 2016, 7, 2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Meng, Q.; Fu, H.; Luo, Q.; Gao, H. Suppression of fabB Mutation by fabF1 Is Mediated by Transcription Read-through in Shewanella oneidensis. J. Bacteriol. 2016, 198, 3060–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Zhang, Y.-M.; Rock, C.O. Transcriptional Regulation of Membrane Lipid Homeostasis in Escherichia coli. J. Boil. Chem. 2009, 284, 34880–34888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Liu, Z.; Meng, D.; Liu, X.; Li, X.; Zhang, M.; Tao, J.; Gu, Y.; Zhong, S.; Yin, H. Comparative Genomic Analysis Reveals the Distribution, Organization, and Evolution of Metal Resistance Genes in the Genus Acidithiobacillus. Appl. Environ. Microbiol. 2018, 85. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Culley, D.E.; Zhang, W. Predicted highly expressed genes in the genomes of Streptomyces coelicolor and Streptomyces avermitilis and the implications for their metabolism. Microbiology 2005, 151, 2175–2187. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Nie, L.; Zhang, W. Predicted highly expressed genes in Nocardia farcinica and the implication for its primary metabolism and nocardial virulence. Antonie van Leeuwenhoek 2006, 89, 135–146. [Google Scholar] [CrossRef]
- Pere, P.; Eduard, G.; Antoni, R.; Santiago, G. OPTIMIZER: A web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007, 35, W126–W131. [Google Scholar] [CrossRef] [Green Version]
- Ramazzotti, M.; Brilli, M.; Fani, R.; Manao, G.; Degl’Innocenti, D. The CAI Analyser Package: Inferring gene expressivity from raw genomic data. Silico Boil. 2007, 7, 507. [Google Scholar]
- Pere, P.; Antoni, R.; Santiago, G. HEG-DB: A database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res. 2008, 36, D524–D527. [Google Scholar] [CrossRef] [Green Version]
- Willenbrock, H.; Friis, C.; Friis, A.S.; Ussery, D. An environmental signature for 323 microbial genomes based on codon adaptation indices. Genome Boil. 2006, 7, R114. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Origin | Phylum | Closest Type Strain | Similarity (%) | Salt Tolerance (%) | COD Removal Percentage (%) |
|---|---|---|---|---|---|---|
| LZH-9 | Activated sludge | Firmicutes | Lysinibacillus pakistanensis JCM 18776T | 100.00 | 14 | 69.8 |
| LZH-13 | Activated sludge | Firmicutes | Pseudomonas hunanensis LVT | 98.87 | 12 | 57.35 |
| LZH-22 | Activated sludge | Firmicutes | Staphylococcus sciuri DSM 20345T | 99.93 | 20 | 54.6 |
| LZH-24 | Activated sludge | Firmicutes | Lysinibacillus pakistanensis JCM 18776T | 100.00 | 12 | 65.43 |
| Organism/Name | Strain | Genbank or IMG Acc. no. | Level | Completeness (%) | Size (Mb) | Coding Density (%) | GC (%) | Gene | Protein | Source | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lysinibacillus pakistanensis | JCM 18776 | GCA_001312325.1 /2728369720 | Draft | 99.1 | 5.01 | 77.7 | 36.3 | 8328 | 8216 | rhizospheric soil of legume | [60] |
| Lysinibacillus contaminans | DSM 25560 | GCA_001278945.1 /2648501842 | Draft | 98.1 | 4.1 | 82.2 | 37.3 | 3879 | 3666 | Surface water | [10] |
| Lysinibacillus xylanilyticus | t26 | GCA_002803495.1 | Draft | 99.4 | 5.69 | 79.9 | 36.8 | 6045 | 5584 | / | / |
| Lysinibacillus sp. | UBA7518 | GCA_002478295.1 | Draft | 88.6 | 4.06 | 71.5 | 37.8 | 3969 | 3915 | / | / |
| Lysinibacillus sphaericus | OT4b.31 | GCA_000392615.1 /2524614537 | Draft | 99.2 | 4.86 | 81.5 | 37.5 | 4687 | 4441 | Beetle larvae | [10] |
| Lysinibacillus mangiferihumi | M-GX18 | GCA_003049665.1 /2791355086 | Draft | 99.4 | 5.31 | 82.6 | 36.7 | 5590 | 5187 | / | / |
| Lysinibacillus pakistanensis | LZH-9 | GCA_009649975.1 /2823662158 | Complete | 100 | 4.99 | 83.7 | 36.6 | 5263 | 4990 | Activated sludge | This study. |
| Lysinibacillus parviboronicapiens | VT1065 | GCA_003049605.1 /2802429645 | Draft | 98.3 | 4.74 | 82 | 37.5 | 4676 | 4351 | / | / |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Zhou, H.; Li, L.; Wang, E.; Zhou, X.; Gu, Y.; Wu, X.; Shen, L.; Zeng, W. Whole Genome Sequencing and Comparative Genomic Analyses of Lysinibacillus pakistanensis LZH-9, a Halotolerant Strain with Excellent COD Removal Capability. Microorganisms 2020, 8, 716. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050716
Wu X, Zhou H, Li L, Wang E, Zhou X, Gu Y, Wu X, Shen L, Zeng W. Whole Genome Sequencing and Comparative Genomic Analyses of Lysinibacillus pakistanensis LZH-9, a Halotolerant Strain with Excellent COD Removal Capability. Microorganisms. 2020; 8(5):716. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050716
Chicago/Turabian StyleWu, Xueling, Han Zhou, Liangzhi Li, Enhui Wang, Xiangyu Zhou, Yichao Gu, Xiaoyan Wu, Li Shen, and Weimin Zeng. 2020. "Whole Genome Sequencing and Comparative Genomic Analyses of Lysinibacillus pakistanensis LZH-9, a Halotolerant Strain with Excellent COD Removal Capability" Microorganisms 8, no. 5: 716. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms8050716