Core and Accessory Genome Analysis of Vibrio mimicus

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Genomes
2.2. Generation of a Coding Core Genome
2.3. Phylogenetic Reconstruction
2.4. Analysis of the Accessory Genome
2.5. Comparative Microbial Genomics (CMG)
2.6. Pan-genome Atlas
2.7. Multilocus Sequence Analysis (MLSA)
3. Results
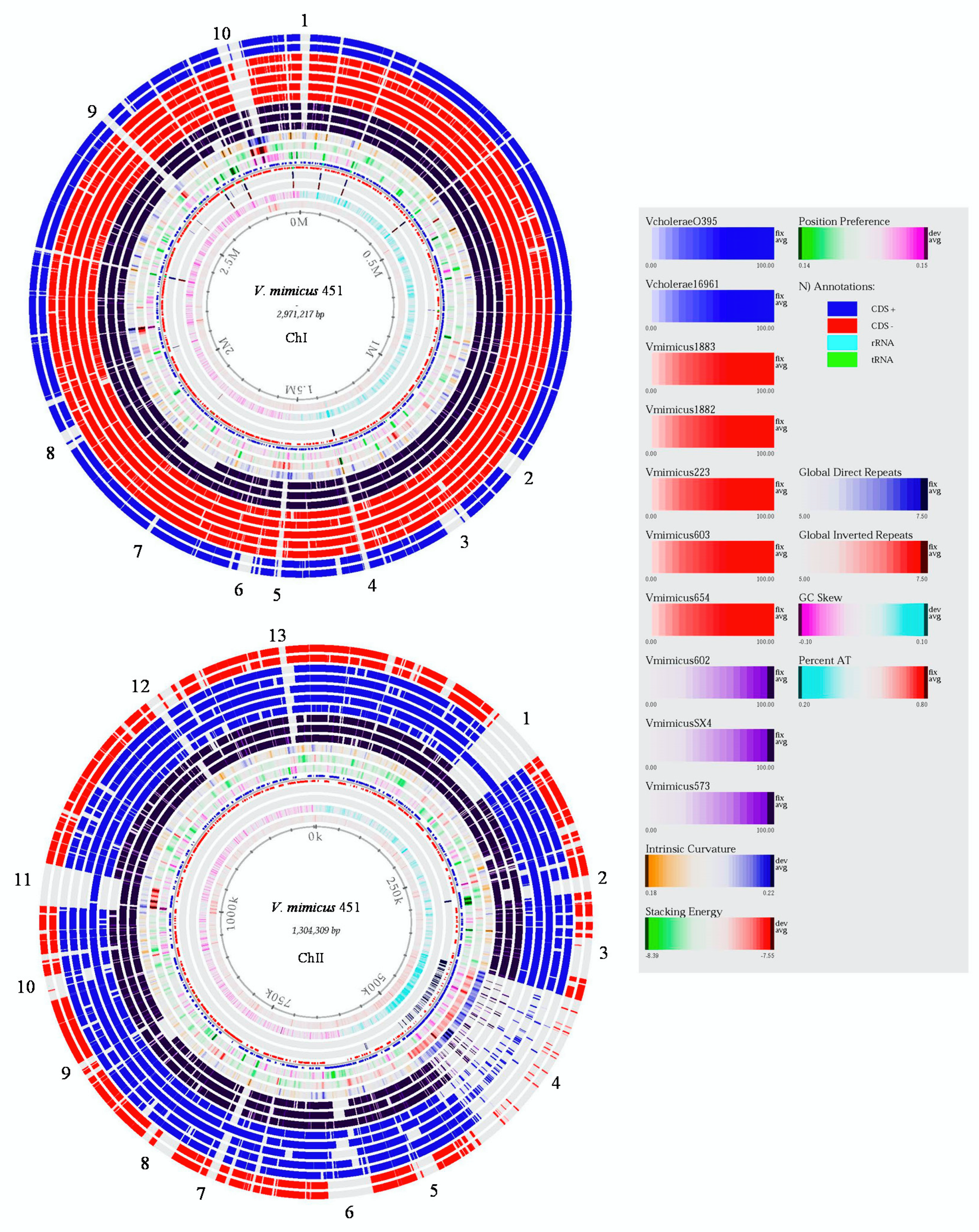
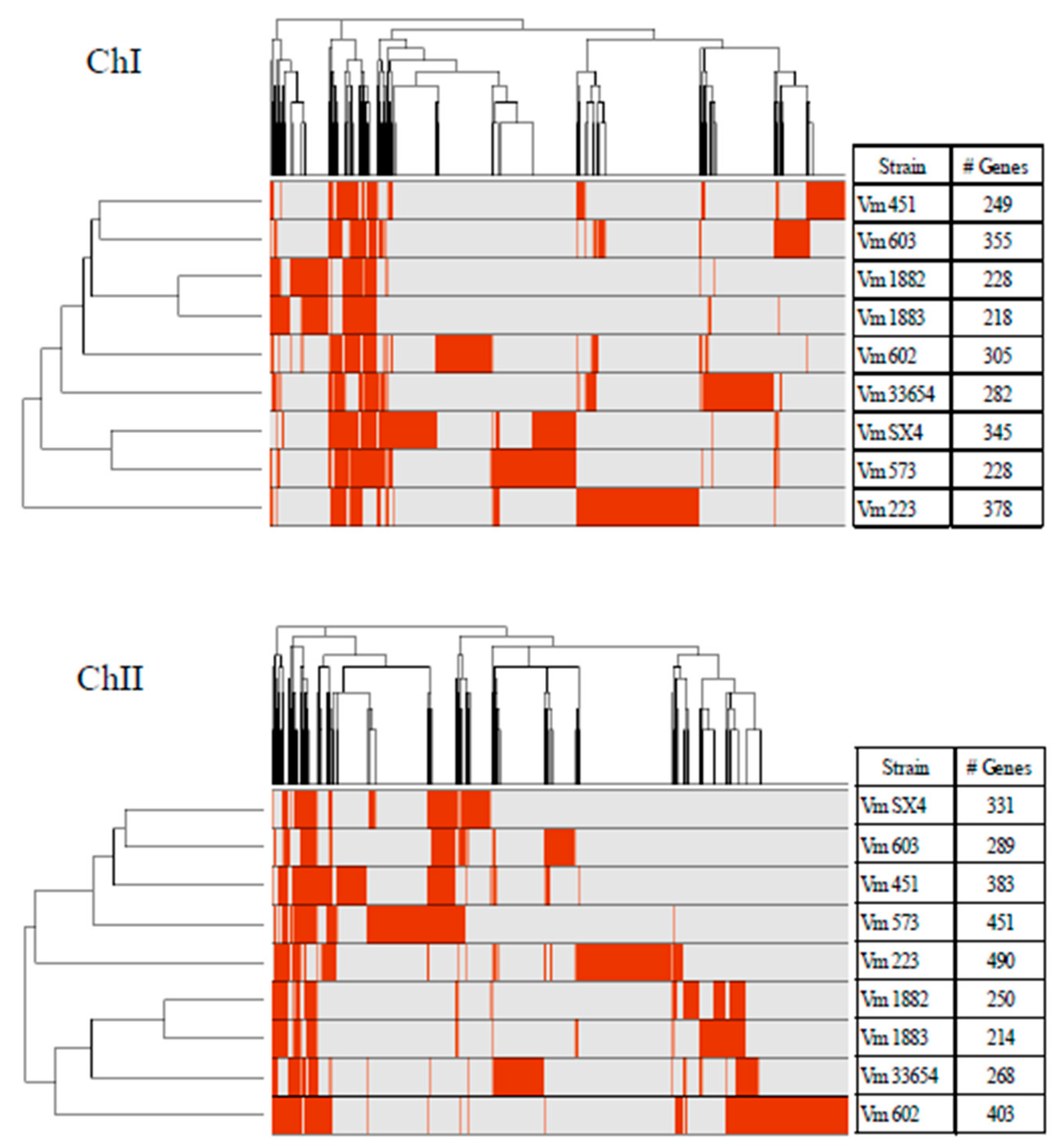
3.1. Pan-Genome of V. mimicus
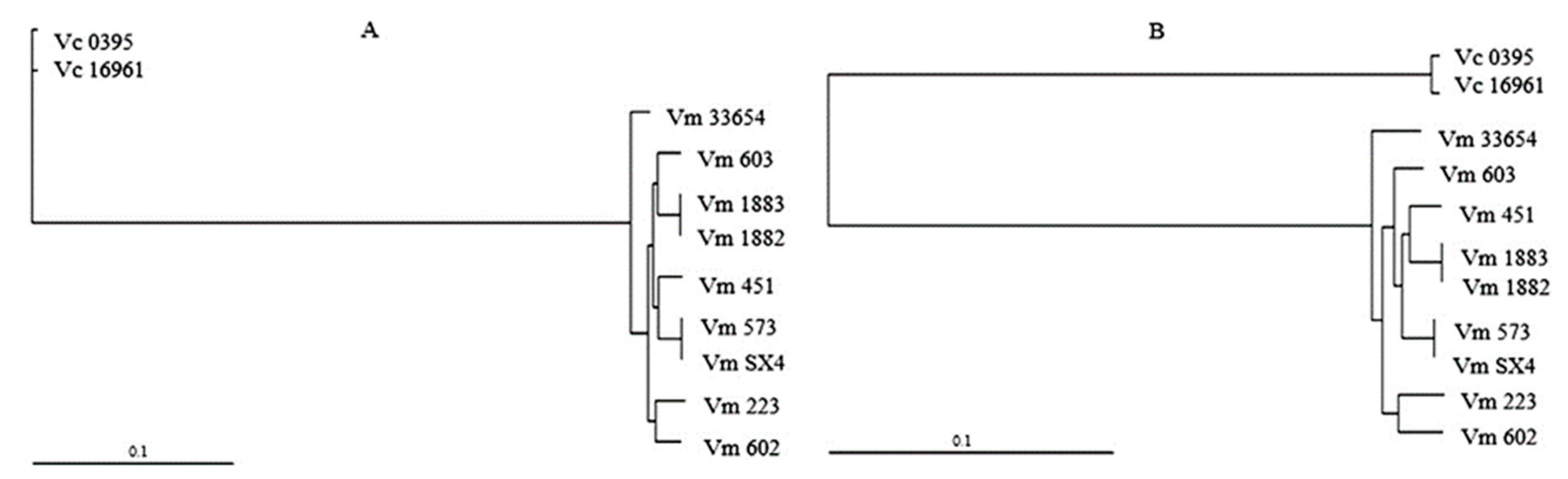
3.2. Core and Accessory Genome of V. mimicus
3.3. Virulence Classification
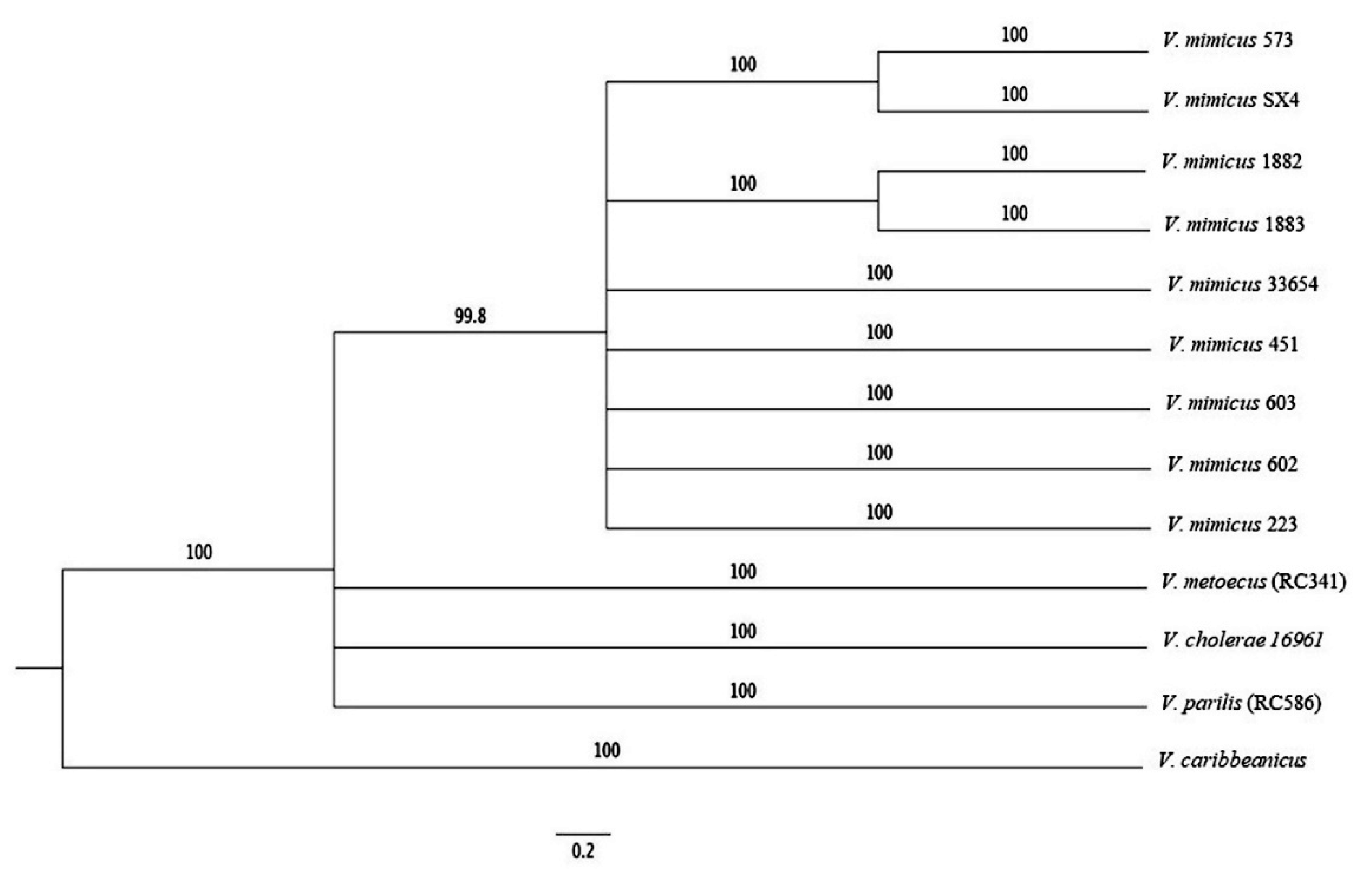
3.4. MLSA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, J., III. The family vibrionaceae. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA; Singapore, 2006; Volume 6, pp. 495–507. ISBN 978-0-387-25496-8. [Google Scholar]
- Davis, B.R.; Fanning, R.; Madden, J.M.; Steigerwalt, A.G.; Bradford, H.B., Jr.; Smith, H.L., Jr.; Brenner, D.J. Characterization of biochemically atypical vibrio cholerae strains and designation of a new pathogenic species, Vibrio mimicus. J. Clin. Microbiol. 1981, 14, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Gil, B.; Thompson, C.C.; Matsumura, Y.; Sawabe, T.; Iida, T.; Christen, R.; Thompson, F.; Sawabe, T. The famlily Vibrionaceae. In The Prokaryotes; Springer: Berlin/Heidelberg, Germany, 2014; pp. 659–747. [Google Scholar]
- Tercero-Alburo, J.J.; González-Márquez, H.; Bonilla-González, E.; Quiñones-Ramírez, E.I.; Vázquez-Salinas, C. Identification of capsule, biofilm, lateral flagellum, and type IV pili in Vibrio mimicus strains. Microb. Pathog. 2014, 76, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.A.; Grim, C.J.; Haley, B.J.; Chun, J.; Alam, M.; Taviani, E.; Hoq, M.; Munk, A.C.; Saunders, E.; Brettin, T.S.; et al. Comparative genomics of clinical and environmental Vibrio mimicus. Proc. Natl. Acad. Sci. USA 2010, 107, 21134–21139. [Google Scholar] [CrossRef] [Green Version]
- Guardiola-Avila, I.; Acedo Félix, E.; Noriega-Orozco, L.; Yepiz-Plascencia, G.; Sifuentes-Romero, I.; Gomez-Gil, B. Draft genome sequence of Vibrio mimicus strain CAIM 602T. Genome Announc. 2013, 1, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Wang, E.; Geng, Y.; Wang, K.; Chen, D.; Huang, X.; Ouyang, P.; Zuo, Z.; Huang, C.; Fang, J.; et al. Complete genome analysis of Vibrio mimicus strain SCCF01, a highly virulent isolate from the freshwater catfish. Virulence 2020, 11, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.C.; Vicente, A.C.P.; Souza, R.C.; Vasconcelos, A.T.R.; Vesth, T.; Alves, N.; Ussery, D.W.; Iida, T.; Thompson, F.L. Genomic taxonomy of vibrios. BMC Evol. Biol. 2009, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Lilburn, T.G.; Gu, J.; Cai, H.; Wang, Y. Comparative genomics of the family Vibrionaceae reveals the wide distribution of genes encoding virulence-associated proteins. BMC Genomics 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Kahlke, T.; Goesmann, A.; Hjerde, E.; Willassen, N.P.; Haugen, P. Unique core genomes of the bacterial family vibrionaceae: Insights into niche adaptation and speciation. BMC Genomics 2012, 13. [Google Scholar] [CrossRef] [Green Version]
- Busschaert, P.; Frans, I.; Crauwels, S.; Zhu, B.; Willems, K.; Bossier, P.; Michiels, C.; Verstrepen, K.; Lievens, B.; Rediers, H. Comparative genome sequencing to assess the genetic diversity and virulence attributes of 15 Vibrio anguillarum isolates. J. Fish. Dis. 2015, 38, 795–807. [Google Scholar] [CrossRef]
- Ceccarelli, D.; Amaro, C.; Romalde, J.L.; Suffredini, E.; Vezzulli, L. Vibrio Species; Wiley: Hoboken, NJ, USA, 2019; ISBN 9781555819972. [Google Scholar]
- Wang, D.; Wang, H.; Zhou, Y.; Zhang, Q.; Zhang, F.; Du, P.; Wang, S.; Chen, C.; Kan, B. Genome sequencing reveals unique mutations in characteristic metabolic pathways and the transfer of virulence genes between V. mimicus and V. cholerae. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, M.A.; Vicente, A.C.P. Architecture of the superintegron in Vibrio cholerae: Identification of core and unique genes. F1000 Res. 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haley, B.J.; Grim, C.J.; Hasan, N.A.; Choi, S.-Y.; Chun, J.; Brettin, T.S.; Bruce, D.C.; Challacombe, J.F.; Detter, J.C.; Han, C.S.; et al. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol. 2010, 10, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, V.V.; Teixeira, L.F.M.; Vicente, A.C.P.; Momen, H.; Salles, C.A. Differentiation of environmental and clinical isolates of Vibrio mimicus from Vibrio cholerae by multilocus enzyme electrophoresis. Appl. Environ. Microbiol. 2001, 67, 2360–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orata, F.D.; Kirchberger, P.C.; Méheust, R.; Barlow, E.J.; Tarr, C.L.; Boucher, Y. The dynamics of genetic interactions between Vibrio metoecus and Vibrio cholerae, two close relatives co-occurring in the environment. Genome Biol. Evol. 2015, 7, 2941–2954. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, P.C.; Turnsek, M.; Hunt, D.E.; Haley, B.J.; Colwell, R.R.; Polz, M.F.; Tarr, C.L.; Boucher, Y. Vibrio metoecus sp. nov., A close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int. J. Syst. Evol. Microbiol. 2014, 64, 3208–3214. [Google Scholar] [CrossRef]
- Guardiola-Avila, I.; Acedo-Felix, E.; Sifuentes-Romero, I.; Yepiz-Plascencia, G.; Gomez-Gil, B.; Noriega-Orozco, L. Molecular and genomic characterization of Vibrio mimicus isolated from a frozen shrimp processing facility in Mexico. PLoS ONE 2016, 11, e0144885. [Google Scholar] [CrossRef]
- Guardiola-Avila, I.; Noriega-Orozco, L.; Gomez-Gil, B.; Acedo-Felix, E. Factores de virulencia de Vibrio mimicus—Vibrio mimicus Virulence factors. Biotecnia 2015, 17, 38–49. [Google Scholar] [CrossRef]
- Lin, H.; Yu, M.; Wang, X.; Zhang, X.H. Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. BMC Genom. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Vesth, T.; Lagesen, K.; Acar, Ö.; Ussery, D. CMG-biotools, a free workbench for basic comparative microbial genomics. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Sjölander, K. Phylogenomic inference of protein molecular function: Advances and challenges. Bioinformatics 2004, 20, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, O.; Verma, M.; Sharma, P.; Kumar, M.; Kumari, K.; Singh, A.; Kumari, H.; Jit, S.; Gupta, S.K.; Khanna, M.; et al. Polyphasic approach of bacterial classification—An overview of recent advances. Ind. J. Microbiol. 2007, 47, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeb, S.; Gulfam, S.M.; Bokhari, H. Comparative core/pan genome analysis of Vibrio cholerae isolates from Pakistan. Infect. Genet. Evol. 2020, 82, 104316. [Google Scholar] [CrossRef] [PubMed]
- Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Clayton, R.A.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Umayam, L.; et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nat. Cell Biol. 2000, 406, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Reeves, P.R.; Lan, R.; Ren, Y.; Gao, C.; Zhou, Z.; Ren, Y.; Cheng, J.; Wang, W.; Wang, J.; et al. A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Busó, L.; Comas, I.; Jorques, G.; González-Candelas, F. Recombination drives genome evolution in outbreak-related Legionella pneumophila isolates. Nat. Genet. 2014, 46, 1205–1211. [Google Scholar] [CrossRef]
- R Team. CR Core Team R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/.
- Pant, A.; Bag, S.; Saha, B.; Verma, J.; Kumar, P.; Banerjee, S.; Kumar, B.; Kumar, Y.; Desigamani, A.; Maiti, S.; et al. Molecular insights into the genome dynamics and interactions between core and acquired genomes of Vibrio cholerae. Proc. Natl. Acad. Sci. USA 2020, 117, 23762–23773. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W. gplots: Various R Programming Tools for Plotting Data. 2013. Available online: https://cran.r-project.org/web/packages/gplots/index.html (accessed on 19 November 2018).
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platformfor ’omics data. Peer J. 2015, 2015, e1319. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Monday, S.R.; Allard, M.W.; Strain, E.A.; Whittaker, P.; Naum, M.; Mccarthy, P.J.; Lopez, J.V.; Fischer, M.; Brown, E.W. Vibrio caribbeanicus sp. nov., isolated from the marine sponge Scleritoderma cyanea. Int. J. Syst. Evol. Microbiol. 2012, 62, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop, New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Sukumaran, J.; Holder, M.T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 2010, 26, 1569–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimes, N.E.; Grim, C.J.; Johnson, W.R.; Hasan, N.A.; Tall, B.D.; Kothary, M.H.; Kiss, H.; Munk, A.C.; Tapia, R.; Green, L.; et al. Temperature regulation of virulence factors in the pathogen Vibrio coralliilyticus. ISME J. 2011, 6, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. [Google Scholar] [CrossRef]
- Petronella, N.; Kundra, P.; Auclair, O.; Hébert, K.; Rao, M.; Kingsley, K.; De Bruyne, K.; Banerjee, S.; Gill, A.; Pagotto, F.; et al. Changes detected in the genome sequences of Escherichia coli, Listeria monocytogenes, Vibrio parahaemolyticus, and Salmonella enterica after serial subculturing. Can. J. Microbiol. 2019, 65, 842–850. [Google Scholar] [CrossRef]
- Zeigler, D.R. Gene sequences useful for predicting relatedness of whole genomes in bacteria. Int. J. Syst. Evol. Microbiol. 2003, 53, 1893–1900. [Google Scholar] [CrossRef]
- Sawabe, T.; Kita-Tsukamoto, K.; Thompson, F.L. Inferring the evolutionary history of vibrios by means of multilocus sequence analysis. J. Bacteriol. 2007, 189, 7932–7936. [Google Scholar] [CrossRef] [Green Version]
- Sawabe, T.; Ogura, Y.; Matsumura, Y.; Feng, G.; Rohul Amin, A.K.M.; Mino, S.; Nakagawa, S.; Sawabe, T.; Kumar, R.; Fukui, Y.; et al. Updating the Vibrio clades defined by multilocus sequence phylogeny: Proposal of eight new clades, and the description of Vibrio tritonius sp. nov. Front. Microbiol. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.M.; Gaastra, W. Bacterial virulence: Can we draw the line? FEMS Microbiol. Lett. 2001, 201, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neogi, S.B.; Chowdhury, N.; Awasthi, S.P.; Asakura, M.; Okuno, K.; Mahmud, Z.H.; Islam, M.S.; Hinenoya, A.; Nair, G.B.; Yamasaki, S. Novel cholera toxin variant and ToxT regulon in environmental Vibrio mimicus isolates: Potential resources for the evolution of Vibrio cholerae hybrid strains. Appl. Environ. Microbiol. 2019, 85, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Tagomori, K.; Iida, T.; Honda, T. Comparison of genome structures of vibrios, bacteria possessing two chromosomes. J. Bacteriol. 2002, 184, 4351–4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V. mimicus | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| CAIM 602T | ATCC 33654 | CAIM 1882 | CAIM 1883 | VM MB451 | VM223 | VM573 | VM603 | SX-4 | |
| Source | Clinical | Environmental | Environmental | Environmental | Clinical | Environmental | Clinical | Environmental | Clinical |
| Origin | Ear infection | River water | Shrimp process water | Shrimp process water | Diarrhoea | Bivalve | Diarrhoea | Fluvial water | Diarrhoea |
| Country | North Carolina, EU | Louisiana, EU | Guaymas, México | Guaymas, México | Shanxi, Chin | Sao Paulo, Brazil | EU | Amazonia, Brazil | China |
| Year | 80s | 80s | 2012 | 2012 | 2009 | - | 90s | 90s | 2009 |
| Size Chr1 (bp) | 2,934,158 | 2,938,455 | 2,819,391 | 2,820,150 | 2,972,217 | 3,055,543 | 2,880,536 | 2,894,575 | 2,997,127 |
| No. subsystems | 434 | 434 | 427 | 426 | 436 | 434 | 415 | 434 | 435 |
| No. CDS | 2729 | 2706 | 2446 | 2642 | 2673 | 2802 | 2652 | 2779 | 2769 |
| No. RNAs | 87 | 85 | 79 | 77 | 115 | 107 | 75 | 49 | 91 |
| % GC | 46.6 | 46.6 | 46.8 | 46.8 | 46.6 | 46.4 | 46.3 | 46.6 | 46.4 |
| Size Chr2 (bp) | 1,268,270 | 1,191,392 | 1,141,600 | 1,115,258 | 1,304,309 | 1,292,428 | 1,460,636 | 1,247,610 | 1,276,483 |
| No. subsystems | 112 | 113 | 110 | 109 | 114 | 108 | 132 | 111 | 113 |
| No. CDS | 1225 | 1090 | 1072 | 1036 | 1205 | 1312 | 1273 | 1111 | 1153 |
| No. RNAs | 6 | 4 | 5 | 6 | 4 | 4 | 18 | 4 | 4 |
| % GC | 45.9 | 46.4 | 46.5 | 46.5 | 45.7 | 45.7 | 45.8 | 45.8 | 45.8 |
| Size Genome | 4,202,428 | 4,129,847 | 3,960,991 | 3,935,408 | 4,276,526 | 4,347,971 | 4,341,172 | 4,142,185 | 4,273,610 |
| Total Genes | 3954 | 3796 | 3518 | 3678 | 3878 | 4114 | 3925 | 3890 | 3922 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guardiola-Avila, I.; Sánchez-Busó, L.; Acedo-Félix, E.; Gomez-Gil, B.; Zúñiga-Cabrera, M.; González-Candelas, F.; Noriega-Orozco, L. Core and Accessory Genome Analysis of Vibrio mimicus. Microorganisms 2021, 9, 191. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010191
Guardiola-Avila I, Sánchez-Busó L, Acedo-Félix E, Gomez-Gil B, Zúñiga-Cabrera M, González-Candelas F, Noriega-Orozco L. Core and Accessory Genome Analysis of Vibrio mimicus. Microorganisms. 2021; 9(1):191. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010191
Chicago/Turabian StyleGuardiola-Avila, Iliana, Leonor Sánchez-Busó, Evelia Acedo-Félix, Bruno Gomez-Gil, Manuel Zúñiga-Cabrera, Fernando González-Candelas, and Lorena Noriega-Orozco. 2021. "Core and Accessory Genome Analysis of Vibrio mimicus" Microorganisms 9, no. 1: 191. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9010191