
Phylogenetic Characterization of Crimean-Congo Hemorrhagic Fever Virus Detected in African Blue Ticks Feeding on Cattle in a Ugandan Abattoir

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
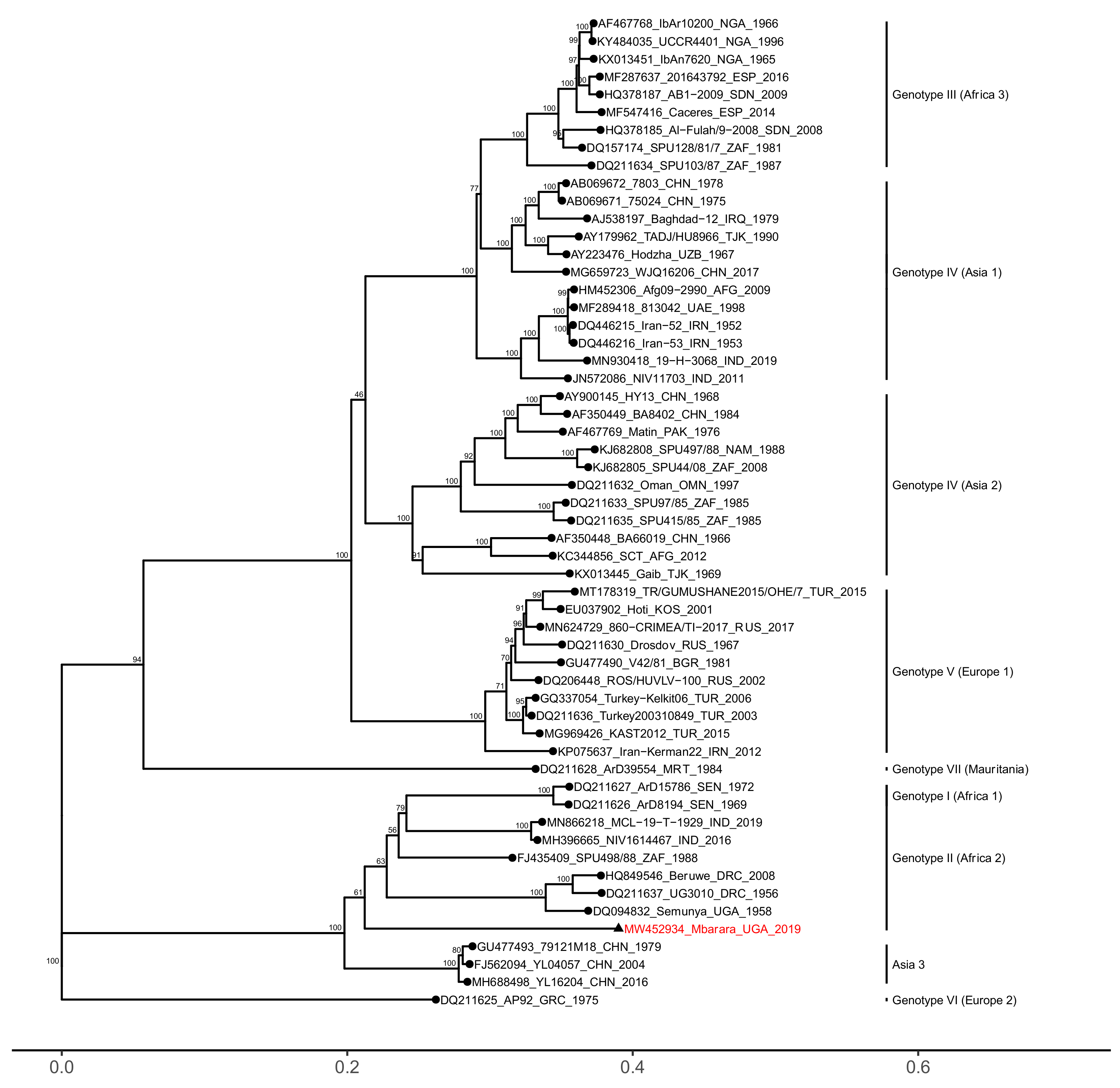
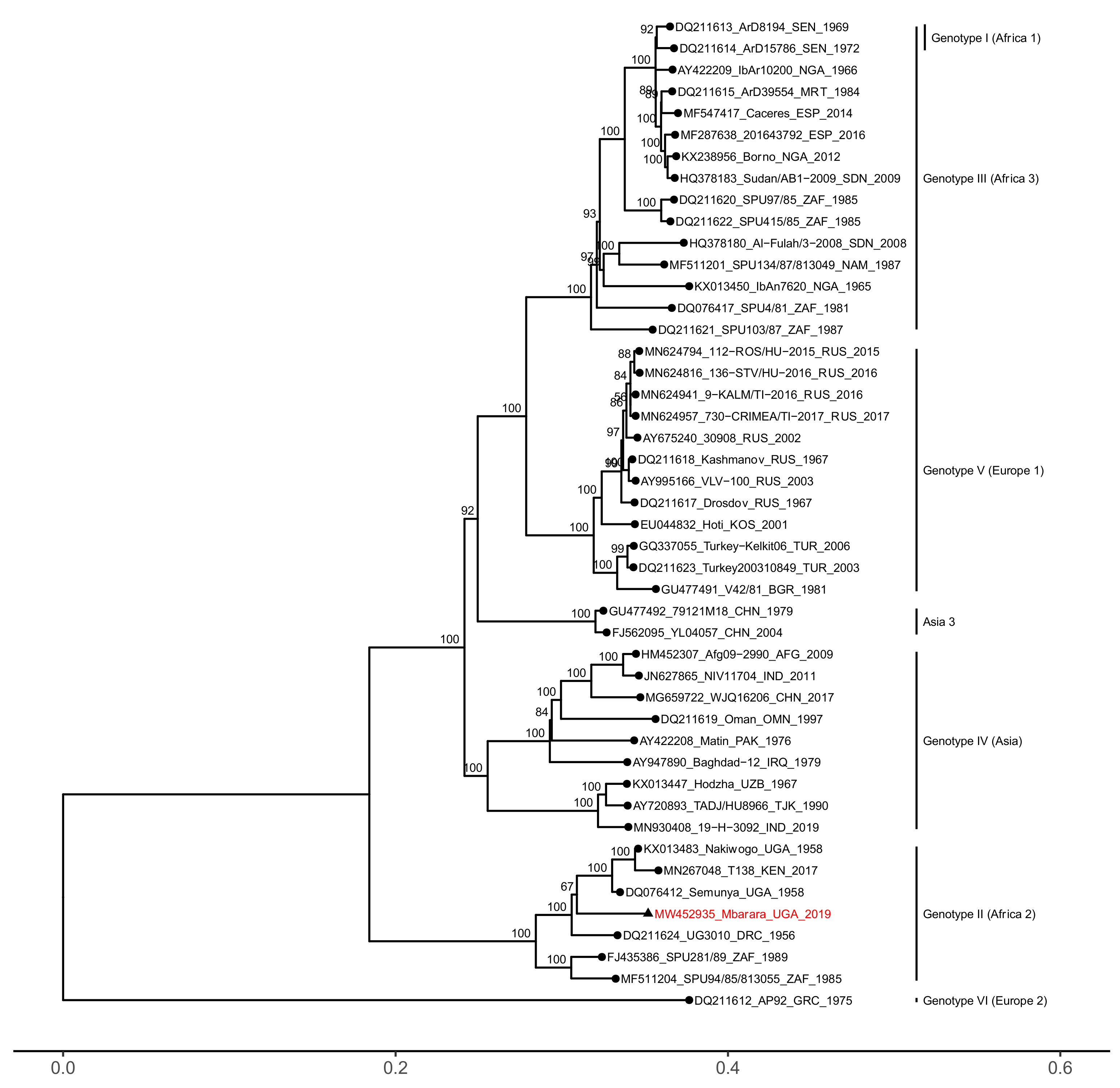
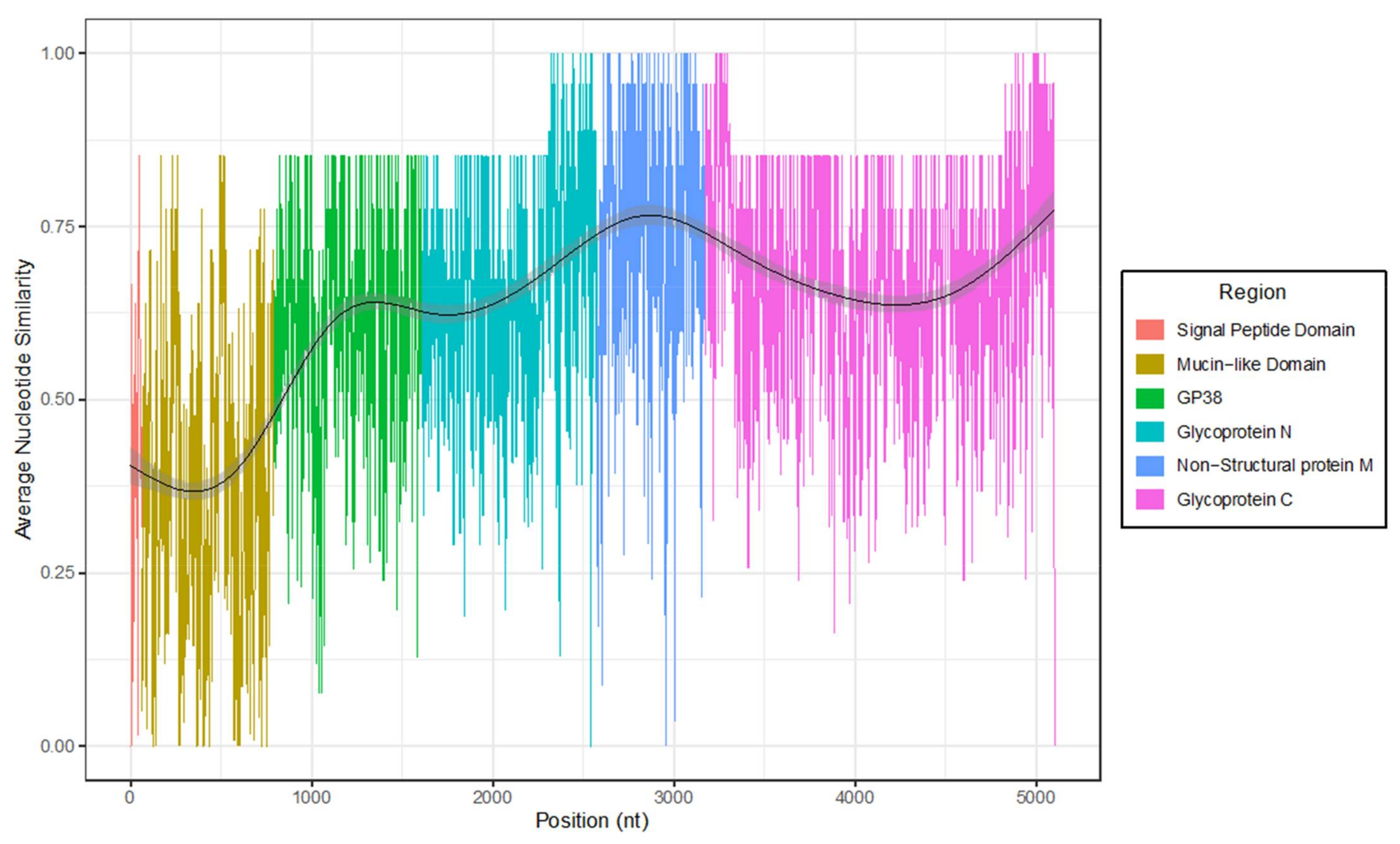
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoogstraal, H. Review Article 1: The Epidemiology of Tick-Borne Crimean-Congo Hemorrhagic Fever in Asia, Europe, and Africa23. J. Med Èntomol. 1979, 15, 307–417. [Google Scholar] [CrossRef] [PubMed]
- Bente, D.A.; Forrester, N.L.; Watts, D.M.; McAuley, A.J.; Whitehouse, C.A.; Bray, M. Crimean-Congo hemorrhagic fever: History, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antivir. Res. 2013, 100, 159–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zivcec, M.; Scholte, F.E.M.; Spiropoulou, C.F.; Spengler, J.R.; Bergeron, É. Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 2016, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyde, V.M.; Khristova, M.L.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Crimean-Congo Hemorrhagic Fever Virus Genomics and Global Diversity. J. Virol. 2006, 80, 8834–8842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N.; Klimentov, A.S.; Smirnova, S.E.; Dzagurova, T.K.; Drexler, J.F.; Gmyl, A.P. Phylogeography of Crimean Congo Hem-orrhagic Fever Virus. PLoS ONE 2016, 11, e0166744. [Google Scholar] [CrossRef] [PubMed]
- Ramírez de Arellano, E.; Hernández, L.; Goyanes, M.J.; Arsuaga, M.; Fernández, C.A.; Negredo, A.; Paz Sánchez-Seco, M. Phylo-genetic characterization of Crimean-Congo hemorrhagic fever virus, Spain. Emerg. Infect. Dis. 2017, 23, 2078–2080. [Google Scholar] [CrossRef] [Green Version]
- Sahay, R.R.; Dhandore, S.; Yadav, P.D.; Chauhan, A.; Bhatt, L.; Garg, V.; Gupta, N.; Nyayanit, D.A.; Shete, A.M.; Singh, R.; et al. Detection of African genotype in Hyalomma tick pools during Crimean Congo hemorrhagic fever outbreak, Rajasthan, India. Virus Res. 2020, 286, 198046. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, S.; Fang, Y.; Liu, J.; Su, Z.; Liang, J.; Zhang, Z.; Wu, Q.; Wang, C.; Abudurexiti, A.; et al. Isolation, Characterization, and Phylogenetic Analysis of Two New Crimean-Congo Hemorrhagic Fever Virus Strains from the Northern Region of Xinjiang Province, China. Virol. Sin. 2018, 33, 74–86. [Google Scholar] [CrossRef]
- Gruber, C.E.M.; Bartolini, B.; Castilletti, C.; Mirazimi, A.; Hewson, R.; Christova, I.; Avšič, T.; Grunow, R.; Papa, A.; Sánchez-Seco, M.P.; et al. Geographical Variability Affects CCHFV Detection by RT–PCR: A Tool for In-Silico Evaluation of Molecular Assays. Viruses 2019, 11, 953. [Google Scholar] [CrossRef] [Green Version]
- Hawman, D.W.; Ahlén, G.; Appelberg, K.S.; Meade-White, K.; Hanley, P.W.; Scott, D.; Monteil, V.; Devignot, S.; Okumura, A.; Weber, F.; et al. A DNA-based vaccine protects against Crimean-Congo haemorrhagic fever virus disease in a Cynomolgus macaque model. Nat. Microbiol. 2021, 6, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Buttigieg, K.R.; Dowall, S.D.; Findlay-Wilson, S.; Miloszewska, A.; Rayner, E.; Hewson, R.; Carroll, M.W. A Novel Vaccine against Crimean-Congo Haemorrhagic Fever Protects 100% of Animals against Lethal Challenge in a Mouse Model. PLoS ONE 2014, 9, e91516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.R.; Bouattour, A.; Camicas, J.-L.; Estrada-Peña, A.; Horak, I.G.; Latif, A.A.; Pegram, R.G.; Preston, P.M. Ticks of Domestic Animals in Africa: A Guide to Identification of Species, 1st ed.; Bioscience Reports: Edinburgh, UK, 2003. [Google Scholar]
- Lambert, A.J.; Lanciotti, R.S. Consensus Amplification and Novel Multiplex Sequencing Method for S Segment Species Identification of 47 Viruses of the Orthobunyavirus, Phlebovirus, and Nairovirus Genera of the Family Bunyaviridae. J. Clin. Microbiol. 2009, 47, 2398–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phy-logenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Compu-ting: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. Ggplot2: Elegrant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analysing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.L.K. Less Is More: An Adaptive Branch-Site Random Effects Model for Efficient Detection of Episodic Diversifying Selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2009, 27, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformation 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Balinandi, S.; Patel, K.; Ojwang, J.; Kyondo, J.; Mulei, S.; Tumusiime, A.; Lubwama, B.; Nyakarahuka, L.; Klena, J.D.; Lutwama, J.; et al. Investigation of an isolated case of human Crimean–Congo hemorrhagic fever in Central Uganda, 2015. Int. J. Infect. Dis. 2018, 68, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Causey, O.R.; Kemp, G.E.; Madbouly, M.H.; David-West, T.S. Congo Virus from Domestic Livestock, African Hedgehog, and Arthropods in Nigeria. Am. J. Trop. Med. Hyg. 1970, 19, 846–850. [Google Scholar] [CrossRef]
- Gargili, A.; Estrada-Peña, A.; Spengler, J.R.; Lukashev, A.; Nuttall, P.A.; Bente, D.A. The role of ticks in the maintenance and trans-mission of Crimean-Congo hemorrhagic fever virus: A review of published field and laboratory studies. Antiviral Res. 2017, 144, 93–119. [Google Scholar] [CrossRef]
- Bukbuk, D.N.; Dowall, S.D.; Lewandowski, K.; Bosworth, A.; Baba, S.S.; Varghese, A.; Watson, R.J.; Bell, A.; Atkinson, B.; Hewson, R. Se-rological and Virological Evidence of Crimean-Congo Haemorrhagic Fever Virus Circulation in the Human Population of Borno State, Northeastern Nigeria. PLoS Negl. Trop. Dis. 2016, 10, e0005126. [Google Scholar] [CrossRef] [Green Version]
- Lindeborg, M.; Barboutis, C.; Ehrenborg, C.; Fransson, T.; Jaenson, T.G.; Lindgren, P.-E.; Lundkvist, Å.; Nyström, F.; Salaneck, E.; Waldenström, J.; et al. Migratory Birds, Ticks, and Crimean-Congo Hemorrhagic Fever Virus. Emerg. Infect. Dis. 2012, 18, 2095–2097. [Google Scholar] [CrossRef]
- Fusco, M.L.; Hashiguchi, T.; Cassan, R.; Biggins, J.E.; Murin, C.D.; Warfield, K.L.; Li, S.; Holtsberg, F.W.; Shulenin, S.; Vu, H.; et al. Protective mAbs and Cross-Reactive mAbs Raised by Immunization with Engineered Marburg Virus GPs. PLoS Pathog. 2015, 11, e1005016. [Google Scholar]
- Fritzen, A.; Risinger, C.; Korukluoglu, G.; Christova, I.; Hitzeroth, A.C.; Viljoen, N.; Burt, F.J.; Mirazimi, A.; Blixt, O. Epitope-mapping of the glycoprotein from Crimean-Congo hemorrhagic fever virus using a microarray approach. PLoS Neglected Trop. Dis. 2018, 12, e0006598. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.W.; Shoemaker, C.J.; Lindquist, M.E.; Zeng, X.; Daye, S.P.; Williams, J.A.; Liu, J.; Coffin, K.M.; Olschner, S.; Flusin, O.; et al. GP38-targeting monoclonal antibodies protect adult mice against lethal Crimean-Congo hemorrhagic fever virus infection. Sci. Adv. 2019, 5, eaaw9535. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Segment | Gene | Number of Sequences (Full Length) | Gene Length (AA) | Polymorphic Sites * (Informative Sites †) | Median Singleton ‡ Count (Range) § | Mbarara Strain Singletons | % Sites Unique to Mbarara Strain |
|---|---|---|---|---|---|---|---|
| S | NP and Ns | 8 (5) | 482 | 11 (3) | 1 (0–2) | 1 | 0.2 |
| M | Signal peptide | 10 (10) | 20 | 16 (13) | 0 (0–1) | 5 | 25.0 |
| MLD | 10 (10) | 242 | 206 (167) | 4 (1–29) | 76 | 31.4 | |
| GP38 | 10 (10) | 271 | 80 (55) | 7 (4–14) | 22 | 8.1 | |
| Gn | 11 (10) | 322 | 58 (41) | 1 (0–4) | 12 | 3.7 | |
| NSm | 11 (11) | 197 | 41 (27) | 1.5 (0–3) | 10 | 5.1 | |
| Gc | 12 (10) | 643 | 103 (53) | 2 (0–12) | 23 | 3.6 | |
| L | RdRp | 7 (5) | 3945 | 153 (32) | 7 (0–60) | 32 | 0.8 |
| S, M, and L | All | - | 6144 | 668 (391) | - | 164 | 2.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wampande, E.M.; Waiswa, P.; Allen, D.J.; Hewson, R.; Frost, S.D.W.; Stubbs, S.C.B. Phylogenetic Characterization of Crimean-Congo Hemorrhagic Fever Virus Detected in African Blue Ticks Feeding on Cattle in a Ugandan Abattoir. Microorganisms 2021, 9, 438. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020438
Wampande EM, Waiswa P, Allen DJ, Hewson R, Frost SDW, Stubbs SCB. Phylogenetic Characterization of Crimean-Congo Hemorrhagic Fever Virus Detected in African Blue Ticks Feeding on Cattle in a Ugandan Abattoir. Microorganisms. 2021; 9(2):438. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020438
Chicago/Turabian StyleWampande, Eddie M., Peter Waiswa, David J. Allen, Roger Hewson, Simon D. W. Frost, and Samuel C. B. Stubbs. 2021. "Phylogenetic Characterization of Crimean-Congo Hemorrhagic Fever Virus Detected in African Blue Ticks Feeding on Cattle in a Ugandan Abattoir" Microorganisms 9, no. 2: 438. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9020438