
Nosocomial Pneumonia in the Era of Multidrug-Resistance: Updates in Diagnosis and Management

 ,
,
Abstract
:1. Introduction
2. Diagnosis of Nosocomial Pneumonia
2.1. Imaging Modalities
2.1.1. Chest X-ray
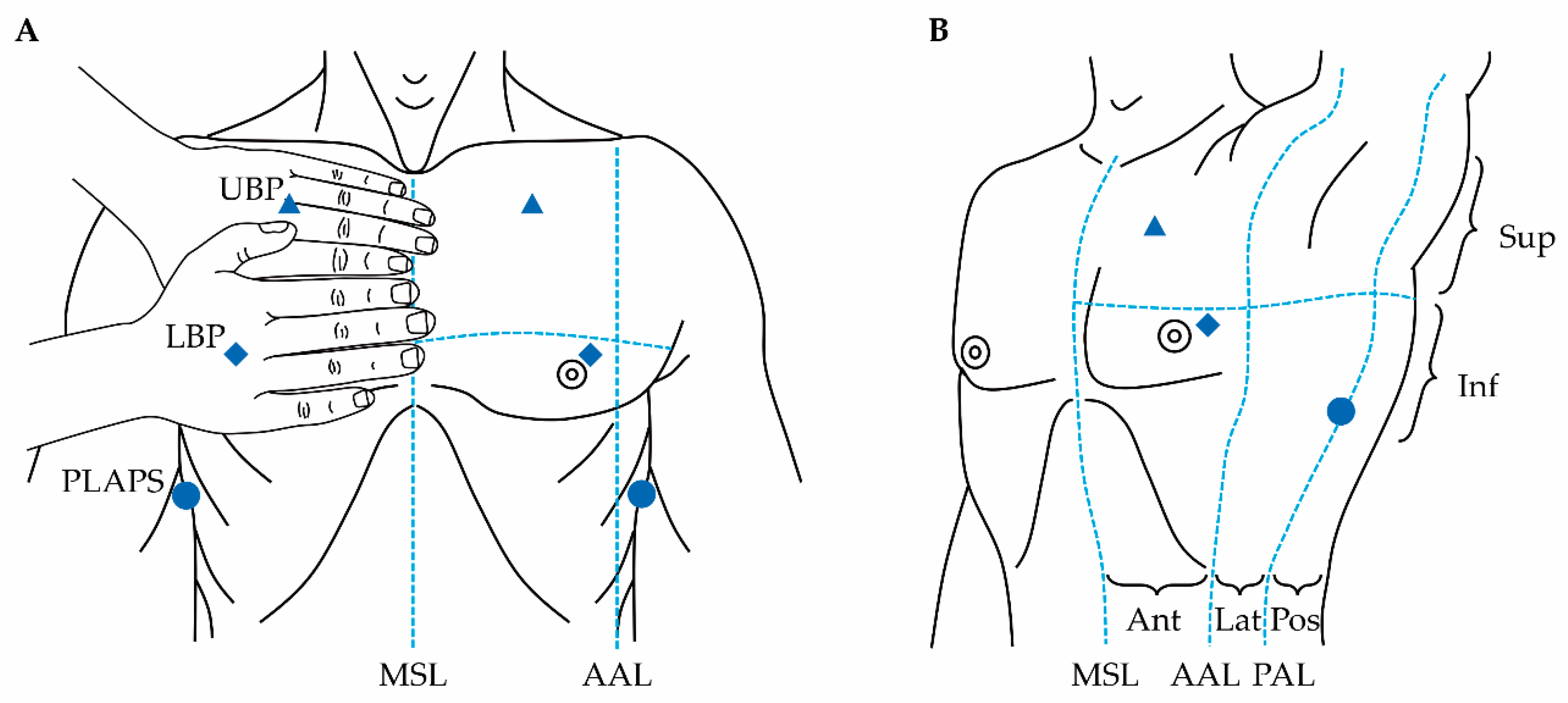
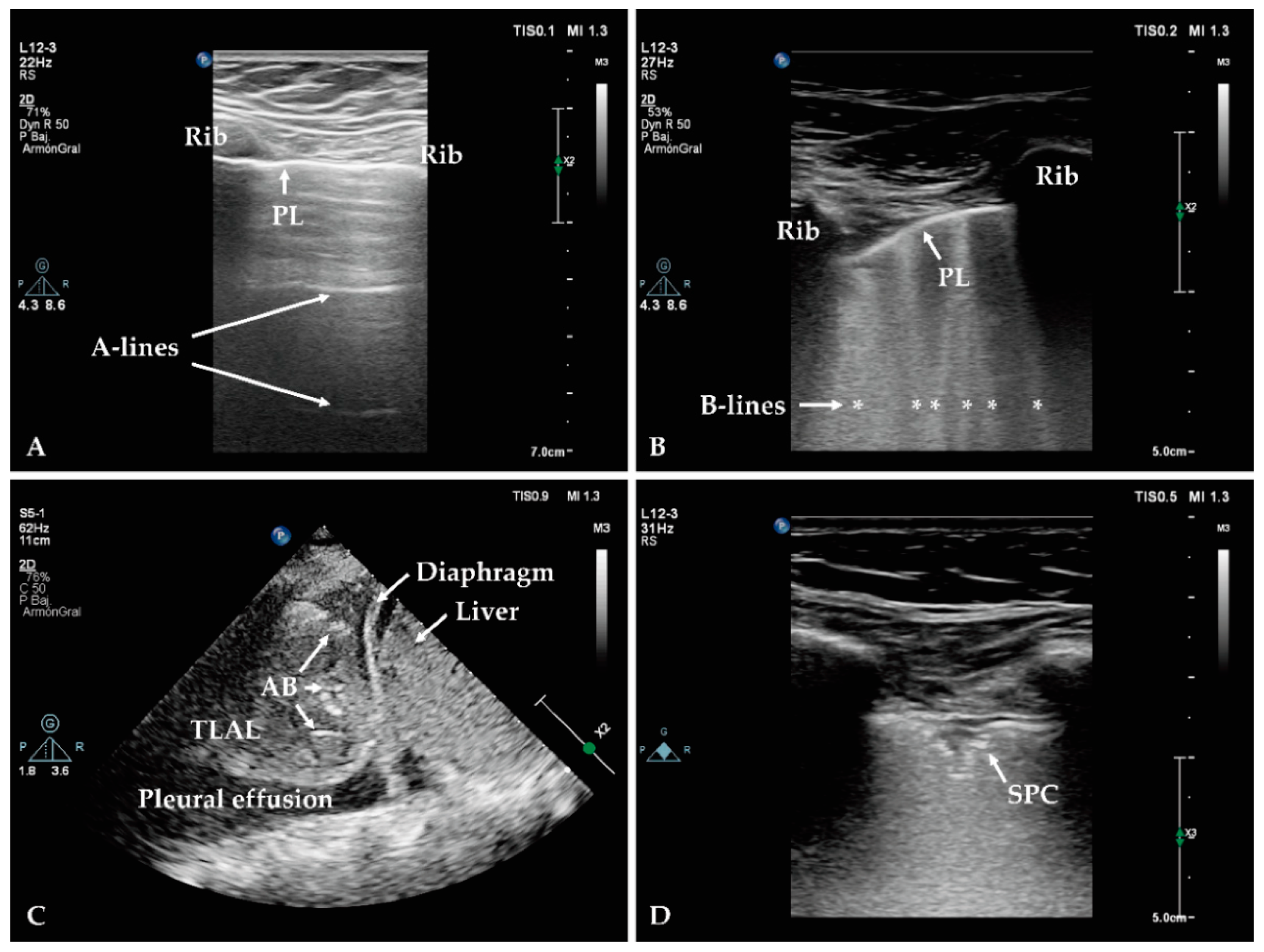
2.1.2. Lung Ultrasound
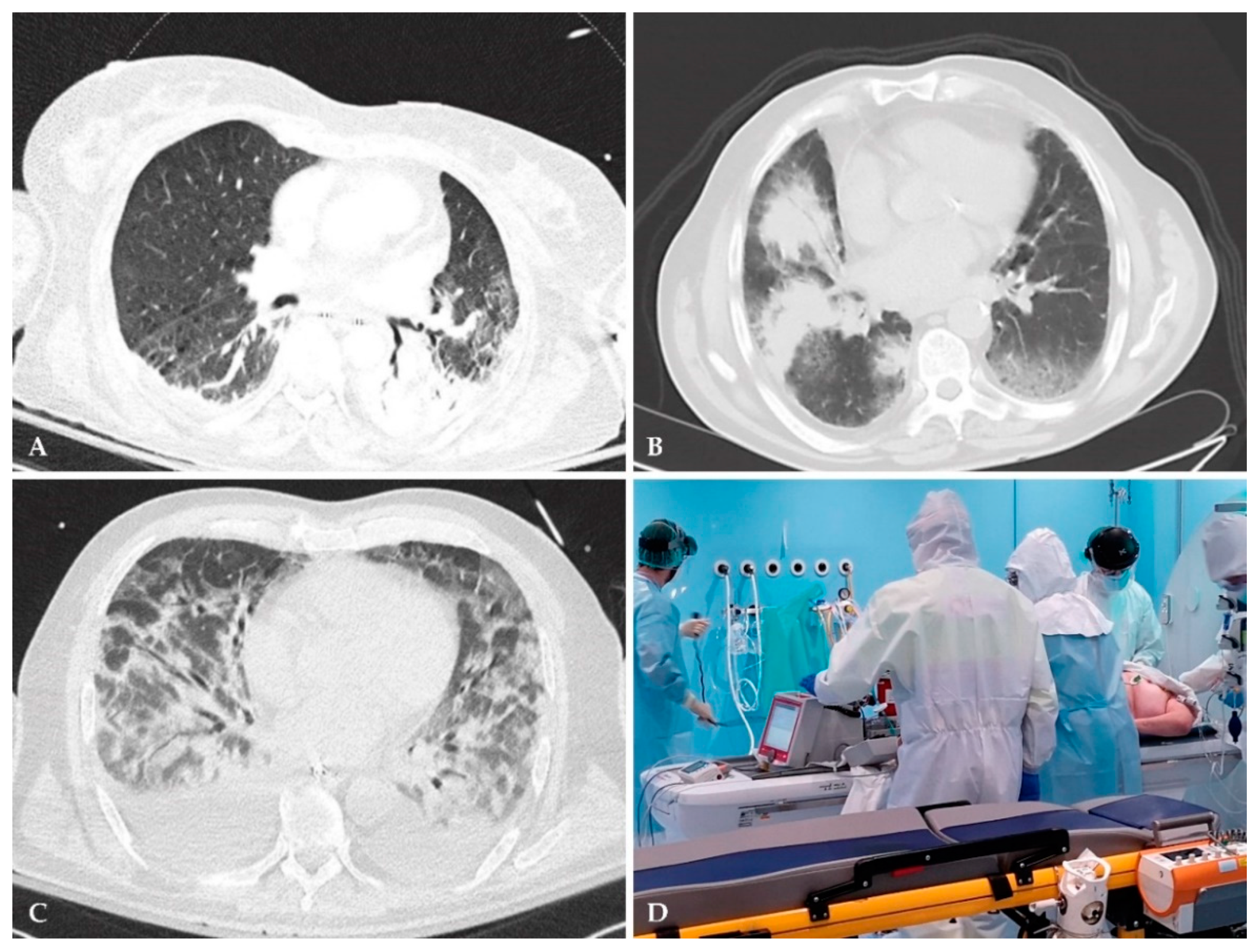
2.1.3. Low-Radiation Computed Tomography
2.2. Aetiological Diagnosis
2.2.1. Conventional Cultures
2.2.2. Syndromic Rapid Multi-Pathogen PCR Panels
- (1)
- BioFire® FilmArray® Pneumonia Panels
- (2)
- Curetis Unyvero multiplex PCR Panels
- (3)
- Other syndromic rapid multi-pathogen PCR panels
2.2.3. Other Rapid Molecular Diagnostics
2.2.4. Volatile Organic Compounds—Electronic Nose
3. Novel Approved Antibiotics for Nosocomial Pneumonia
3.1. Ceftobiprole Medocaril
3.2. Telavancin
3.3. Ceftolozane/Tazobactam
3.4. Ceftazidime/Avibactam
3.5. Meropenem/Vaborbactam
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 388. [Google Scholar]
- Kalil, A.C.; Metersky, M.L.; Klompas, M.; Muscedere, J.; Sweeney, D.A.; Palmer, L.B.; Napolitano, L.M.; O’Grady, N.P.; Bartlett, J.G.; Carratalà, J. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin. Infect. Dis. 2016, 63, e61–e111. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.; Koch, O.; Laurenson, I.; O’Shea, D.; Sutherland, R.; Mackintosh, C. Diagnosis and features of hospital-acquired pneumonia: A retrospective cohort study. J. Hosp. Infect. 2016, 92, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papazian, L.; Klompas, M.; Luyt, C.-E. Ventilator-associated pneumonia in adults: A narrative review. Intensiv. Care Med. 2020, 46, 888–906. [Google Scholar] [CrossRef] [Green Version]
- Vallecoccia, M.S.; Dominedò, C.; Cutuli, S.L.; Martin-Loeches, I.; Torres, A.; De Pascale, G. Is ventilated hospital-acquired pneumonia a worse entity than ventilator-associated pneumonia? Eur. Respir. Rev. 2020, 29, 157. [Google Scholar] [CrossRef]
- Masterton, R.; Galloway, A.; French, G.; Street, M.; Armstrong, J.; Brown, E.; Cleverley, J.; Dilworth, P.; Fry, C.; Gascoigne, A. Guidelines for the management of hospital-acquired pneumonia in the UK: Report of the working party on hospital-acquired pneumonia of the British Society for Antimicrobial Chemotherapy. J. Antimicrob. Chemother. 2008, 62, 5–34. [Google Scholar] [CrossRef]
- Koulenti, D.; Tsigou, E.; Rello, J. Nosocomial pneumonia in 27 ICUs in Europe: Perspectives from the EU-VAP/CAP study. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Lisboa, T.; Brun-Buisson, C.; Krueger, W.; Macor, A.; Sole-Violan, J.; Diaz, E.; Topeli, A.; DeWaele, J.; Carneiro, A. Spectrum of practice in the diagnosis of nosocomial pneumonia in patients requiring mechanical ventilation in European intensive care units. Crit. Care Med. 2009, 37, 2360–2369. [Google Scholar] [CrossRef]
- Melsen, W.G.; Rovers, M.M.; Groenwold, R.H.; Bergmans, D.C.; Camus, C.; Bauer, T.T.; Hanisch, E.W.; Klarin, B.; Koeman, M.; Krueger, W.A. Attributable mortality of ventilator-associated pneumonia: A meta-analysis of individual patient data from randomised prevention studies. Lancet Infect. Dis. 2013, 13, 665–671. [Google Scholar] [CrossRef]
- Sopena, N.; Sabrià, M.; Group, N.S. Multicenter study of hospital-acquired pneumonia in non-ICU patients. Chest 2005, 127, 213–219. [Google Scholar] [CrossRef]
- Kollef, M.H.; Hamilton, C.W.; Ernst, F.R. Economic impact of ventilator-associated pneumonia in a large matched cohort. Infect. Control Hospit. Epidemiol. 2012, 33, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zimlichman, E.; Henderson, D.; Tamir, O.; Franz, C.; Song, P.; Yamin, C.K.; Keohane, C.; Denham, C.R.; Bates, D.W. Health care-associated infections: A meta-analysis of costs and financial impact on the US health care system. JAMA Intern. Med. 2013, 173, 2039–2046. [Google Scholar] [CrossRef]
- Agada, U.; Chaudhri, N.; Conlon, M.; Dada, C.; Green, L. Diagnosis and management of hospital-acquired pneumonia in older adults. Evaluation 2020, 14, 34. [Google Scholar]
- Blot, S.; Koulenti, D.; Dimopoulos, G.; Martin, C.; Komnos, A.; Krueger, W.A.; Spina, G.; Armaganidis, A.; Rello, J.; Investigators, E.-V.S. Prevalence, risk factors, and mortality for ventilator-associated pneumonia in middle-aged, old, and very old critically ill patients. Crit. Care Med. 2014, 42, 601–609. [Google Scholar] [CrossRef]
- Koulenti, D.; Zhang, Y.; Fragkou, P.C. Nosocomial pneumonia diagnosis revisited. Curr. Opin. Crit. Care 2020, 26, 442–449. [Google Scholar] [CrossRef]
- Iregui, M.; Ward, S.; Sherman, G.; Fraser, V.J.; Kollef, M.H. Clinical importance of delays in the initiation of appropriate antibiotic treatment for ventilator-associated pneumonia. Chest 2002, 122, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Yoo, I.Y.; Huh, K.; Shim, H.J.; Yun, S.A.; Chung, Y.N.; Kang, O.K.; Huh, H.J.; Lee, N.Y. Evaluation of the BioFire® FilmArray® Pneumonia Panel for rapid detection of respiratory bacterial pathogens and antibiotic resistance genes in sputum and endotracheal aspirate specimens. Int. J. Infect. Dis. 2020, 95, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Song, A.; Ellingboe, A.; Abdul-Aziz, M.H.; Harris, P.; Gavey, E.; Lipman, J. Infections by multidrug-resistant Gram-negative Bacteria: What’s new in our arsenal and what’s in the pipeline? Int. J. Antimicrob. Agents 2019, 53, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Xu, E.; Yin Sum Mok, I.; Song, A.; Karageorgopoulos, D.E.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel antibiotics for multidrug-resistant gram-positive microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Wielandner, A. NosokomialePneumonieausradiologischer Sicht. Der Radiol. 2017, 57, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Goodman, L.R. Felson’s Principles of Chest Roentgenology, A Programmed Text, 5th ed.; Elsevier: Philadelphia, PA, USA, 2021. [Google Scholar]
- Ferreira-Coimbra, J.; Ardanuy, C.; Diaz, E.; Leone, M.; De Pascale, G.; Póvoa, P.; Prat-Aymerich, C.; Serrano-Garcia, R.; Solé-Violan, J.; Zaragoza, R. Ventilator-associated pneumonia diagnosis: A prioritization exercise based on multi-criteria decision analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Klompas, M. Does this patient have ventilator-associated pneumonia? JAMA 2007, 297, 1583–1593. [Google Scholar] [CrossRef]
- Franquet, T. Imaging of pneumonia: Trends and algorithms. Eur. Respir. J. 2001, 18, 196–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polverino, E.; Torres, A. Diagnostic strategies for healthcare-associated pneumonia. Semin. Respir. Crit. Care Med. 2009, 30, 036–045. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Maycher, B.; Eschun, G. Radiological imaging in pneumonia: Recent innovations. Curr. Opin. Pulm. Med. 2007, 13, 159–169. [Google Scholar] [CrossRef]
- Greenbaum, D.M.; Marschall, K.E. The value of routine daily chest X-rays in intubated patients in the medical intensive care unit. Crit. Care Med. 1982, 10, 29–30. [Google Scholar] [CrossRef]
- Gruden, J.F.; Huang, L.; Turner, J.; Webb, W.R.; Merrifield, C.; Stansell, J.D.; Gamsu, G.; Hopewell, P.C. High-resolution CT in the evaluation of clinically suspected Pneumocystis carinii pneumonia in AIDS patients with normal, equivocal, or nonspecific radiographic findings. AJR Am. J. Roentgenol. 1997, 169, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Ely, E.W.; Baker, A.M.; Dunagan, D.P.; Burke, H.L.; Smith, A.C.; Kelly, P.T.; Johnson, M.M.; Browder, R.W.; Bowton, D.L.; Haponik, E.F. Effect on the duration of mechanical ventilation of identifying patients capable of breathing spontaneously. N. Engl. J. Med. 1996, 335, 1864–1869. [Google Scholar] [CrossRef]
- Wunderink, R.G.; Woldenberg, L.S.; Zeiss, J.; Day, C.M.; Ciemins, J.; Lacher, D.A. The radiologic diagnosis of autopsyproven ventilator-associated pneumonia. Chest 1992, 101, 458–463. [Google Scholar] [CrossRef]
- Fernando, S.M.; Tran, A.; Cheng, W.; Klompas, M.; Kyeremanteng, K.; Mehta, S.; English, S.W.; Muscedere, J.; Cook, D.J.; Torres, A. Diagnosis of ventilator-associated pneumonia in critically ill adult patients—A systematic review and meta-analysis. Intensiv. Care Med. 2020, 46, 1–10. [Google Scholar] [CrossRef]
- Bouhemad, B.; Mongodi, S.; Via, G.; Rouquette, I. Ultrasound for “lung monitoring” of ventilated patients. Anesthesiology 2015, 122, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Mayo, P.; Copetti, R.; Feller-Kopman, D.; Mathis, G.; Maury, E.; Mongodi, S.; Mojoli, F.; Volpicelli, G.; Zanobetti, M. Thoracic ultrasonography: A narrative review. Intensiv. Care Med. 2019, 45, 1–12. [Google Scholar] [CrossRef]
- Lichtenstein, D.A.; Mezière, G.A. The BLUE-points: Three standardized points used in the BLUE-protocol for ultrasound assessment of the lung in acute respiratory failure. Crit. Ultrasound J. 2011, 3, 109–110. [Google Scholar] [CrossRef] [Green Version]
- Wiersema, R.; Forte, J.N.C.; Kaufmann, T.; de Haas, R.J.; Koster, G.; Hummel, Y.M.; Koeze, J.; Franssen, C.F.; Vos, M.E.; Hiemstra, B. Observational study protocol for repeated clinical examination and critical care ultrasonography within the simple intensive care studies. JoVE J. Vis. Exp. 2019, e58802. [Google Scholar] [CrossRef]
- Cox, E.G.; Wiersema, R.; Wong, A.; van der Horst, I.C. Six versus eight and twenty-eight scan sites for B-line assessment: Differences in examination time and findings. Intensiv. Care Med. 2020, 46, 1063–1064. [Google Scholar] [CrossRef]
- Lichtenstein, D.A. The Pleural Line. In Lung Ultrasound in the Critically Ill; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; p. 376. [Google Scholar]
- Mojoli, F.; Bouhemad, B.; Mongodi, S.; Lichtenstein, D. Lung ultrasound for critically ill patients. Am. J. Respir. Crit. Care Med. 2019, 199, 701–714. [Google Scholar] [CrossRef]
- Lichtenstein, D.A.; Menu, Y. A bedside ultrasound sign ruling out pneumothorax in the critically III: Lung sliding. Chest 1995, 108, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, D.A.; Mezière, G.A.; Lagoueyte, J.-F.; Biderman, P.; Goldstein, I.; Gepner, A. A-lines and B-lines: Lung ultrasound as a bedside tool for predicting pulmonary artery occlusion pressure in the critically ill. Chest 2009, 136, 1014–1020. [Google Scholar] [CrossRef] [Green Version]
- Chiumello, D.; Mongodi, S.; Algieri, I.; Vergani, G.L.; Orlando, A.; Via, G.; Crimella, F.; Cressoni, M.; Mojoli, F. Assessment of lung aeration and recruitment by CT scan and ultrasound in acute respiratory distress syndrome patients. Crit. Care Med. 2018, 46, 1761–1768. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, C.F.; Mathis, G.; Blaivas, M.; Volpicelli, G.; Seibel, A.; Wastl, D.; Atkinson, N.S.; Cui, X.-W.; Fan, M.; Yi, D. Lung B-line artefacts and their use. J. Thorac. Dis. 2016, 8, 1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenstein, D.; Mezière, G.; Seitz, J. The dynamic air bronchogram: A lung ultrasound sign of alveolar consolidation ruling out atelectasis. Chest 2009, 135, 1421–1425. [Google Scholar] [CrossRef]
- Staub, L.J.; Biscaro, R.R.M.; Maurici, R. Emergence of Alveolar Consolidations in Serial Lung Ultrasound and Diagnosis of Ventilator-Associated Pneumonia. J. Intensiv. Care Med. 2019, 36, 088506661989427. [Google Scholar] [CrossRef]
- Bouhemad, B.; Dransart-Rayé, O.; Mojoli, F.; Mongodi, S. Lung ultrasound for diagnosis and monitoring of ventilator-associated pneumonia. Ann. Transl. Med. 2018, 6, 418. [Google Scholar] [CrossRef]
- Zagli, G.; Cozzolino, M.; Terreni, A.; Biagioli, T.; Caldini, A.L.; Peris, A. Diagnosis of ventilator-associated pneumonia: A pilot, exploratory analysis of a new score based on procalcitonin and chest echography. Chest 2014, 146, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Song, J.; Gong, S.; Hu, W.; Wang, M.; Xiao, A.; Zhang, C.; Dong, Z. Lung ultrasound combined with procalcitonin for a diagnosis of ventilator-associated pneumonia. Respir. Care 2019, 64, 519–527. [Google Scholar] [CrossRef]
- Xia, Y.; Ying, Y.; Wang, S.; Li, W.; Shen, H. Effectiveness of lung ultrasonography for diagnosis of pneumonia in adults: A systematic review and meta-analysis. J. Thorac. Dis. 2016, 8, 2822. [Google Scholar] [CrossRef] [Green Version]
- Fraile-Ribot, P.A.; Mulet, X.; Cabot, G.; Del Barrio-Tofiño, E.; Juan, C.; Pérez, J.L.; Oliver, A. In vivo emergence of resistance to novel cephalosporin–β-lactamase inhibitor combinations through the duplication of amino acid D149 from OXA-2 β-lactamase (OXA-539) in sequence type 235 Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.H.; Touw, H.R.; van de Ven, P.M.; Twisk, J.; Tuinman, P.R. Diagnostic accuracy of chest radiograph, and when concomitantly studied lung ultrasound, in critically ill patients with respiratory symptoms: A systematic review and meta-analysis. Crit. Care Med. 2018, 46, e707–e714. [Google Scholar] [CrossRef] [PubMed]
- Knight, P.H.; Maheshwari, N.; Hussain, J.; Scholl, M.; Hughes, M.; Papadimos, T.J.; Guo, W.A.; Cipolla, J.; Stawicki, S.P.; Latchana, N. Complications during intrahospital transport of critically ill patients: Focus on risk identification and prevention. Int. J. Crit. Illn. Inj. Sci. 2015, 5, 256. [Google Scholar]
- Fred, H.L. Drawbacks and limitations of computed tomography: Views from a medical educator. Tex. Heart Inst. J. 2004, 31, 345. [Google Scholar]
- Hoesein, F.M. Low-dose computed tomography instead of radiography in suspected pneumonia. Breathe 2019, 15, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Prendki, V.; Scheffler, M.; Huttner, B.; Garin, N.; Herrmann, F.; Janssens, J.-P.; Marti, C.; Carballo, S.; Roux, X.; Serratrice, C. Low-dose computed tomography for the diagnosis of pneumonia in elderly patients: A prospective, interventional cohort study. Eur. Respir. J. 2018, 51, 1702375. [Google Scholar] [CrossRef]
- Kroft, L.J.; van der Velden, L.; Girón, I.H.; Roelofs, J.J.; de Roos, A.; Geleijns, J. Added value of ultra-low-dose computed tomography, dose Equivalent to chest x-ray radiography, for diagnosing chest pathology. J. Thorac. Imaging 2019, 34, 179. [Google Scholar] [CrossRef] [PubMed]
- Endimiani, A.; Hujer, K.M.; Hujer, A.M.; Kurz, S.; Jacobs, M.R.; Perlin, D.S.; Bonomo, R.A. Are we ready for novel detection methods to treat respiratory pathogens in hospital-acquired pneumonia? Clin. Infect. Dis. 2011, 52, S373–S383. [Google Scholar] [CrossRef] [Green Version]
- Cilloniz, C.; Liapikou, A.; Torres, A. Advances in molecular diagnostic tests for pneumonia. Curr. Opin. Pulm. Med. 2020, 26, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.-Y.; Chiang-Ni, C.; Teng, S.-H. Current status of MALDI-TOF mass spectrometry in clinical microbiology. J. Food Drug Anal. 2019, 27, 404–414. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [Green Version]
- Liapikou, A.; Cillóniz, C.; Torres, A. Emerging strategies for the noninvasive diagnosis of nosocomial pneumonia. Expert Rev. Anti-Infect. Ther. 2019, 17, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Seng, P.; Rolain, J.-M.; Fournier, P.E.; La Scola, B.; Drancourt, M.; Raoult, D. MALDI-TOF-mass spectrometry applications in clinical microbiology. Future Microbiol. 2010, 5, 1733–1754. [Google Scholar] [CrossRef] [PubMed]
- Seng, P.; Drancourt, M.; Gouriet, F.; La Scola, B.; Fournier, P.-E.; Rolain, J.M.; Raoult, D. Ongoing revolution in bacteriology: Routine identification of bacteria by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Infect. Dis. 2009, 49, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.M.; Newton, D.; Kunapuli, A.; Gandhi, T.N.; Washer, L.L.; Isip, J.; Collins, C.D.; Nagel, J.L. Impact of rapid organism identification via matrix-assisted laser desorption/ionization time-of-flight combined with antimicrobial stewardship team intervention in adult patients with bacteremia and candidemia. Clin. Infect. Dis. 2013, 57, 1237–1245. [Google Scholar] [CrossRef]
- Mok, J.H.; Eom, J.S.; Jo, E.J.; Kim, M.H.; Lee, K.; Kim, K.U.; Park, H.K.; Yi, J.; Lee, M.K. Clinical utility of rapid pathogen identification using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry in ventilated patients with pneumonia: A pilot study. Respirology 2016, 21, 321–328. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.N.; Fowler, R.; Balada-Llasat, J.M.; Carroll, A.; Stone, H.; Akerele, O.; Buchan, B.; Windham, S.; Hopp, A.; Ronen, S. Multicenter Evaluation of the BioFire® FilmArray® Pneumonia/Pneumonia plus Panel for the Detection and Quantification of Agents of Lower Respiratory Tract Infection. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef] [PubMed]
- BioFire by Biomerieux. The BioFire® FilmArray®Pneumonia (PN) Panel. Available online: https://www.biofiredx.com/products/the-filmarray-panels/filmarray-pneumonia/ (accessed on 26 December 2020).
- Biomerieux. BIOFIRE® FILMARRAY® Pneumonia Plus Panel. Available online: https://www.biomerieux-diagnostics.com/biofire-filmarray-pneumonia-panel (accessed on 26 December 2020).
- Edin, A.; Eilers, H.; Allard, A. Evaluation of the BiofireFilmarray Pneumonia panel plus for lower respiratory tract infections. Infect. Dis. 2020, 52, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Ruan, S.-Y.; Pan, S.-C.; Lee, T.-F.; Chien, J.-Y.; Hsueh, P.-R. Performance of a multiplex PCR pneumonia panel for the identification of respiratory pathogens and the main determinants of resistance from the lower respiratory tract specimens of adult patients in intensive care units. J. Microbiol. Immunol. Infect. 2019, 52, 920–928. [Google Scholar] [CrossRef]
- Yugueros-Marcos, J.; Barraud, O.; Iannello, A.; Ploy, M.C.; Ginocchio, C.; Rogatcheva, M.; Alberti-Segui, C.; Pachot, A.; Moucadel, V.; François, B. New molecular semi-quantification tool provides reliable microbiological evidence for pulmonary infection. Intensiv. Care Med. 2018, 44, 2302–2304. [Google Scholar] [CrossRef] [PubMed]
- Luyt, C.-E.; Hékimian, G.; Bonnet, I.; Bréchot, N.; Schmidt, M.; Robert, J.; Combes, A.; Aubry, A. Usefulness of point-of-care multiplex PCR to rapidly identify pathogens responsible for ventilator-associated pneumonia and their resistance to antibiotics: An observational study. Crit. Care 2020, 24, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ozongwu, C.; Personne, Y.; Platt, G.; Jeanes, C.; Aydin, S.; Kozato, N.; Gant, V.; O’Grady, J.; Enne, V. The Unyvero P55 ‘sample-in, answer-out’pneumonia assay: A performance evaluation. Biomol. Detect. Quantif. 2017, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, N.J.; McHugh, M.P.; Forbes, C.; MacKenzie, L.; Hamilton, S.K.; Griffith, D.M.; Templeton, K.E. Comparison of Unyvero P55 Pneumonia Cartridge, in-house PCR and culture for the identification of respiratory pathogens and antibiotic resistance in bronchoalveolar lavage fluids in the critical care setting. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Peiffer-Smadja, N.; Bouadma, L.; Mathy, V.; Allouche, K.; Patrier, J.; Reboul, M.; Montravers, P.; Timsit, J.-F.; Armand-Lefevre, L. Performance and impact of a multiplex PCR in ICU patients with ventilator-associated pneumonia or ventilated hospital-acquired pneumonia. Crit. Care 2020, 24, 1–10. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Impact of a Strategy Based on Bacterial DNA Detection to Optimize Antibiotics in Patients with Hospital-Acquired Pneumonia (VAPERO). Available online: https://clinicaltrials.gov/ct2/show/NCT03711331?recrs=ab&cond=Pneumonia%2C+Ventilator-Associated&draw=3&rank=20 (accessed on 16 September 2020).
- Enne, V.I.; Aydin, A.; Baldan, R.; Owen, D.R.; Richardson, H.; Ricciardi, F.; Russell, C.; Nomamiukor-Ikeji, B.O.; Swart, A.M.; High, J. Multicentre evaluation of two multiplex PCR platforms for the rapid microbiological investigation of nosocomial pneumonia in UK ICUs: The INHALE WP1 study. medRxiv 2020. [Google Scholar] [CrossRef]
- Morris, A.C.; Gadsby, N.; McKenna, J.P.; Hellyer, T.P.; Dark, P.; Singh, S.; Walsh, T.S.; McAuley, D.F.; Templeton, K.; Simpson, A.J. 16S pan-bacterial PCR can accurately identify patients with ventilator-associated pneumonia. Thorax 2017, 72, 1046–1048. [Google Scholar] [CrossRef] [Green Version]
- Navapurkar, V.; Bartholdson-Scott, J.; Maes, M.; Higginson, E.; Forrest, S.; Dias, J.P.; Parmar, S.; Heasman-Hunt, E.; Polgarova, P.; Brown, J. Development and implementation of a customised rapid syndromic diagnostic test for severe pneumonia. medRxiv 2020. [Google Scholar] [CrossRef]
- Maes, M.; Higginson, E.; Pereira-Dias, J.; Curran, M.D.; Parmar, S.; Khokhar, F.; Cuchet-Lourenço, D.; Lux, J.; Sharma-Hajela, S.; Ravenhill, B. Ventilator-associated pneumonia in critically ill patients with COVID-19. Crit. Care 2021, 25, 1–11. [Google Scholar] [CrossRef]
- Fast Track Diagnostics. Human Line FTD Respiratory Pathogens 21. Available online: http://www.fast-trackdiagnostics.com/human-line/products/ftd-respiratory-pathogens-21/ (accessed on 27 December 2020).
- Reijans, M.; Dingemans, G.; Klaassen, C.H.; Meis, J.F.; Keijdener, J.; Mulders, B.; Eadie, K.; Van Leeuwen, W.; Van Belkum, A.; Horrevorts, A.M. RespiFinder: A new multiparameter test to differentially identify fifteen respiratory viruses. J. Clin. Microbiol. 2008, 46, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, L.; Ozog, Y.; Mansour, S.; Loens, K.; Reijans, M.; Ieven, M.; Simons, G. RespiFinder SMART 22 FAST: Detection of 22 respiratory pathogens within four hours. In Proceedings of the 23rd ECCMID 2013, Berlin, Germany, 27–30 April 2013. [Google Scholar]
- Luminex Corporation. VERIGENE® Respiratory Pathogens Flex Test. Available online: https://www.luminexcorp.com/respiratory-pathogens-flex-test/#documentation (accessed on 27 December 2020).
- Cepheid International. The New GeneXpert® System. Available online: https://p.widencdn.net/bia0nv/Cepheid-GeneXpert-System-Brochure-CE-IVD-0309-English (accessed on 4 January 2020).
- Cepheid International. New Ways. New Innovations. New Paths towards Disease Management. Available online: https://www.cepheid.com/en/impact/improved-oncology-diagnostics (accessed on 5 January 2020).
- Cepheid. Xpert® Carba-R. Available online: https://www.cepheid.com/en/tests/Healthcare-Associated-Infections/Xpert-Carba-R (accessed on 27 December 2020).
- Ko, Y.J.; Kim, J.; Kim, H.-N.; Yoon, S.-Y.; Lim, C.S.; Lee, C.K. Diagnostic performance of the XpertCarba-R assay for active surveillance of rectal carbapenemase-producing organisms in intensive care unit patients. Antimicrob. Resist. Infect. Control 2019, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Cortegiani, A.; Russotto, V.; Graziano, G.; Geraci, D.; Saporito, L.; Cocorullo, G.; Raineri, S.M.; Mammina, C.; Giarratano, A. Use of cepheid xpertcarba-r® for rapid detection of carbapenemase-producing bacteria in abdominal septic patients admitted to intensive care unit. PLoS ONE 2016, 11, e0160643. [Google Scholar] [CrossRef]
- Bauer, K.A.; West, J.E.; Balada-Llasat, J.-M.; Pancholi, P.; Stevenson, K.B.; Goff, D.A. An antimicrobial stewardship program’s impact. Clin. Infect. Dis. 2010, 51, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Coppens, J.; Van Heirstraeten, L.; Ruzin, A.; Yu, L.; Timbermont, L.; Lammens, C.; Matheeussen, V.; McCarthy, M.; Jorens, P.; Ieven, M. Comparison of GeneXpert MRSA/SA ETA assay with semi-quantitative and quantitative cultures and nuc gene-based qPCR for detection of Staphylococcus aureus in endotracheal aspirate samples. Antimicrob. Resist. Infect. Control 2019, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.; Fijten, R.; Smolinska, A.; Dallinga, J.; Boumans, M.-L.; Stobberingh, E.; Boots, A.; Roekaerts, P.; Bergmans, D.; van Schooten, F.J. Analysis of volatile organic compounds in exhaled breath to diagnose ventilator-associated pneumonia. Sci. Rep. 2015, 5, 17179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, W.; Wilde, M.; Cordell, R.; Salman, D.; Ruszkiewicz, D.; Bryant, L.; Richardson, M.; Free, R.C.; Zhao, B.; Yousuf, A. Assessment of breath volatile organic compounds in acute cardiorespiratory breathlessness: A protocol describing a prospective real-world observational study. BMJ Open 2019, 9, e025486. [Google Scholar] [CrossRef]
- Amann, A.; de Lacy Costello, B.; Miekisch, W.; Schubert, J.; Buszewski, B.; Pleil, J.; Ratcliffe, N.; Risby, T. The human volatilome: Volatile organic compounds (VOCs) in exhaled breath, skin emanations, urine, feces and saliva. J. Breath Res. 2014, 8, 034001. [Google Scholar] [CrossRef]
- Kort, S.; Tiggeloven, M.; Brusse-Keizer, M.; Gerritsen, J.; Schouwink, J.; Citgez, E.; de Jongh, F.; Samii, S.; van der Maten, J.; van den Bogart, M. Multi-centre prospective study on diagnosing subtypes of lung cancer by exhaled-breath analysis. Lung Cancer 2018, 125, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.; Lin, W.-C.; Yang, H.-Y. Diagnosis of ventilator-associated pneumonia using electronic nose sensor array signals: Solutions to improve the application of machine learning in respiratory research. Respir. Res. 2020, 21, 45. [Google Scholar] [CrossRef]
- Krauss, E.; Haberer, J.; Maurer, O.; Barreto, G.; Drakopanagiotakis, F.; Degen, M.; Seeger, W.; Guenther, A. Exploring the Ability of Electronic Nose Technology to Recognize Interstitial Lung Diseases (ILD) by Non-Invasive Breath Screening of Exhaled Volatile Compounds (VOC): A Pilot Study from the European IPF Registry (eurIPFreg) and Biobank. J. Clin. Med. 2019, 8, 1698. [Google Scholar] [CrossRef]
- Boots, A.W.; Bos, L.D.; van der Schee, M.P.; van Schooten, F.-J.; Sterk, P.J. Exhaled molecular fingerprinting in diagnosis and monitoring: Validating volatile promises. Trends Mol. Med. 2015, 21, 633–644. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Wujak, L.; Wygrecka, M.; Markart, P. Biomarkers in idiopathic pulmonary fibrosis. Matrix Biol. 2018, 68, 404–421. [Google Scholar] [CrossRef]
- Wojnowski, W.; Majchrzak, T.; Dymerski, T.; Gębicki, J.; Namieśnik, J. Portable electronic nose based on electrochemical sensors for food quality assessment. Sensors 2017, 17, 2715. [Google Scholar] [CrossRef] [Green Version]
- Van Geffen, W.H.; Bruins, M.; Kerstjens, H.A. Diagnosing viral and bacterial respiratory infections in acute COPD exacerbations by an electronic nose: A pilot study. J. Breath Res. 2016, 10, 036001. [Google Scholar] [CrossRef] [PubMed]
- De Heer, K.; van der Schee, M.P.; Zwinderman, K.; van den Berk, I.A.; Visser, C.E.; van Oers, R.; Sterk, P.J. Electronic nose technology for detection of invasive pulmonary aspergillosis in prolonged chemotherapy-induced neutropenia: A proof-of-principle study. J. Clin. Microbiol. 2013, 51, 1490–1495. [Google Scholar] [CrossRef] [Green Version]
- Filipiak, W.; Sponring, A.; Baur, M.M.; Ager, C.; Filipiak, A.; Wiesenhofer, H.; Nagl, M.; Troppmair, J.; Amann, A. Characterization of volatile metabolites taken up by or released from Streptococcus pneumoniae and Haemophilus influenzae by using GC-MS. Microbiology 2012, 158, 3044–3053. [Google Scholar] [CrossRef] [Green Version]
- Fend, R.; Kolk, A.H.; Bessant, C.; Buijtels, P.; Klatser, P.R.; Woodman, A.C. Prospects for clinical application of electronic-nose technology to early detection of Mycobacterium tuberculosis in culture and sputum. J. Clin. Microbiol. 2006, 44, 2039–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.Y.; Deffenderfer, O.F.; Hanson, W.; Phillips, M.P.; Thaler, E.R. Identification of upper respiratory bacterial pathogens with the electronic nose. Laryngoscope 2002, 112, 975–979. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; Hines, E.L.; Gardner, J.W.; Boilot, P. Bacteria classification using Cyranose 320 electronic nose. Biomed. Eng. Online 2002, 1, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipiak, W.; Beer, R.; Sponring, A.; Filipiak, A.; Ager, C.; Schiefecker, A.; Lanthaler, S.; Helbok, R.; Nagl, M.; Troppmair, J. Breath analysis for in vivo detection of pathogens related to ventilator-associated pneumonia in intensive care patients: A prospective pilot study. J. Breath Res. 2015, 9, 016004. [Google Scholar] [CrossRef]
- Gao, J.; Zou, Y.; Wang, Y.; Wang, F.; Lang, L.; Wang, P.; Zhou, Y.; Ying, K. Breath analysis for noninvasively differentiating Acinetobacter baumannii ventilator-associated pneumonia from its respiratory tract colonization of ventilated patients. J. Breath Res. 2016, 10, 027102. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, R.; Boumans, M.; Smolinska, A.; Stobberingh, E.; Kaufmann, R.; Roekaerts, P.; Bergmans, D. Electronic nose analysis of exhaled breath to diagnose ventilator-associated pneumonia. Respir. Med. 2015, 109, 1454–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. National Library of Medicine. Serial, Non-Invasive Analysis of Exhaled Breath Condensate in Ventilated Trauma Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02652247?recrs=ab&cond=Pneumonia%2C+Ventilator-Associated&draw=4&rank=21 (accessed on 16 September 2020).
- Electronic Medicines Compendium. Zevtera: Summary of Product Characteristics. Available online: https://www.medicines.org.uk/emc/product/9164 (accessed on 10 December 2020).
- Morosini, M.I.; Díez-Aguilar, M.; Cantón, R. Mechanisms of action and antimicrobial activity of ceftobiprole. Rev. Esp. Quimioter. 2019, 32, 3–10. [Google Scholar]
- Mensa, J.; Soriano, A.; García-Sánchez, J.E.; Marco, F.; Letang, E.; Llinares, P.; López-Suñé, E.; Barberán, J. Guía de Terapéutica Antimicrobiana 2020, 30th ed.; Editorial Antares, Ediciones Escofet Zamora S.L.: Barcelona, Spain, 2019; p. 975. [Google Scholar]
- Farrell, D.J.; Flamm, R.K.; Sader, H.S.; Jones, R.N. Ceftobiprole activity against over 60,000 clinical bacterial pathogens isolated in Europe, Turkey, and Israel from 2005 to 2010. Antimicrob. Agents Chemother. 2014, 58, 3882–3888. [Google Scholar] [CrossRef] [Green Version]
- Walkty, A.; Adam, H.J.; Laverdière, M.; Karlowsky, J.A.; Hoban, D.J.; Zhanel, G.G.; Alliance, C.A.R. In vitro activity of ceftobiprole against frequently encountered aerobic and facultative Gram-positive and Gram-negative bacterial pathogens: Results of the CANWARD 2007–2009 study. Diagn. Microbiol. Infect. Dis. 2011, 69, 348–355. [Google Scholar] [CrossRef]
- Pfaller, M.; Flamm, R.; Duncan, L.; Streit, J.; Castanheira, M.; Sader, H. Antimicrobial activity of ceftobiprole and comparator agents when tested against contemporary Gram-positive and-negative organisms collected from Europe (2015). Diagn. Microbiol. Infect. Dis. 2018, 91, 77–84. [Google Scholar] [CrossRef]
- Agencia Española de Medicamentos y Productos Sanitarios. Zevtera: Fichatécnica. Available online: https://cima.aemps.es/cima/pdfs/es/ft/78691/78691_ft.pdf (accessed on 10 December 2020).
- Banerjee, R.; Gretes, M.; Basuino, L.; Strynadka, N.; Chambers, H.F. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2008, 52, 2089–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, S.M.; Alexander, J.A.N.; Choo, E.J.; Basuino, L.; Da Costa, T.M.; Severin, A.; Chung, M.; Aedo, S.; Strynadka, N.C.J.; Tomasz, A.; et al. High-level resistance of Staphylococcus aureus to β-Lactam antibiotics mediated by penicillin-binding protein 4 (PBP4). Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Greninger, A.L.; Chatterjee, S.S.; Chan, L.C.; Hamilton, S.M.; Chambers, H.F.; Chiu, C.Y. Whole-genome sequencing of methicillin-resistant Staphylococcus aureus resistant to fifth-generation cephalosporins reveals potential non-mecA mechanisms of resistance. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Flamm, R.K.; Duncan, L.R.; Shortridge, D.; Smart, J.I.; Hamed, K.A.; Mendes, R.E.; Sader, H.S. Ceftobiprole activity when tested against contemporary bacteria causing bloodstream infections in the United States (2016–2017). Diagn. Microbiol. Infect. Dis. 2019, 94, 304–313. [Google Scholar] [CrossRef]
- Morroni, G.; Brenciani, A.; Brescini, L.; Fioriti, S.; Simoni, S.; Pocognoli, A.; Mingoia, M.; Giovanetti, E.; Barchiesi, F.; Giacometti, A.; et al. High rate of ceftobiprole resistance among clinical methicillin-resistant staphylococcus aureus isolates from a Hospital in central Italy. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Mouton, J.W.; Pea, F. Pharmacokinetics and Dosing of Ceftobiprole Medocaril for the Treatment of Hospital- and Community-Acquired Pneumonia in Different Patient Populations. Clin. Pharmacokinet. 2016, 55, 1507–1520. [Google Scholar] [CrossRef] [Green Version]
- Barbour, A.; Schmidt, S.; Rand, K.H.; Derendorf, H. Ceftobiprole: A novel cephalosporin with activity against Gram-positive and Gram-negative pathogens, including methicillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2009, 34, 1–7. [Google Scholar] [CrossRef]
- Rodvold, K.A.; Nicolau, D.P.; Lodise, T.P.; Khashab, M.; Noel, G.J.; Kahn, J.B.; Gotfried, M.; Murray, S.A.; Nicholson, S.; Laohavaleeson, S.; et al. Identifying exposure targets for treatment of staphylococcal pneumonia with ceftobiprole. Antimicrob. Agents Chemother. 2009, 53, 3294–3301. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A.; George, J.M.; Yoo, L. Penetration of anti-infective agents into pulmonary epithelial lining fluid: Focus on antibacterial agents. Clin. Pharmacokinet. 2011, 50, 637–664. [Google Scholar] [CrossRef] [PubMed]
- Cojutti, P.G.; Merelli, M.; De Stefanis, P.; Fregonese, C.; Lucchese, F.; Bassetti, M.; Pea, F. Disposition of ceftobiprole during continuous venous-venous hemodiafiltration (CVVHDF) in a single critically ill patient. Clin. Pharmacokinet. 2018, 74, 1671–1672. [Google Scholar] [CrossRef]
- Nicholson, S.C.; Welte, T.; File, T.M.; Strauss, R.S.; Michiels, B.; Kaul, P.; Balis, D.; Arbit, D.; Amsler, K.; Noel, G.J. A randomised, double-blind trial comparing ceftobiprole medocaril with ceftriaxone with or without linezolid for the treatment of patients with community-acquired pneumonia requiring hospitalisation. Int. J. Antimicrob. Agents 2012, 39, 240–246. [Google Scholar] [CrossRef]
- Awad, S.S.; Rodriguez, A.H.; Chuang, Y.C.; Marjanek, Z.; Pareigis, A.J.; Reis, G.; Scheeren, T.W.L.; Sánchez, A.S.; Zhou, X.; Saulay, M.; et al. A phase 3 randomized double-blind comparison of ceftobiprole medocaril versus ceftazidime plus linezolid for the treatment of hospital-acquired pneumonia. Clin. Infect. Dis. 2014, 59, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.E.; Werth, B.J.; Ireland, C.E.; Stone, N.E.; Nonejuie, P.; Sakoulas, G.; Pogliano, J.; Rybak, M.J. Potent synergy of ceftobiprole plus daptomycin against multiple strains of Staphylococcus aureus with various resistance phenotypes. J. Antimicrob. Chemother. 2014, 69, 3006–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Jalali, V.; Zeitlinger, M. Clinical pharmacokinetics and pharmacodynamics of telavancin compared with the other glycopeptides. Clin. Pharmacokinet. 2018, 57, 797–816. [Google Scholar] [CrossRef] [Green Version]
- Theravance Biopharma. Theravance Biopharma Announces FDA Approval of Expanded Label for Vibativ(R) (Telavancin). Available online: https://investor.theravance.com/news-releases/news-release-details/theravance-biopharma-announces-fda-approval-expanded-label (accessed on 31 December 2020).
- European Medicines Agency. Vibativ. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vibativ-0 (accessed on 20 December 2020).
- Cumberland Pharmaceuticals, Inc. Cumberland Pharmaceuticals to Acquire VIBATIV®. Available online: https://www.biospace.com/article/releases/cumberland-pharmaceuticals-to-acquire-vibativ-/ (accessed on 5 January 2021).
- Cumberland Pharmaceuticals, Inc. Cumberland Pharmaceuticals Announces Initiative To Expand Availability Of VIBATIV® To Treat Hospital-Acquired & Ventilator-Associated Pneumonia Resulting From Coronavirus Infections. Available online: https://www.prnewswire.com/news-releases/cumberland-pharmaceuticals-announces-initiative-to-expand-availability-of-vibativ-to-treat-hospital-acquired--ventilator-associated-pneumonia-resulting-from-coronavirus-infections-301028435.html (accessed on 5 January 2021).
- Judice, J.K.; Pace, J.L. Semi-synthetic glycopeptide antibacterials. Bioorg. Med. Chem. Lett. 2003, 13, 4165–4168. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Nichol, K.; Zhanel, G.G. Telavancin: Mechanisms of action, in vitro activity, and mechanisms of resistance. Clin. Infect. Dis. 2015, 61, S58–S68. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.L.; Chang, R.; Debabov, D.V.; Leung, J.; Wu, T.; Krause, K.M.; Sandvik, E.; Hubbard, J.M.; Kaniga, K.; Schmidt, D.E. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Attwood, R.J.; LaPlante, K.L. Telavancin: A novel lipoglycopeptide antimicrobial agent. Am. J. Health-Syst. Pharm. 2007, 64, 2335–2348. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Phillips, I.; Kaniga, K. Comparative in vitro activity of telavancin (TD-6424), a rapidly bactericidal, concentration-dependent anti-infective with multiple mechanisms of action against Gram-positive bacteria. J. Antimicrob. Chemother. 2004, 53, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosowska-Shick, K.; Clark, C.; Pankuch, G.A.; McGhee, P.; Dewasse, B.; Beachel, L.; Appelbaum, P.C. Activity of telavancin against staphylococci and enterococci determined by MIC and resistance selection studies. Antimicrob. Agents Chemother. 2009, 53, 4217–4224. [Google Scholar] [CrossRef] [Green Version]
- Bugg, T.D.; Wright, G.D.; Dutka-Malen, S.; Arthur, M.; Courvalin, P.; Walsh, C.T. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: Biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Vibativ: Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/022110s011lbl.pdf (accessed on 31 December 2020).
- Wong, S.L.; Goldberg, M.R.; Ballow, C.H.; Kitt, M.M.; Barriere, S.L. Effect of Telavancin on the pharmacokinetics of the cytochrome P450 3A probe substrate midazolam: A randomized, double-blind, crossover study in healthy subjects. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2010, 30, 136–143. [Google Scholar] [CrossRef]
- Wong, S.L.; Sörgel, F.; Kinzig, M.; Goldberg, M.R.; Kitt, M.M.; Barriere, S.L. Lack of pharmacokinetic drug interactions following concomitant administration of telavancin with aztreonam or piperacillin/tazobactam in healthy participants. J. Clin. Pharmacol. 2009, 49, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, S.; Madhan, S. Telavancin: A novel lipoglycopeptide antibiotic. J. Pharmacol. Pharmacother. 2011, 2, 135. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library of Medicine. Comparison of Telavancin and Vancomycin for Hospital-Acquired Pneumonia Due to Methicillin-Resistant Staphylococcus aureus (ATTAIN1). Available online: https://clinicaltrials.gov/ct2/show/study/NCT00107952?cond=telavancin+pneumonia&draw=2&rank=1 (accessed on 17 January 2020).
- Rubinstein, E.; Corey, G.; Boucher, H.; Niederman, M.; Shorr, A.; Torres, A.; Barriere, S.; Friedland, H. Telavancin for the treatment of hospital-acquired pneumonia in severely ill and older patients: The ATTAIN studies. Crit. Care 2009, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- McKinnell, J.A.; Corman, S.; Patel, D.; Leung, G.H.; Gordon, L.M.; Lodise, T.P. Effective antimicrobial stewardship strategies for cost-effective utilization of telavancin for the treatment of patients with hospital-acquired bacterial pneumonia caused by Staphylococcus aureus. Clin. Ther. 2018, 40, 406–414.e2. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Zerbaxa: Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206829lbl.pdf (accessed on 29 December 2020).
- European Medicines Agency. Zerbaxa: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/zerbaxa#product-information-section (accessed on 29 December 2020).
- Zhanel, G.G.; Chung, P.; Adam, H.; Zelenitsky, S.; Denisuik, A.; Schweizer, F.; Lagacé-Wiens, P.R.; Rubinstein, E.; Gin, A.S.; Walkty, A. Ceftolozane/tazobactam: A novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 2014, 74, 31–51. [Google Scholar] [CrossRef]
- Takeda, S.; Nakai, T.; Wakai, Y.; Ikeda, F.; Hatano, K. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2007, 51, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PubChem. Ceftolozane. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ceftolozane (accessed on 5 January 2021).
- Giacobbe, D.R.; Bassetti, M.; De Rosa, F.G.; Del Bono, V.; Grossi, P.A.; Menichetti, F.; Pea, F.; Rossolini, G.M.; Tumbarello, M.; Viale, P.; et al. Ceftolozane/tazobactam: Place in therapy. Expert Rev. Anti-Infect. Ther. 2018, 16, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Lob, S.H.; Hoban, D.J.; Young, K.; Motyl, M.R.; Sahm, D.F. Activity of ceftolozane–tazobactam and comparators against Pseudomonas aeruginosa from patients in different risk strata—SMART United States 2016–2017. J. Glob. Antimicrob. Resist. 2020, 20, 209–213. [Google Scholar] [CrossRef]
- Sader, H.S.; Carvalhaes, C.G.; Duncan, L.R.; Shortridge, D. Antimicrobial Activity of Ceftolozane-Tazobactam and Comparators against Clinical Isolates of Haemophilus influenzae from the United States and Europe. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Mazer, D.M.; Young, C.; Kalikin, L.M.; Spilker, T.; LiPuma, J.J. In vitro activity of ceftolozane-tazobactam and other antimicrobial agents against Burkholderiacepacia complex and Burkholderia gladioli. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodlet, K.J.; Nicolau, D.P.; Nailor, M.D. Ceftolozane/tazobactam and ceftazidime/avibactam for the treatment of complicated intra-abdominal infections. Ther. Clin. Risk Manag. 2016, 12, 1811. [Google Scholar] [CrossRef] [Green Version]
- Snydman, D.R.; McDermott, L.A.; Jacobus, N.V. Activity of ceftolozane-tazobactam against a broad spectrum of recent clinical anaerobic isolates. Antimicrob. Agents Chemother. 2014, 58, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M.; Mushtaq, S.; Meunier, D.; Hopkins, K.L.; Hill, R.; Adkin, R.; Chaudhry, A.; Pike, R.; Staves, P.; Woodford, N.; et al. Activity of ceftolozane/tazobactam against surveillance and ‘problem’ Enterobacteriaceae, Pseudomonas aeruginosa and non-fermenters from the British Isles. J. Antimicrob. Chemother. 2017, 72, 2278–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaumburg, F.; Bletz, S.; Mellmann, A.; Becker, K.; Idelevich, E.A. Susceptibility of MDR Pseudomonas aeruginosa to ceftolozane/tazobactam and comparison of different susceptibility testing methods. J. Antimicrob. Chemother. 2017, 72, 3079–3084. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; Bruchmann, S.; Mulet, X.; Zamorano, L.; Moyá, B.; Juan, C.; Haussler, S.; Olivera, A. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of ampc. Antimicrob. Agents Chemother. 2014, 58, 3091–3099. [Google Scholar] [CrossRef] [Green Version]
- Berrazeg, M.; Jeannot, K.; Enguéné, V.Y.N.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef] [Green Version]
- Haidar, G.; Philips, N.J.; Shields, R.K.; Snyder, D.; Cheng, S.; Potoski, B.A.; Hao, B.; Press, E.G.; Cooper, V.S.; Clancy, C.J. Ceftolozane-tazobactam for the treatment of multidrug-resistant Pseudomonas aeruginosa infections: Clinical effectiveness and evolution of resistance. Clin. Infect. Dis. 2017, 65, 110–120. [Google Scholar] [CrossRef]
- MacVane, S.; Pandey, R.; Steed, L.; Kreiswirth, B.; Chen, L. Emergence of ceftolozane-tazobactam-resistant Pseudomonas aeruginosa during treatment is mediated by a single AmpC structural mutation. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.; Hershberger, E.; Benziger, D.; Trinh, M.M.; Friedland, I. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob. Agents Chemother. 2012, 56, 3086–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Aziz, M.H.; Alffenaar, J.W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper#. Intensiv. Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Chandorkar, G.; Huntington, J.A.; Gotfried, M.H.; Rodvold, K.A.; Umeh, O. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J. Antimicrob. Chemother. 2012, 67, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Kollef, M.H.; Nováček, M.; Kivistik, Ü.; Réa-Neto, Á.; Shime, N.; Martin-Loeches, I.; Timsit, J.F.; Wunderink, R.G.; Bruno, C.J.; Huntington, J.A.; et al. Ceftolozane-tazobactam versus meropenem for treatment of nosocomial pneumonia (ASPECT-NP): A randomised, controlled, double-blind, phase 3, non-inferiority trial. Lancet Infect. Dis. 2019, 19, 1299–1311. [Google Scholar] [CrossRef]
- Xiao, A.J.; Miller, B.W.; Huntington, J.A.; Nicolau, D.P. Ceftolozane/tazobactam pharmacokinetic/pharmacodynamic-derived dose justification for phase 3 studies in patients with nosocomial pneumonia. J. Clin. Pharmacol. 2016, 56, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Caro, L.; Nicolau, D.P.; De Waele, J.J.; Kuti, J.L.; Larson, K.B.; Gadzicki, E.; Yu, B.; Zeng, Z.; Adedoyin, A.; Rhee, E.G. Lung penetration, bronchopulmonary pharmacokinetic/pharmacodynamic profile and safety of 3 g of ceftolozane/tazobactam administered to ventilated, critically ill patients with pneumonia. J. Antimicrob. Chemother. 2020, 75, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, G.; Ferriols, R.; Martínez-Castro, S.; Ezquer, C.; Pastor, E.; Carbonell, J.A.; Alós, M.; Navarro, D. Optimizing ceftolozane-tazobactam dosage during continuous renal replacement therapy: Some nuances. Crit. Care 2020, 24. [Google Scholar] [CrossRef]
- Bassetti, M.; Castaldo, N.; Cattelan, A.; Mussini, C.; Righi, E.; Tascini, C.; Menichetti, F.; Mastroianni, C.M.; Tumbarello, M.; Grossi, P.; et al. Ceftolozane/tazobactam for the treatment of serious Pseudomonas aeruginosa infections: A multicentre nationwide clinical experience. Int. J. Antimicrob. Agents 2019, 53, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.M.; Mugisha, A.; Barreto Gutierrez, L.; Redant, S.; Kaefer, K.; Gallerani, A.; De Bels, D. Optimizing ceftolozane-tazobactam dosage during continuous renal replacement therapy: Additional insights. Crit. Care 2019, 23. [Google Scholar] [CrossRef] [Green Version]
- Mané, C.; Delmas, C.; Porterie, J.; Jourdan, G.; Verwaerde, P.; Marcheix, B.; Concordet, D.; Georges, B.; Ruiz, S.; Gandia, P. Influence of extracorporeal membrane oxygenation on the pharmacokinetics of ceftolozane/tazobactam: An ex vivo and in vivo study. J. Transl. Med. 2020, 18. [Google Scholar] [CrossRef]
- Arena, F.; De Angelis, L.H.; Maglioni, E.; Contorni, M.; Cassetta, M.I.; Novelli, A.; Rossolini, G.M. Ceftolozane-tazobactam pharmacokinetics during extracorporeal membrane oxygenation in a lung transplant recipient. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomkin, J.; Hershberger, E.; Miller, B.; Popejoy, M.; Friedland, I.; Steenbergen, J.; Yoon, M.; Collins, S.; Yuan, G.; Barie, P.; et al. Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: Results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin. Infect. Dis. 2015, 60, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Zavicefta: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/zavicefta-epar-product-information_en.pdf (accessed on 29 December 2020).
- U.S. Food and Drug Administration. Avycaz: Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/206494s005,s006lbl.pdf (accessed on 29 December 2020).
- Rains, C.P.; Bryson, H.M.; Peters, D.H. Ceftazidime: An Update of its Antibacterial Activity, Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1995, 49, 577–617. [Google Scholar] [CrossRef]
- Shirley, M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef]
- Bonnefoy, A.; Dupuis-Hamelin, C.; Steier, V.; Delachaume, C.; Seys, C.; Stachyra, T.; Fairley, M.; Guitton, M.; Lampilas, M. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J. Antimicrob. Chemother. 2004, 54, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non–β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [Green Version]
- Lizana, I.; Delgado, E.J. Molecular insights on the release of avibactam from the acyl-enzyme complex. Biophys. J. 2019, 116, 1650–1657. [Google Scholar] [CrossRef]
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to β-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Aktaş, Z.; Kayacan, C.; Oncul, O. In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 2012, 39, 86–89. [Google Scholar] [CrossRef]
- De Jonge, B.L.M.; Karlowsky, J.A.; Kazmierczak, K.M.; Biedenbach, D.J.; Sahm, D.F.; Nichols, W.W. In vitro susceptibility to ceftazidime-avibactam of carbapenem-nonsusceptibleenterobacteriaceae isolates collected during the INFORM global surveillance study (2012 to 2014). Antimicrob. Agents Chemother. 2016, 60, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Bajaksouzian, S.; Abdelhamed, A.M.; Foster, A.N.; Winkler, M.L.; Gatta, J.A.; Nichols, W.W.; Testa, R.; Bonomo, R.A.; Jacobs, M.R. Activities of ceftazidime, ceftaroline, and aztreonam alone and combined with avibactam against isogenic Escherichia coli strains expressing selected single β-lactamases. Diagn. Microbiol. Infect. Dis. 2015, 82, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlowsky, J.A.; Biedenbach, D.J.; Kazmierczak, K.M.; Stone, G.G.; Sahm, D.F. Activity of ceftazidime-avibactam against extended-spectrum- and AmpC β-lactamase-producing Enterobacteriaceae collected in the INFORM global surveillance study from 2012 to 2014. Antimicrob. Agents Chemother. 2016, 60, 2849–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, W.W.; De Jonge, B.L.M.; Kazmierczak, K.M.; Karlowsky, J.A.; Sahm, D.F. In vitro susceptibility of global surveillance isolates of Pseudomonas aeruginosa to ceftazidime-avibactam (INFORM 2012 to 2014). Antimicrob. Agents Chemother. 2016, 60, 4743–4749. [Google Scholar] [CrossRef] [Green Version]
- Kazmierczak, K.M.; Biedenbach, D.J.; Hackel, M.; Rabine, S.; De Jonge, B.L.M.; Bouchillon, S.K.; Sahm, D.F.; Bradford, P.A. Global dissemination of blaKPC into bacterial species beyond Klebsiella pneumoniae and in vitro susceptibility to ceftazidime-avibactam and aztreonam-avibactam. Antimicrob. Agents Chemother. 2016, 60, 4490–4500. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, E.T.; Becka, S.A.; Wilson, B.M.; Barnes, M.D.; LiPuma, J.J.; Papp-Wallace, K.M. “Switching partners”: Piperacillin-Avibactam Is a Highly Potent Combination against Multidrug-Resistant Burkholderiacepacia Complex and Burkholderia gladioli Cystic Fibrosis Isolates. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [Green Version]
- Testa, R.; Cantón, R.; Giani, T.; Morosini, M.-I.; Nichols, W.W.; Seifert, H.; Stefanik, D.; Rossolini, G.M.; Nordmann, P. In vitro activity of ceftazidime, ceftaroline and aztreonam alone and in combination with avibactam against European Gram-negative and Gram-positive clinical isolates. Int. J. Antimicrob. Agents 2015, 45, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Alatoom, A.; Elsayed, H.; Lawlor, K.; AbdelWareth, L.; El-Lababidi, R.; Cardona, L.; Mooty, M.; Bonilla, M.F.; Nusair, A.; Mirza, I. Comparison of antimicrobial activity between ceftolozane-tazobactam and ceftazidime-avibactam against multidrug-resistant isolates of Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa. Int. J. Infect. Dis. 2017, 62, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Buehrle, D.J.; Shields, R.K.; Chen, L.; Hao, B.; Press, E.G.; Alkrouk, A.; Potoski, B.A.; Kreiswirth, B.N.; Clancy, C.J.; Nguyen, M.H. Evaluation of the in vitro activity of ceftazidime-avibactam and ceftolozane-tazobactam against meropenem-resistant Pseudomonas aeruginosa isolates. Antimicrob. Agents Chemother. 2016, 60, 3227–3231. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, S.D.; Walkup, G.K.; Whiteaker, J.D.; Palmer, T.; McCormack, K.; Angela Tanudra, M.; Nash, T.J.; Thresher, J.; Johnstone, M.R.; Hajec, L.; et al. Selection and molecular characterization of ceftazidime/avibactamresistant mutants in Pseudomonas aeruginosa strains containing derepressed AmpC. J. Antimicrob. Chemother. 2014, 70, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M.; Mushtaq, S.; Barker, K.; Hope, R.; Warner, M.; Woodford, N. Characterization of β-lactamase and porin mutants of enterobacteriaceae selected with ceftaroline+avibactam (NXL104). J. Antimicrob. Chemother. 2012, 67, 1354–1358. [Google Scholar] [CrossRef]
- Nelson, K.; Hemarajata, P.; Sun, D.; Rubio-Aparicio, D.; Tsivkovski, R.; Yang, S.; Sebra, R.; Kasarskis, A.; Nguyen, H.; Hanson, B.M. Resistance to ceftazidime-avibactam is due to transposition of KPC in a porin-deficient strain of Klebsiella pneumoniae with increased efflux activity. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Humphries, R.M.; Hemarajata, P. Resistance to ceftazidime-avibactam in Klebsiella pneumoniae due to porin mutations and the increased expression of KPC-3. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galani, I.; Karaiskos, I.; Angelidis, E.; Papoutsaki, V.; Galani, L.; Souli, M.; Antoniadou, A.; Giamarellou, H. Emergence of ceftazidime-avibactam resistance through distinct genomic adaptations in KPC-2-producing Klebsiella pneumoniae of sequence type 39 during treatment. Eur. J. Clin. Microbiol. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Galani, I.; Karaiskos, I.; Souli, M.; Papoutsaki, V.; Galani, L.; Gkoufa, A.; Antoniadou, A.; Giamarellou, H. Outbreak of KPC-2-producing Klebsiella pneumoniae endowed with ceftazidime-avibactam resistance mediated through a VEB-1-mutant (VEB-25), Greece, September to October 2019. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Haidar, G.; Clancy, C.J.; Shields, R.K.; Hao, B.; Cheng, S.; Nguyen, M.H. Mutations in blaKPC-3 that confer ceftazidime-avibactam resistance encode novel KPC-3 variants that function as extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.K.; Chen, L.; Cheng, S.; Chavda, K.D.; Press, E.G.; Snyder, A.; Pandey, R.; Doi, Y.; Kreiswirth, B.N.; Nguyen, M.H.; et al. Emergence of ceftazidime-avibactam resistance due to plasmid-borne blaKPC-3 mutations during treatment of carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Göttig, S.; Frank, D.; Mungo, E.; Nolte, A.; Hogardt, M.; Besier, S.; Wichelhaus, T.A. Emergence of ceftazidime/avibactam resistance in KPC-3-producing Klebsiella pneumoniae in vivo. J. Antimicrob. Chemother. 2019, 74, 3211–3216. [Google Scholar] [CrossRef]
- Nicolau, D.P.; Siew, L.; Armstrong, J.; Li, J.; Edeki, T.; Learoyd, M.; Das, S. Phase 1 study assessing the steady-state concentration of ceftazidime and avibactam in plasma and epithelial lining fluid following two dosing regimens. J. Antimicrob. Chemother. 2015, 70, 2862–2869. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, X.; Xia, Y.; Chu, Y.; Zhong, H.; Li, J.; Liang, P.; Bu, Y.; Zhao, R.; Liao, Y.; et al. Recommendation of Antimicrobial Dosing Optimization During Continuous Renal Replacement Therapy. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Soukup, P.; Faust, A.C.; Edpuganti, V.; Putnam, W.C.; McKinnell, J.A. Steady-State Ceftazidime-Avibactam Serum Concentrations and Dosing Recommendations in a Critically Ill Patient Being Treated for Pseudomonas aeruginosa Pneumonia and Undergoing Continuous Venovenous Hemodiafiltration. Pharmacotherapy 2019, 39, 1216–1222. [Google Scholar] [CrossRef]
- Wenzler, E.; Bunnell, K.L.; Bleasdale, S.C.; Benken, S.; Danziger, L.H.; Rodvold, K.A. Pharmacokinetics and dialytic clearance of ceftazidime-avibactam in a critically ill patient on continuous venovenous hemofiltration. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Zhong, N.; Pachl, J.; Timsit, J.-F.; Kollef, M.; Chen, Z.; Song, J.; Taylor, D.; Laud, P.J.; Stone, G.G. Ceftazidime-avibactam versus meropenem in nosocomial pneumonia, including ventilator-associated pneumonia (REPROVE): A randomised, double-blind, phase 3 non-inferiority trial. Lancet Infect. Dis. 2018, 18, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, S.; Singh, N.B.; Kebriaei, R.; Rice, S.A.; Stamper, K.C.; Castanheira, M.; Rybak, M.J. Evaluation of the synergy of ceftazidime-avibactam in combination with meropenem, amikacin, aztreonam, colistin, or fosfomycin against well-characterized multidrug-resistant Klebsiella pneumoniae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00779-19. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Vaborem: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/vaborem-epar-product-information_en.pdf (accessed on 29 December 2020).
- U.S. Food Drug and Administration. Vabomere: Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209776lbl.pdf (accessed on 29 December 2020).
- Novelli, A.; Del Giacomo, P.; Rossolini, G.M.; Tumbarello, M. Meropenem/vaborbactam: A next generation β-lactam β-lactamase inhibitor combination. Expert Rev. Anti-Infect. Ther. 2020, 18, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Wiebe, R.; Dilay, L.; Thomson, K.; Rubinstein, E.; Hoban, D.J.; Noreddin, A.M.; Karlowsky, J.A. Comparative review of the carbapenems. Drugs 2007, 67, 1027–1052. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Tsivkovski, R.; Griffith, D.C.; Dudley, M.N. Vaborbactam: Spectrum of beta-lactamase inhibition and impact of resistance mechanisms on activity in enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Dudley, M.N.; Lomovskaya, O. Meropenem-vaborbactam resistance selection, resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Quale, J.; Landman, D. Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against gram-negative clinical isolates in New York City. Antimicrob. Agents Chemother. 2015, 59, 4856–4860. [Google Scholar] [CrossRef] [Green Version]
- Patel, T.S.; Pogue, J.M.; Mills, J.P.; Kaye, K.S. Meropenem-vaborbactam: A new weapon in the war against infections due to resistant Gram-negative bacteria. Future Microbiol. 2018, 13, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Kline, E.G.; Jones, C.E.; Morder, K.T.; Mettus, R.T.; Doi, Y.; Nguyen, M.H.; Clancy, C.J.; Shields, R.K. Effects of KPC variant and porin genotype on the in vitro activity of meropenem-vaborbactam against carbapenem-resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubino, C.M.; Bhavnani, S.M.; Loutit, J.S.; Morgan, E.E.; White, D.; Dudley, M.N.; Griffith, D.C. Phase 1 study of the safety, tolerability, and pharmacokinetics of vaborbactam and meropenem alone and in combination following single and multiple doses in healthy adult subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Meropenem/Vaborbactam: A Review in Complicated Urinary Tract Infections. Drugs 2018, 78, 1259–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzler, E.; Gotfried, M.H.; Loutit, J.S.; Durso, S.; Griffith, D.C.; Dudley, M.N.; Rodvold, K.A. Meropenem-RPX7009 concentrations in plasma, epithelial lining fluid, and alveolar macrophages of healthy adult subjects. Antimicrob. Agents Chemother. 2015, 59, 7232–7239. [Google Scholar] [CrossRef] [Green Version]
- Sime, F.B.; Pandey, S.; Karamujic, N.; Parker, S.; Alexander, E.; Loutit, J.; Durso, S.; Griffith, D.; Lipman, J.; Wallis, S.C.; et al. Ex vivo characterization of effects of renal replacement therapy modalities and settings on pharmacokinetics of meropenem and vaborbactam. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderink, R.G.; Giamarellos-Bourboulis, E.J.; Rahav, G.; Mathers, A.J.; Bassetti, M.; Vazquez, J.; Cornely, O.A.; Solomkin, J.; Bhowmick, T.; Bishara, J.; et al. Effect and Safety of Meropenem-Vaborbactam versus Best-Available Therapy in Patients with Carbapenem-Resistant Enterobacteriaceae Infections: The TANGO II Randomized Clinical Trial. Infect. Dis. Ther. 2018, 7, 439–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vélez-Díaz-Pallarés, M.; Silveira, E.D.; Díaz, A.Á.; Menéndez-Conde, C.P.; Oliveros, N.V.; Vicedo, T.B. Analysis of the valproic acid-meropenem interaction in hospitalised patients. Neurología 2012, 27, 34–38. [Google Scholar]
- Kaye, K.S.; Bhowmick, T.; Metallidis, S.; Bleasdale, S.C.; Sagan, O.S.; Stus, V.; Vazquez, J.; Zaitsev, V.; Bidair, M.; Chorvat, E. Effect of meropenem-vaborbactam vs piperacillin-tazobactam on clinical cure or improvement and microbial eradication in complicated urinary tract infection: The TANGO I randomized clinical trial. JAMA 2018, 319, 788–799. [Google Scholar] [CrossRef]
- Petty, L.A.; Henig, O.; Patel, T.S.; Pogue, J.M.; Kaye, K.S. Overview of meropenem-vaborbactam and newer antimicrobial agents for the treatment of carbapenem-resistant enterobacteriaceae. Infect. Drug Resist. 2018, 11, 1461–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.; Niederman, M.S.; Chastre, J.; Ewig, S.; Fernandez-Vandellos, P.; Hanberger, H.; Kollef, M.; Li Bassi, G.; Luna, C.M.; Martin-Loeches, I.; et al. International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: Guidelines for the management of hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) of the European Respiratory Society (ERS), European Society of Intensive Care Medicine (ESICM), European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and Asociación Latinoamericana del Tórax (ALAT). Eur. Respir. J. 2017, 50, 1700582. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Multiplex PCR Panel | Type of Sample and Time to Results | Performance § | Pathogens/Markers of Resistance Genes Detected | |
|---|---|---|---|---|
| BioFire® FilmArray® Pneumonia Panel and Pneumonia Panel Plus (bioMérieux SA, France) [https://www.biofiredx.com/products/the-filmarray-panels/filmarray-pneumonia; Access date 20 February 2021, https://www.biomerieux-diagnostics.com/biofire-filmarray-pneumonia-panel; Access date 20 February 2021] | BAL/mini-BAL, tracheal aspirate, induced and expectorated sputum Time to results: 1 h | Both panels: BAL/BAL-like: Sens/Spec= 96.2%/98.3% Sputum: Sens/Spec= 96.3%/97.2% | Bacteria Acinetobacter calcoaceticus-baumannii complex Enterobacter cloacae Escherichia coli Haemophilus influenzae Klebsiella aerogenes Klebsiella oxytoca Klebsiella pneumoniae group Moraxella catarrhalis Proteus spp. Pseudomonas aeruginosa Serratia marcescens Staphylococcus aureus Streptococcus agalactiae Streptococcus pneumoniae Streptococcus pyogenes Legionella pneumophila Mycoplasma pneumoniae Chlamydia pneumoniae | Viruses Influenza A and B Adenovirus Coronavirus Parainfluenza virus Respiratory Syncytial virus Human Rhinovirus/Enterovirus Human Metapneumovirus Middle East Respiratory Syndrome Coronavirus (MERS-CoV) * Antibiotic resistance genes CTX-M, KPC, NDM Oxa48-like, VIM, IMP, mecA/mecC and MREJ |
| Unyvero Lower Respiratory Tract (LRT) Panel and LRT BAL (Curetis AG, USA) ** [https://www.curetisusa.com/wp-content/uploads/Unyvero-Pneumonia-Panel-Flyer-PN3677A.pdf; Access date 20 February 2021] | BAL/mini-BAL or tracheal aspirate Time to results: 5 h | Both panels: Sens/Spec = 91.4%/99.5% | Bacteria Acinetobacter spp. Chlamydia pneumoniae Citrobacter freundii Enterobacter cloacae complex Escherichia coli Haemophilus influenzae Klebsiella oxytoca Klebsiella pneumoniae Klebsiella variicola Legionella pneumophila Moraxella catarrhalis Morganella morganii Mycoplasma pneumoniae Proteus spp. Pseudomonas aeruginosa Serratia marcescens Staphylococcus aureus Stenotrophomonas maltophilia Streptococcus pneumoniae | Other/Fungi *** Pneumocystis jirovecii Antibiotic resistance genes KPC, NDM, OXA-23, OXA-24, OXA-48, OXA-58, VIM, CTX-M, mecA, TEM |
| Unyvero Hospitalised Pneumonia (HPN) Cartridge (Curetis AG, USA) [https://www.curetisusa.com/wp-content/uploads/Unyvero-LRT-Pneumonia-Brochure.pdf; Access date 20 February 2021] | BAL/mini-BAL, tracheal aspirate, sputum Time to results: 4–5 h | For microorganisms: Sens/Spec= 92.5%/97.4% For AMR markers Sens/Spec = 93%/98.8% | Bacteria Same as Unyvero LRT BAL Panel and additionally Chlamydophila pneumoniae | Other/Fungi Pneumocystis jirovecii Antibiotic resistance genes ERMB, mecA/mecC, TEM SHV, CTX-M, KPC, NDM, OXA-23, OXA-24/40, OXA-48, OXA-58, VIM, SUL1, gyrA83, gyrA87 |
| Unyvero P55 panel (Curetis AG, USA) [https://curetis.com/wp-content/uploads/20150416_Curetis_P55_study_completion_EN_FINAL_APPROVED.pdf;Access date 20 February 2021] | BAL/mini-BAL, tracheal aspirate, sputum Time to results: 4–5 h | Sens/Spec= 94%/99.4% | Bacteria Same as Unyvero LRT BAL Panel and additionally: Klebsiella aerogenes (previously known as Enterobacter aerogenes) | Other/Fungi Pneumocystis jirovecii Antibiotic resistance genes ERMB, mecA/mecC, TEM SHV, CTX-M, IMP, KPC, NDM, OXA-23, OXA-24, OXA-48, OXA-58, VIM, SUL1, gyrA83 gyrA87 |
| Advantages | Disadvantages |
|---|---|
| Exceptionally faster time to results for pathogen and resistance profiles: major utility for prompt treatment modification and effective patient management | Over-detection of microbial and viral genome: problem in results interpretation: pathogen or coloniser? (may be partially solved with semi-quantification of bacterial targets) |
| Multiple targets detection at the same and Detection of viral and atypical pathogens as well | The presence of a resistance gene marker may not be linked to the detected microorganism, but to other co-existent organisms either undetectable or below the detection limit, thus making culture-based techniques still necessary in many cases |
| Detection of pathogens even when antimicrobial treatment has been initiated | Initial cost to buy the equipment |
| Potential for better antibiotic utilisation and positive impact on: -nosocomial pneumonia management, shortening hospital stay and decreasing healthcare costs, -antibiotic stewardship programs | Not widely available among different institutions yet |
| Early identification of MDR pathogens should facilitate enhanced infection control practices and reduce spread | Further validation versus traditional diagnostic techniques needed and determination of the effect on antimicrobial prescribing, patient outcomes and resistance is needed |
| NP (HAP and/or VAP): Dosage and Treatment Duration for NP | Other Approved Indications | |
|---|---|---|
| Ceftolozane/tazobactam | HAP and VAP 1 Dosage: 3 g (2/1) every 8 h (h), 1-h IV infusion, (Note: double dose compared to other indications) Duration: 8–14 days (d) | cIAIs cUTIs (including acute pyelonephritis) |
| Ceftazidime/avibactam | HAP and VAP, including bacteraemic cases (bacteraemia associated with or suspected to be associated with HAP/VAP) 1 Dosage: 2.5 g (2/0.5) every 8 h, 2-h IV infusion Duration: 7–14 d | cIAI (in combination with metronidazole), cUTI (including pyelonephritis), Bacteraemia associated with or suspected to be associated with cIAI or cUTI Infections due to aerobic Gram-negative organisms in patients with limited treatment options |
| Meropenem/vaborbactam | HAP and VAP, including bacteraemic cases (bacteraemia associated with or suspected to be associated with HAP/VAP) 1 Dosage: 4 g (2/2) every 8 h, 3-h IV infusion Duration: 7–14 d | cIAI cUTI (including pyelonephritis), Bacteraemia associated with or suspected to be associated with cIAI or cUTI Infections due to aerobic Gram-negative organisms in patients with limited treatment option |
| Ceftobiprol medocaril | HAP (not for VAP) 1 Dosage: 500 mg every 8 h, 2-h IV infusion Duration: 7–14 d | CAP |
| Telavancin | HAP and VAP caused by S. aureus including bacteraemic cases (when no alternative treatment available) 2 Dosage: 10 mg/kg every 24 h, 1-h IV infusion Duration: 7–21 d | cSSSI caused by S. aureus including bacteraemic cases (when no alternative treatment available) |
| ESBL | AmpC | KPC | OXA | MBL | Carb-R A.B. | MRSA | |
|---|---|---|---|---|---|---|---|
| Ceftolozane/tazobactam 1 | + | +/− | − | − | − | − | − |
| Ceftazidime/avibactam 2 | ++ | + | + | + | − | − | − |
| Meropenem/vaborbactam 3 | + | + | + | − | − | − | − |
| Ceftobiprol medocaril | − | − | − | − | − | − | + |
| Telavancin | − | − | − | − | − | − | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, E.; Pérez-Torres, D.; Fragkou, P.C.; Zahar, J.-R.; Koulenti, D. Nosocomial Pneumonia in the Era of Multidrug-Resistance: Updates in Diagnosis and Management. Microorganisms 2021, 9, 534. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030534
Xu E, Pérez-Torres D, Fragkou PC, Zahar J-R, Koulenti D. Nosocomial Pneumonia in the Era of Multidrug-Resistance: Updates in Diagnosis and Management. Microorganisms. 2021; 9(3):534. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030534
Chicago/Turabian StyleXu, Elena, David Pérez-Torres, Paraskevi C. Fragkou, Jean-Ralph Zahar, and Despoina Koulenti. 2021. "Nosocomial Pneumonia in the Era of Multidrug-Resistance: Updates in Diagnosis and Management" Microorganisms 9, no. 3: 534. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030534