
Targeting the DEAD-Box RNA Helicase eIF4A with Rocaglates—A Pan-Antiviral Strategy for Minimizing the Impact of Future RNA Virus Pandemics


 , ,
, ,
Abstract
:1. Introduction
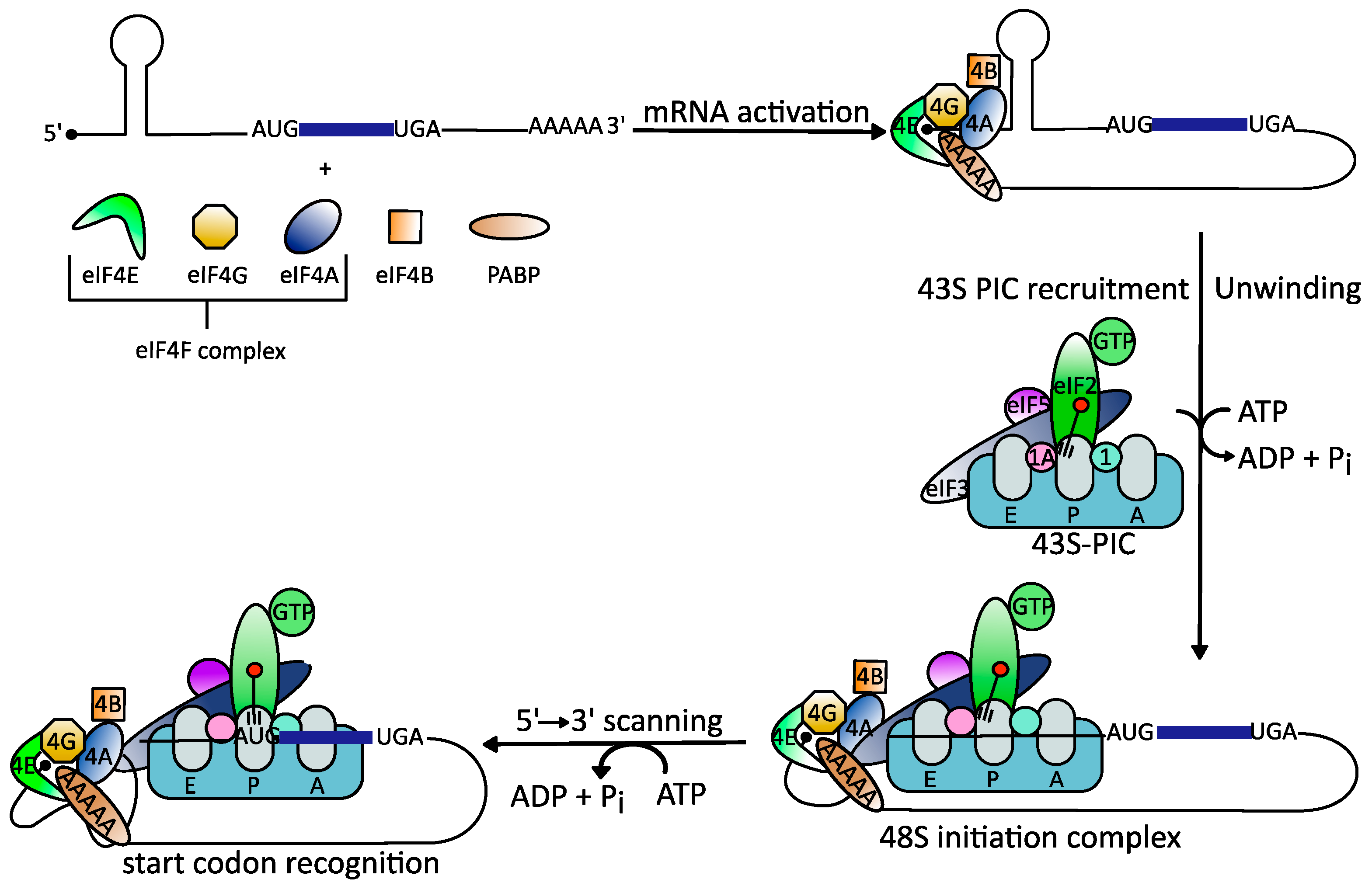
2. The Role of the DEAD-Box RNA Helicase eIF4A in RNA Virus Translation
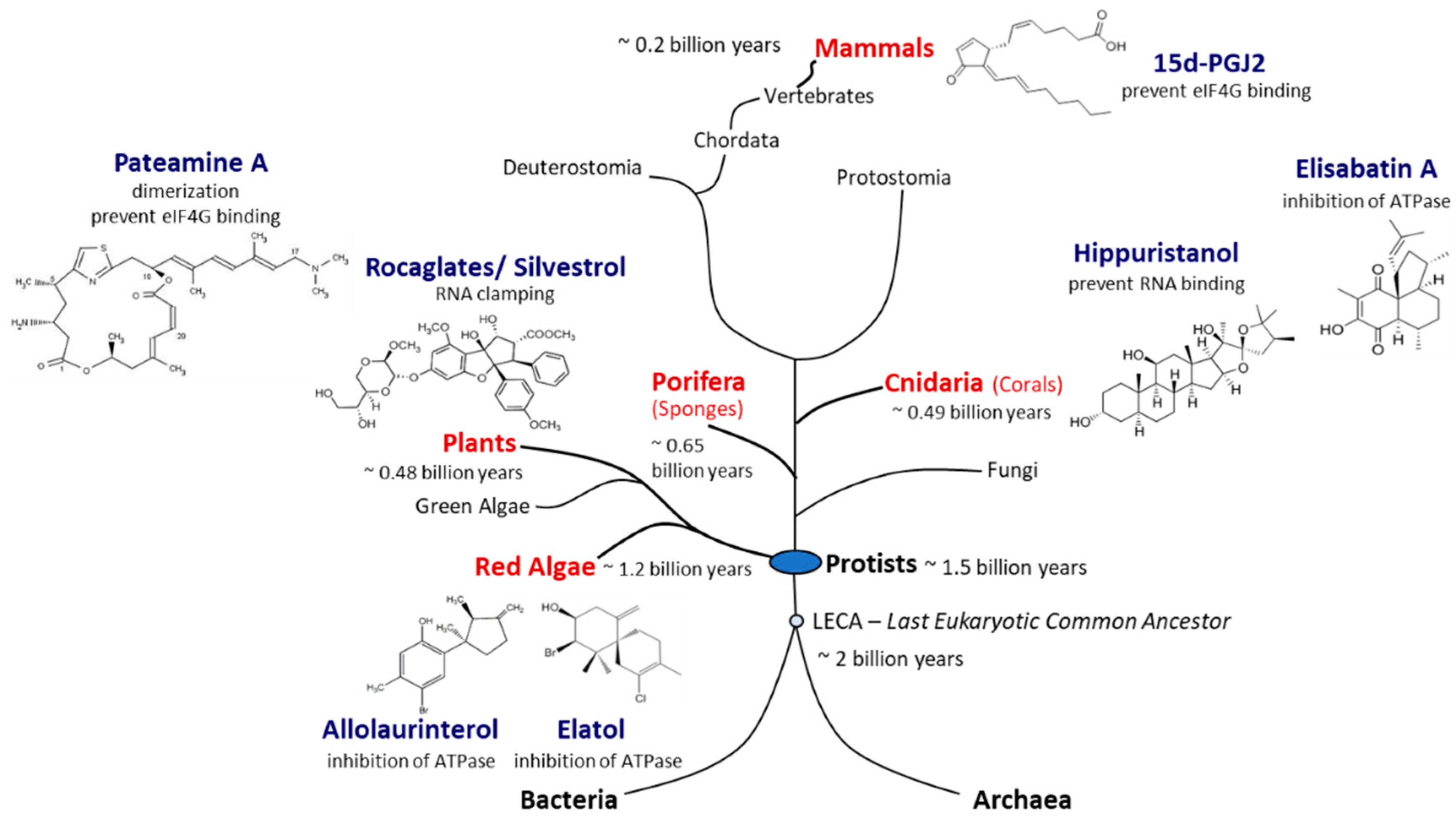
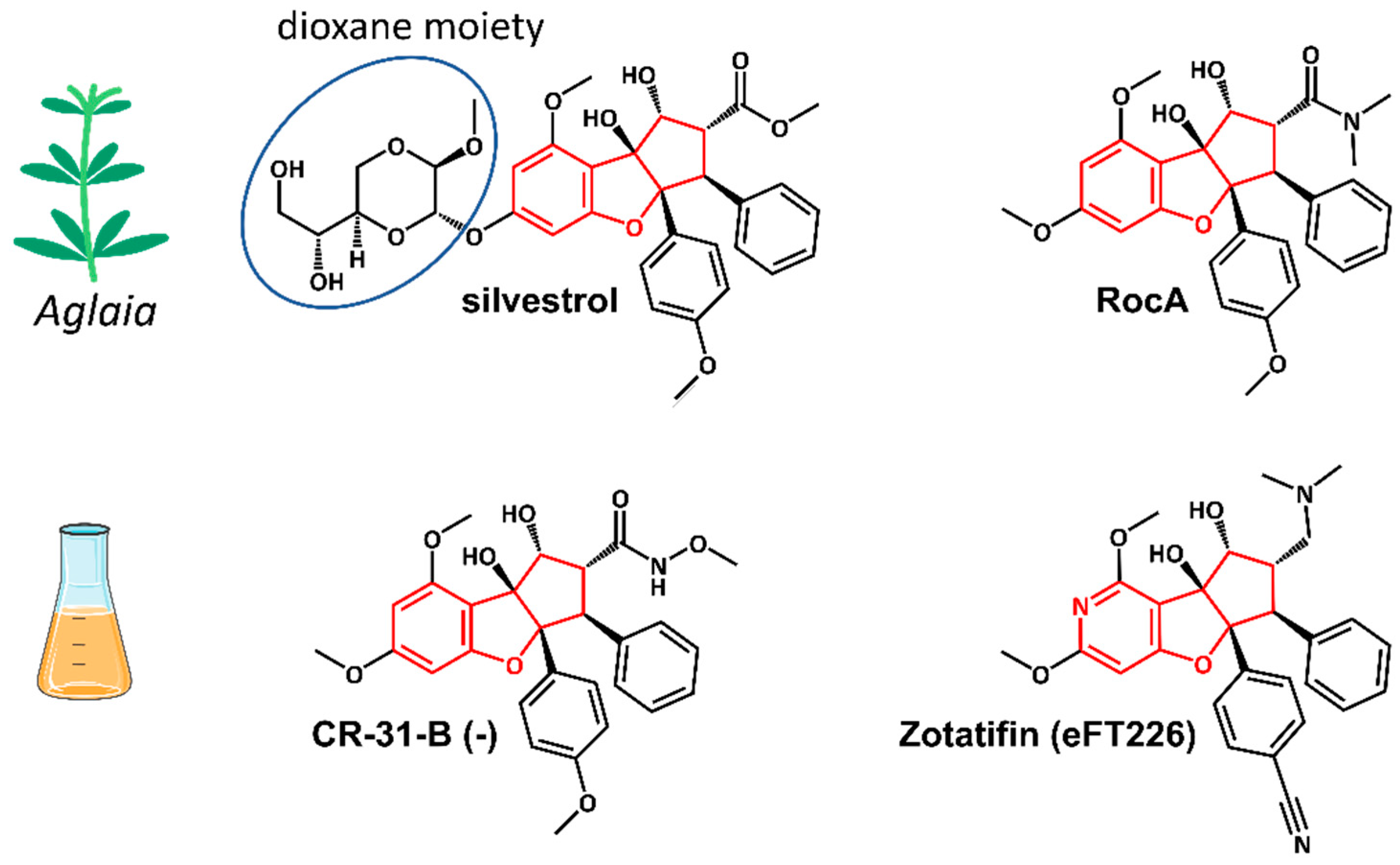
3. eIF4A Inhibitors
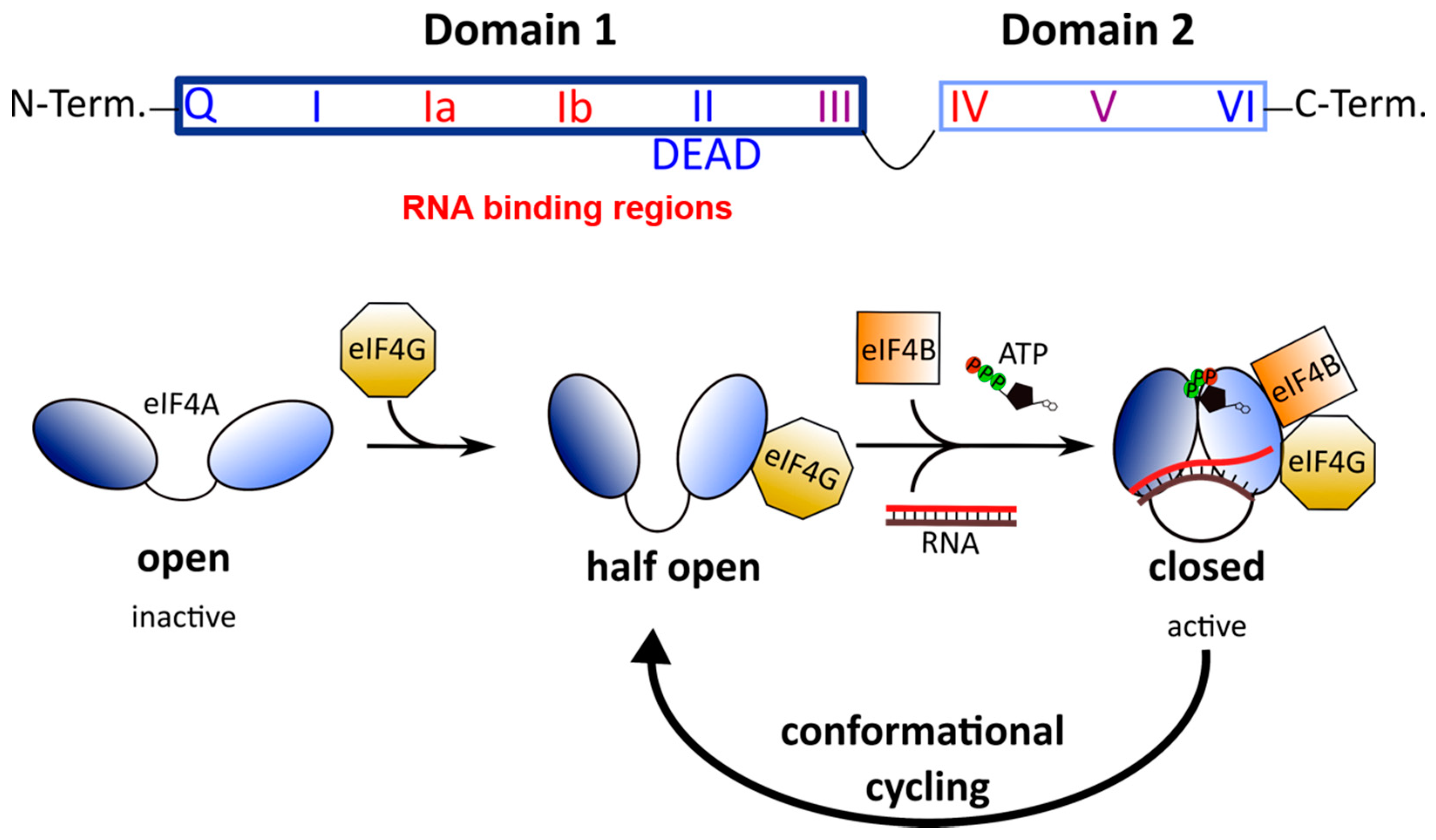
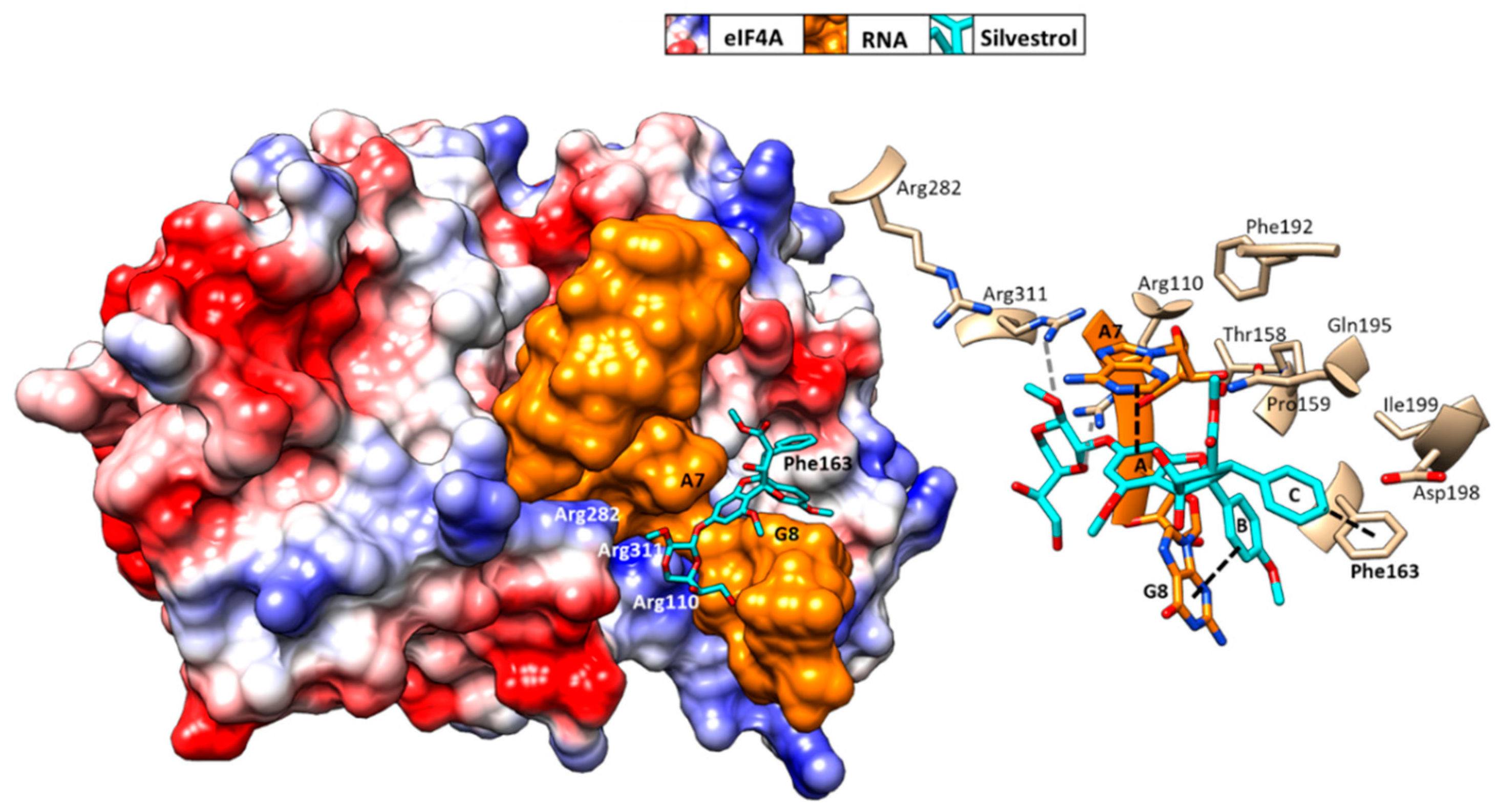
4. Mechanism of Action of Rocaglates
5. eIF4A as an Antiviral Rocaglate Target
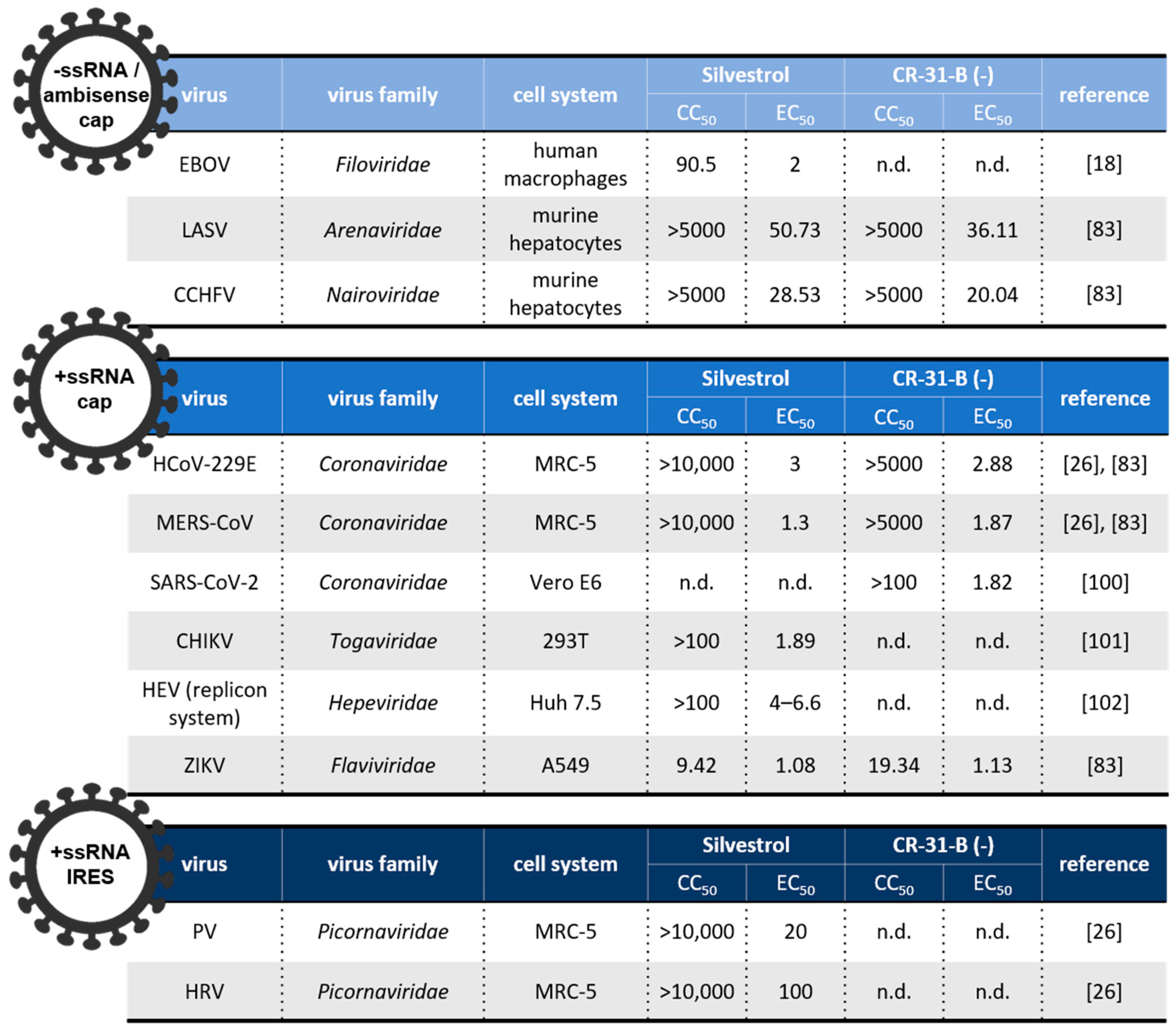
5.1. Negative (−)-Stranded and Ambisense RNA Viruses
5.2. Positive (+)-Stranded RNA Viruses
5.2.1. Coronaviridae
5.2.2. Togaviridae
5.2.3. Hepeviridae
5.2.4. Picornaviridae
5.2.5. Flaviviridae
6. A Pan-Antiviral Translational Roadmap for Rocaglates
6.1. Foundational Research
6.2. Preclinical Research
6.3. Clinical Studies
6.4. Other Considerations
7. Outlook and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Muehlenbein, M.P. Human-Wildlife Contact and Emerging Infectious Diseases. In Human-Environment Interactions; Springer: Dordrecht, The Netherlands, 2013; pp. 79–94. [Google Scholar]
- Karesh, W.B.; Dobson, A.; Lloyd-Smith, J.O.; Lubroth, J.; Dixon, M.A.; Bennett, M.; Aldrich, S.; Harrington, T.; Formenty, P.; Loh, E.H.; et al. Ecology of zoonoses: Natural and unnatural histories. Lancet 2012, 380, 1936–1945. [Google Scholar] [CrossRef]
- Pak, A.; Adegboye, O.A.; Adekunle, A.I.; Rahman, K.M.; McBryde, E.S.; Eisen, D.P. Economic Consequences of the COVID-19 Outbreak: The Need for Epidemic Preparedness. Front. Public Health 2020, 8, 241. [Google Scholar] [CrossRef]
- Faust, J.S.; Krumholz, H.M.; Du, C.; Mayes, K.D.; Lin, Z.; Gilman, C.; Walensky, R.P. All-Cause Excess Mortality and COVID-19–Related Mortality Among US Adults Aged 25–44 Years, March-July 2020. JAMA 2020. [Google Scholar] [CrossRef]
- Jester, B.; Uyeki, T.; Jernigan, D. Readiness for Responding to a Severe Pandemic 100 Years After 1918. Am. J. Epidemiol. 2018, 187, 2596–2602. [Google Scholar] [CrossRef]
- Smith, K.F.; Goldberg, M.; Rosenthal, S.; Carlson, L.; Chen, J.; Chen, C.; Ramachandran, S. Global rise in human infectious disease outbreaks. J. R. Soc. Interface 2014, 11, 20140950. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.; Gaunt, E. Ecological Origins of Novel Human Pathogens. Crit. Rev. Microbiol. 2007, 33, 231–242. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Woolhouse, M.E.J.; Brierley, L.; McCaffery, C.; Lycett, S. Assessing the Epidemic Potential of RNA and DNA Viruses. Emerg. Infect. Dis. 2016, 22, 2037–2044. [Google Scholar] [CrossRef]
- Tse, L.V.; Meganck, R.M.; Graham, R.L.; Baric, R.S. The Current and Future State of Vaccines, Antivirals and Gene Therapies Against Emerging Coronaviruses. Front. Microbiol. 2020, 11, 658. [Google Scholar] [CrossRef]
- Lou, Z.; Sun, Y.; Rao, Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Elzayat, M.T.; Wang, L.; Graham, B.S.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J. Virol. 2019, 93, e01381-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, W.; Wang, Y.; Fett, C.A.; Zhao, G.; Li, F.; Perlman, S.; Jiang, S.; Zhou, Y.; Du, L. Recombinant Receptor-Binding Domains of Multiple Middle East Respiratory Syndrome Coronaviruses (MERS-CoVs) Induce Cross-Neutralizing Antibodies against Divergent Human and Camel MERS-CoVs and Antibody Escape Mutants. J. Virol. 2016, 91, e01651-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Sharma, S.; Kumar, R.; Tripathi, B.N.; Barua, S.; Ly, H.; Rouse, B.T. Host-directed antiviral therapy. Clin. Microbiol. Rev. 2020, 33, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Biedenkopf, N.; Lange-Grünweller, K.; Schulte, F.W.; Weißer, A.; Müller, C.; Becker, D.; Becker, S.; Hartmann, R.K.; Grünweller, A. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antivir. Res. 2017, 137, 76–81. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov Identifier NCT04382066. Available online: https://clinicaltrials.gov/ct2/show/NCT04382066 (accessed on 28 January 2021).
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, eabf4058. [Google Scholar] [CrossRef]
- Clinicaltrials.gov Identifier NCT04632381. Available online: https://clinicaltrials.gov/ct2/show/NCT04632381 (accessed on 28 January 2021).
- Ernst, J.T.; Thompson, P.A.; Nilewski, C.; Sprengeler, P.A.; Sperry, S.; Packard, G.; Michels, T.; Xiang, A.; Tran, C.; Wegerski, C.J.; et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Schulz, G.; Victoria, C.; Kirschning, A.; Steinmann, E. Rocaglamide and silvestrol: A long story from anti-tumor to anti-coronavirus compounds. Nat. Prod. Rep. 2020. [Google Scholar] [CrossRef]
- Toribio, R.; Díaz-López, I.; Ventoso, I. New insights into the topology of the scanning ribosome during translation initiation: Lessons from viruses. RNA Biol. 2016, 13, 1223–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, C.; Schulte, F.W.; Lange-Grünweller, K.; Obermann, W.; Madhugiri, R.; Pleschka, S.; Ziebuhr, J.; Hartmann, R.K.; Grünweller, A. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antivir. Res. 2018, 150, 123–129. [Google Scholar] [CrossRef]
- Nebigil, C.G.; Moog, C.; Vagner, S.; Benkirane-Jessel, N.; Smith, D.R.; Désaubry, L. Flavaglines as natural products targeting eIF4A and prohibitins: From traditional Chinese medicine to antiviral activity against coronaviruses. Eur. J. Med. Chem. 2020, 203, 1–9. [Google Scholar] [CrossRef]
- Kim, S.; Bang, Y.H.; Su, B.N.; Chai, H.; Mi, Q.; Kinghorn, A.D.; Wild, R.; Swanson, S.M. Silvestrol, a potential anticancer rocaglate derivative from Aglaia foveolata, induces apoptosis in LNCaP cells through the mitochondrial/apoptosome pathway without activation of executioner caspase-3 or -7. Anticancer Res. 2007, 27, 2175–2183. [Google Scholar]
- Shen, L.; Pelletier, J. Selective targeting of the DEAD-box RNA helicase eukaryotic initiation factor (eIF) 4A by natural products. Nat. Prod. Rep. 2020, 37, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Hershey, J.W.B.; Sonenberg, N.; Mathews, M.B. Principles of translational control: An overview. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; García-Román, R.; Mora, S.I. eIF4E as a control target for viruses. Viruses 2015, 7, 739–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.W.P.; Gray, N.K. Poly(A)-binding protein (PABP): A common viral target. Biochem. J. 2010, 426, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D. Manipulation of the host translation initiation complex eIF4F by DNA viruses. Biochem. Soc. Trans. 2010, 38, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Kapp, L.D.; Lorsch, J.R. The Molecular Mechanics of Eukaryotic Translation. Annu. Rev. Biochem. 2004, 73, 657–704. [Google Scholar] [CrossRef]
- Galicia-Vazquez, G.; Cencic, R.; Robert, F.; Agenor, A.Q.; Pelletier, J. A cellular response linking eIF4AI activity to eIF4AII transcription. RNA 2012, 18, 1373–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özeş, A.R.; Feoktistova, K.; Avanzino, B.C.; Fraser, C.S. Duplex unwinding and ATPase activities of the DEAD-box helicase eIF4A are coupled by eIF4G and eIF4B. J. Mol. Biol. 2011, 412, 674–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestova, T.V.; Kolupaeva, V.G.; Lomakin, I.B.; Pilipenko, E.V.; Shatsky, I.N.; Agol, V.I.; Hellen, C.U.T. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 7029–7036. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.W.; Komar, A.A.; Merrick, W.C. eIF4A: The godfather of the DEAD box helicases. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 307–331. [Google Scholar] [CrossRef]
- Lawson, T.G.; Ray, B.K.; Dodds, J.T.; Grifo, J.A.; Abramson, R.D.; Merrick, W.C.; Betsch, D.F.; Weith, H.L.; Thach, R.E. Influence of 5′ proximal secondary structure on the translational efficiency of eukaryotic mRNAs and on their interaction with initiation factors. J. Biol. Chem. 1986, 261, 13979–13989. [Google Scholar] [CrossRef]
- Villa, N.; Fraser, C.S. Mechanism of Translation in Eukaryotes. In Translation and Its Regulation in Cancer Biology and Medicine; Parsyan, A., Ed.; Springer: Dordrecht, The Netherlands, 2014; Volume 2, pp. 7–27. ISBN 978-94-017-9077-2. [Google Scholar]
- Brina, D.; Grosso, S.; Miluzio, A.; Biffo, S. Translational control by 80S formation and 60S availability: The central role of eIF6, a rate limiting factor in cell cycle progression and tumorigenesis. Cell Cycle 2011, 10, 3441–3446. [Google Scholar] [CrossRef]
- Cigan, A.; Feng, L.; Donahue, T. tRNAi(met) functions in directing the scanning ribosome to the start site of translation. Science 1988, 242, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Eliseev, B.; Yeramala, L.; Leitner, A.; Karuppasamy, M.; Raimondeau, E.; Huard, K.; Alkalaeva, E.; Aebersold, R.; Schaffitzel, C. Structure of a human cap-dependent 48S translation pre-initiation complex. Nucleic Acids Res. 2018, 46, 2678–2689. [Google Scholar] [CrossRef]
- Martinez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Embarek, A.M. Insights into structural and mechanistic features of viral IRES elements. Front. Microbiol. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mailliot, J.; Martin, F. Viral internal ribosomal entry sites: Four classes for one goal. Wiley Interdiscip. Rev. RNA 2017, 9. [Google Scholar] [CrossRef]
- de Breyne, S.; Yu, Y.; Unbehaun, A.; Pestova, T.V.; Hellen, C.U.T. Direct functional interaction of initiation factor eIF4G with type 1 internal ribosomal entry sites. Proc. Natl. Acad. Sci. USA 2009, 106, 9197–9202. [Google Scholar] [CrossRef] [Green Version]
- Kolupaeva, V.G.; Pestova, T.V.; Hellen, C.U.T.; Shatsky, I.N. Translation Eukaryotic Initiation Factor 4G Recognizes a Specific Structural Element within the Internal Ribosome Entry Site of Encephalomyocarditis Virus RNA. J. Biol. Chem. 1998, 273, 18599–18604. [Google Scholar] [CrossRef] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Andreou, A.Z.; Klostermeier, D. The DEAD-box helicase eIF4A: Paradigm or the odd one out? RNA Biol. 2013, 10, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Williams-Hill, D.M.; Duncan, R.F.; Nielsen, P.J.; Tahara, S.M. Differential Expression of the Murine Eukaryotic Translation Initiation Factor Isogenes eIF4AI and eIF4AIIIs Dependent upon Cellular Growth Status. Arch. Biochem. Biophys. 1997, 338, 111–120. [Google Scholar] [CrossRef]
- Putnam, A.A.; Jankowsky, E. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta-Gene Regul. Mech. 2013, 1829, 884–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, U.; Andreou, A.Z.; Gubaev, A.; Klostermeier, D. EIF4B, eIF4G and RNA regulate eIF4A activity in translation initiation by modulating the eIF4A conformational cycle. Nucleic Acids Res. 2014, 42, 7911–7922. [Google Scholar] [CrossRef] [Green Version]
- Kukhanova, M.K.; Karpenko, I.L.; Ivanov, A.V. DEAD-box RNA Helicase DDX3: Functional Properties and Development of DDX3 Inhibitors as Antiviral and Anticancer Drugs. Molecules 2020, 25, 1015. [Google Scholar] [CrossRef] [Green Version]
- Heerma van Voss, M.R.; van Diest, P.J.; Raman, V. Targeting RNA helicases in cancer: The translation trap. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 510–520. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.-J.; Liu, X.; Jiang, L.-N.; Zhou, J.-H.; Xiong, Y.-Q.; Ding, H.; Chen, Q. Immunogenicity and safety of currently available Japanese encephalitis vaccines: A systematic review. Hum. Vaccin. Immunother. 2014, 10, 3579–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, C.G.; Chen, Y.-L.; Dong, H.; Gu, F.; Lim, S.P.; Schul, W.; Wang, Q.-Y.; Shi, P.-Y. Strategies for development of dengue virus inhibitors. Antivir. Res. 2010, 85, 450–462. [Google Scholar] [CrossRef]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brai, A.; Ronzini, S.; Riva, V.; Botta, L.; Zamperini, C.; Borgini, M.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Boccuto, A.; et al. Synthesis and Antiviral Activity of Novel 1,3,4-Thiadiazole Inhibitors of DDX3X. Molecules 2019, 24, 3988. [Google Scholar] [CrossRef] [Green Version]
- Brai, A.; Fazi, R.; Tintori, C.; Zamperini, C.; Bugli, F.; Sanguinetti, M.; Stigliano, E.; Esté, J.; Badia, R.; Franco, S.; et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl. Acad. Sci. USA 2016, 113, 5388–5393. [Google Scholar] [CrossRef] [Green Version]
- Brai, A.; Riva, V.; Saladini, F.; Zamperini, C.; Trivisani, C.I.; Garbelli, A.; Pennisi, C.; Giannini, A.; Boccuto, A.; Bugli, F.; et al. DDX3X inhibitors, an effective way to overcome HIV-1 resistance targeting host proteins. Eur. J. Med. Chem. 2020, 200, 112319. [Google Scholar] [CrossRef] [PubMed]
- Riva, V.; Garbelli, A.; Casiraghi, F.; Arena, F.; Trivisani, C.I.; Gagliardi, A.; Bini, L.; Schroeder, M.; Maffia, A.; Sabbioneda, S.; et al. Novel alternative ribonucleotide excision repair pathways in human cells by DDX3X and specialized DNA polymerases. Nucleic Acids Res. 2020, 48, 11551–11565. [Google Scholar] [CrossRef]
- Cunningham, T.A.; Chapman, E.; Schatz, J.H. EIF4A inhibition: Ready for primetime? Oncotarget 2018, 9, 35515–35516. [Google Scholar] [CrossRef]
- Novac, O.; Guenier, A.S.; Pelletier, J. Inhibitors of protein synthesis identifed by a high throughput multiplexed translation screen. Nucleic Acids Res. 2004, 32, 902–915. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, M.-E.; Robert, F.; Gerard, B.; Lindqvist, L.; Chen, S.M.H.; Wendel, H.-G.; Brem, B.; Greger, H.; Lowe, S.W.; Porco, J.A.; et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [Green Version]
- Cencic, R.; Galicia-Vázquez, G.; Pelletier, J. Inhibitors of translation targeting eukaryotic translation initiation factor 4A. Methods Enzymol. 2012, 511, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Rao, B.G.; Jeang, K.T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005, 4, 845–853. [Google Scholar] [CrossRef]
- Bordeleau, M.-E.; Matthews, J.; Wojnar, J.M.; Lindqvist, L.; Novac, O.; Jankowsky, E.; Sonenberg, N.; Northcote, P.; Teesdale-Spittle, P.; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci. USA 2005, 102, 10460–10465. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, M.-E.; Cencic, R.; Lindqvist, L.; Oberer, M.; Northcote, P.; Wagner, G.; Pelletier, J. RNA-Mediated Sequestration of the RNA Helicase eIF4A by Pateamine A Inhibits Translation Initiation. Chem. Biol. 2006, 13, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, M.-E.; Mori, A.; Oberer, M.; Lindqvist, L.; Chard, L.S.; Higa, T.; Belsham, G.J.; Wagner, G.; Tanaka, J.; Pelletier, J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–220. [Google Scholar] [CrossRef]
- Lindqvist, L.; Oberer, M.; Reibarkh, M.; Cencic, R.; Bordeleau, M.-E.; Vogt, E.; Marintchev, A.; Tanaka, J.; Fagotto, F.; Altmann, M.; et al. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS ONE 2008, 3, e0001583. [Google Scholar] [CrossRef] [Green Version]
- Cencic, R.; Pelletier, J. Hippuristanol—A potent steroid inhibitor of eukaryotic initiation factor 4A. Translation 2016, 4, e1137381. [Google Scholar] [CrossRef] [Green Version]
- Brocard, M.; Iadevaia, V.; Klein, P.; Hall, B.; Lewis, G.; Lu, J.; Burke, J.; Willcocks, M.M.; Parker, R.; Goodfellow, I.G.; et al. Norovirus infection results in eIF2α independent host translation shut-off and remodels the G3BP1 interactome evading stress granule formation. PLoS Pathog. 2020, 16, e1008250. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Ishikawa, C.; Machijima, Y.; Nakachi, S.; Senba, M.; Tanaka, J.; Mori, N. Effects of hippuristanol, an inhibitor of eIF4A, on adult T-cell leukemia. Biochem. Pharmacol. 2011, 81, 713–722. [Google Scholar] [CrossRef]
- Low, W.-K.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of Eukaryotic Translation Initiation by the Marine Natural Product Pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef]
- Northcote, P.T.; Blunt, J.W.; Munro, M.H.G. Pateamine: A potent cytotoxin from the New Zealand Marine sponge, mycale sp. Tetrahedron Lett. 1991, 32, 6411–6414. [Google Scholar] [CrossRef]
- Slaine, P.; Kleer, M.; Smith, N.; Khaperskyy, D.; McCormick, C. Stress Granule-Inducing Eukaryotic Translation Initiation Factor 4A Inhibitors Block Influenza A Virus Replication. Viruses 2017, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Zhang, W.; Cencic, R.; O’Connor, P.B.F.; Robert, F.; Devine, W.G.; Selznick, A.; Henkel, T.; Merrick, W.C.; Brown, L.E.; et al. Rocaglates Induce Gain-of-Function Alterations to eIF4A and eIF4F. Cell Rep. 2020, 30, 2481–2488.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, B.Y.; Su, B.-N.; Chai, H.; Mi, Q.; Kardono, L.B.S.; Afriastini, J.J.; Riswan, S.; Santarsiero, B.D.; Mesecar, A.D.; Wild, R.; et al. Silvestrol and Episilvestrol, Potential Anticancer Rocaglate Derivatives from Aglaia silvestris. J. Org. Chem. 2004, 69, 3350–3358. [Google Scholar] [CrossRef]
- King, M.L.; Chiang, C.-C.; Ling, H.-C.; Fujita, E.; Ochiai, M.; McPhail, A.T. X-Ray Crystal Structure of Rocaglamide, a Novel Antileukemic 1H-Cyclopenta[b]benzofuran from Aglaia elliptifolia. J. Chem. Soc. 1982, 260, 1150–1151. [Google Scholar]
- Trost, B.M.; Greenspan, P.D.; Yang, B.V.; Saulnier, M.G. An unusual oxidative cyclization. A synthesis and absolute stereochemical assignment of (-)-rocaglamide. J. Am. Chem. Soc. 1990, 112, 9022–9024. [Google Scholar] [CrossRef]
- Zhang, W.; Chu, J.; Cyr, A.M.; Yueh, H.; Brown, L.E.; Wang, T.T.; Pelletier, J.; Porco, J.A. Intercepted Retro-Nazarov Reaction: Syntheses of Amidino-Rocaglate Derivatives and Their Biological Evaluation as eIF4A Inhibitors. J. Am. Chem. Soc. 2019, 141, 12891–12900. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Obermann, W.; Schulte, F.W.; Lange-Grünweller, K.; Oestereich, L.; Elgner, F.; Glitscher, M.; Hildt, E.; Singh, K.; Wendel, H.G.; et al. Comparison of broad-spectrum antiviral activities of the synthetic rocaglate CR-31-B (−) and the eIF4A-inhibitor Silvestrol. Antivir. Res. 2020, 175, 104706. [Google Scholar] [CrossRef]
- Rodrigo, C.M.; Cencic, R.; Roche, S.P.; Pelletier, J.; Porco, J.A. Synthesis of Rocaglamide Hydroxamates and Related Compounds as Eukaryotic Translation Inhibitors: Synthetic and Biological Studies. J. Med. Chem. 2012, 55, 558–562. [Google Scholar] [CrossRef]
- Naineni, S.K.; Maïga, R.I.; Cencic, R.; Putnam, A.A.; Amador, L.A.; Rodriguez, A.D.; Jankowsky, E.; Pelletier, J. A comparative study of small molecules targeting eIF4A. RNA 2020, 26, 541–549. [Google Scholar] [CrossRef]
- Abdelkrim, Y.Z.; Harigua-Souiai, E.; Barhoumi, M.; Banroques, J.; Blondel, A.; Guizani, I.; Tanner, N.K. The steroid derivative 6-aminocholestanol inhibits the DEAD-box helicase eIF4A (LieIF4A) from the Trypanosomatid parasite Leishmania by perturbing the RNA and ATP binding sites. Mol. Biochem. Parasitol. 2018, 226, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Tang, Y.; Ding, L.; Tan, R.; Li, X.; Lu, J.; Jiang, J.; Cui, Z.; Tang, Z.; Li, W.; et al. Targeting the N Terminus of eIF4AI for Inhibition of Its Catalytic Recycling. Cell Chem. Biol. 2019, 26, 1417–1426.e5. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, J.H.; Jang, S.K. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J. 2007, 26, 5020–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, T.L.; Tillotson, J.; Yeomans, A.M.; Wilmore, S.; Lemm, E.; Jiménez-Romero, C.; Amador, L.A.; Li, L.; Amin, A.D.; Pongtornpipat, P.; et al. Target-Based Screening against eIF4A1 Reveals the Marine Natural Product Elatol as a Novel Inhibitor of Translation Initiation with In Vivo Antitumor Activity. Clin. Cancer Res. 2018, 24, 4256–4270. [Google Scholar] [CrossRef] [Green Version]
- Tillotson, J.; Kedzior, M.; Guimarães, L.; Ross, A.B.; Peters, T.L.; Ambrose, A.J.; Schmidlin, C.J.; Zhang, D.D.; Costa-Lotufo, L.V.; Rodríguez, A.D.; et al. ATP-competitive, marine derived natural products that target the DEAD box helicase, eIF4A. Bioorg. Med. Chem. Lett. 2017, 27, 4082–4085. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, S.; Iwasaki, W.; Takahashi, M.; Sakamoto, A.; Watanabe, C.; Shichino, Y.; Floor, S.N.; Fujiwara, K.; Mito, M.; Dodo, K.; et al. The Translation Inhibitor Rocaglamide Targets a Bimolecular Cavity between eIF4A and Polypurine RNA. Mol. Cell 2019, 73, 738–748.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PDB: 5ZC9. Available online: https://www.rcsb.org/structure/5ZC9 (accessed on 28 January 2021).
- Chu, J.; Zhang, W.; Cencic, R.; Devine, W.G.; Beglov, D.; Henkel, T.; Brown, L.E.; Vajda, S.; Porco, J.A.; Pelletier, J. Amidino-Rocaglates: A Potent Class of eIF4A Inhibitors. Cell Chem. Biol. 2019, 26, 1586–1593.e3. [Google Scholar] [CrossRef]
- Chen, M.; Asanuma, M.; Takahashi, M.; Shichino, Y.; Mito, M.; Fujiwara, K.; Saito, H.; Floor, S.N.; Ingolia, N.T.; Sodeoka, M.; et al. Dual targeting of DDX3 and eIF4A by the translation inhibitor rocaglamide A. Cell Chem. Biol. 2020, 1–12. [Google Scholar] [CrossRef]
- Winnard, P.T., Jr.; Vesuna, F.; Raman, V. Targeting host DEAD-box RNA helicase DDX3X for treating viral infections. Antivir. Res. 2021, 185, 104994. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Robert, F.; Oertlin, C.; Kapeller-Libermann, D.; Avizonis, D.; Gutierrez, J.; Handly-Santana, A.; Doubrovin, M.; Park, J.; Schoepfer, C.; et al. eIF4A supports an oncogenic translation program in pancreatic ductal adenocarcinoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.A.; Weisburd, B.; Holderfield, M.; Arias, C.; Fang, E.; DeRisi, J.L.; Fanidi, A. Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 2014, 15, 476. [Google Scholar] [CrossRef]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, L.; Geisslinger, G.; Parnham, M.J.; Grünweller, A.; Schiffmann, S. Natural antiviral compound silvestrol modulates human monocyte-derived macrophages and dendritic cells. J. Cell. Mol. Med. 2020, 24, 6988–6999. [Google Scholar] [CrossRef]
- Müller, C.; Obermann, W.; Karl, N.; Wendel, H.-G.; Taroncher-Oldenburg, G.; Pleschka, S.; Hartmann, R.K.; Grünweller, A.; Ziebuhr, J. The rocaglate CR-31-B (−) inhibits SARS-CoV-2 replication at non-cytotoxic, low nanomolar concentrations in vitro and ex vivo. Antivir. Res. 2021, 186, 105012. [Google Scholar] [CrossRef] [PubMed]
- Henss, L.; Scholz, T.; Grünweller, A.; Schnierle, B. Silvestrol Inhibits Chikungunya Virus Replication. Viruses 2018, 10, 592. [Google Scholar] [CrossRef] [Green Version]
- Todt, D.; Moeller, N.; Praditya, D.; Kinast, V.; Friesland, M.; Engelmann, M.; Verhoye, L.; Sayed, I.M.; Behrendt, P.; Dao Thi, V.L.; et al. The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antivir. Res. 2018, 157, 151–158. [Google Scholar] [CrossRef]
- Pigott, D.C. Hemorrhagic Fever Viruses. Crit. Care Clin. 2005, 21, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.; Bollinger, L.; Johnson, J.C.; Wada, J.; Radoshitzky, S.R.; Palacios, G.; Bavari, S.; Jahrling, P.B.; Kuhn, J.H. Neglected filoviruses. FEMS Microbiol. Rev. 2016, 40, 494–519. [Google Scholar] [CrossRef] [Green Version]
- Mühlberger, E.; Trommer, S.; Funke, C.; Volchkov, V.; Klenk, H.-D.; Becker, S. Termini of All mRNA Species of Marburg Virus: Sequence and Secondary Structure. Virology 1996, 223, 376–380. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- PROPEL Forwarding COVID-19 Research. Available online: https://propelcovidclinicaltrial.com/ (accessed on 22 January 2021).
- Marklewitz, M.; Junglen, S. Evolutionary and ecological insights into the emergence of arthropod-borne viruses. Acta Trop. 2019, 190, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef]
- Goh, V.S.L.; Mok, C.-K.; Chu, J.J.H. Antiviral Natural Products for Arbovirus Infections. Molecules 2020, 25, 2796. [Google Scholar] [CrossRef]
- Madden, E.A.; Plante, K.S.; Morrison, C.R.; Kutchko, K.M.; Sanders, W.; Long, K.M.; Taft-Benz, S.; Cruz Cisneros, M.C.; White, A.M.; Sarkar, S.; et al. Using SHAPE-MaP To Model RNA Secondary Structure and Identify 3′UTR Variation in Chikungunya Virus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Weaver, S.C.; Chen, R.; Diallo, M. Chikungunya Virus: Role of Vectors in Emergence from Enzootic Cycles. Annu. Rev. Entomol. 2020, 65, 313–332. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xu, L.; Wang, Y.; Wang, W.; Sprengers, D.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Requirement of the eukaryotic translation initiation factor 4F complex in hepatitis E virus replication. Antivir. Res. 2015, 124, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Johne, R.; Reuter, A.; Spiric, J.; Schwaben, L.; Grünweller, A.; Hildt, E. Inhibition of hepatitis E virus spread by the natural compound silvestrol. Viruses 2018, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D. Virion Assembly and Release. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Bartenschlager, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 199–218. [Google Scholar]
- Edgil, D.; Polacek, C.; Harris, E. Dengue Virus Utilizes a Novel Strategy for Translation Initiation When Cap-Dependent Translation Is Inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Mugavero, J.; Stauft, C.B.; Wimmer, E. Dengue and Zika Virus 5′ Untranslated Regions Harbor Internal Ribosomal Entry Site Functions. MBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Elgner, F.; Sabino, C.; Basic, M.; Ploen, D.; Grünweller, A.; Hildt, E. Inhibition of zika virus replication by silvestrol. Viruses 2018, 10, 149. [Google Scholar] [CrossRef] [Green Version]
- Adalja, A.; Inglesby, T. Broad-Spectrum Antiviral Agents: A Crucial Pandemic Tool. Expert Rev. Anti. Infect. Ther. 2019, 17, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Bekerman, E.; Einav, S. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Oprea, T.I.; Bauman, J.E.; Bologa, C.G.; Buranda, T.; Chigaev, A.; Edwards, B.S.; Jarvik, J.W.; Gresham, H.D.; Haynes, M.K.; Hjelle, B.; et al. Drug repurposing from an academic perspective. Drug Discov. Today Ther. Strateg. 2011, 8, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.S.; Brown, C.M. Know Your Enemy: Successful Bioinformatic Approaches to Predict Functional RNA Structures in Viral RNAs. Front. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Wang, J.; Gribskov, M. IRESpy: An XGBoost model for prediction of internal ribosome entry sites. BMC Bioinform. 2019, 20, 409. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Chatterjee, S.; Devine, W.G.; Kobzik, L.; Beeler, A.B.; Porco, J.A.; Kramnik, I. Fine-tuning of macrophage activation using synthetic rocaglate derivatives. Sci. Rep. 2016, 6, 24409. [Google Scholar] [CrossRef] [Green Version]
- Louz, D.; Bergmans, H.E.; Loos, B.P.; Hoeben, R.C. Animal models in virus research: Their utility and limitations. Crit. Rev. Microbiol. 2012, 39, 325–361. [Google Scholar] [CrossRef]
- Pellegatti, M. Preclinical in vivo ADME studies in drug development: A critical review. Expert Opin. Drug Metab. Toxicol. 2012, 8, 161–172. [Google Scholar] [CrossRef]
- Saradhi, U.V.R.V.; Gupta, S.V.; Chiu, M.; Wang, J.; Ling, Y.; Liu, Z.; Newman, D.J.; Covey, J.M.; Kinghorn, A.D.; Marcucci, G.; et al. Characterization of silvestrol pharmacokinetics in mice using liquid chromatography-tandem mass spectrometry. AAPS J. 2011, 13, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grobler, J.A.; Anderson, A.S.; Fernandes, P.; Diamond, M.S.; Colvis, C.M.; Menetski, J.P.; Alvarez, R.M.; Young, J.A.T.; Carter, K.L. Accelerated Preclinical Paths to Support Rapid Development of COVID-19 Therapeutics. Cell Host Microbe 2020, 28, 638–645. [Google Scholar] [CrossRef]
- Andersen, P.I.; Ianevski, A.; Lysvand, H.; Vitkauskiene, A.; Oksenych, V.; Bjørås, M.; Telling, K.; Lutsar, I.; Dumpis, U.; Irie, Y.; et al. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 2020, 93, 268–276. [Google Scholar] [CrossRef]
- Fragkou, P.C.; Belhadi, D.; Peiffer-Smadja, N.; Moschopoulos, C.D.; Lescure, F.-X.; Janocha, H.; Karofylakis, E.; Yazdanpanah, Y.; Mentré, F.; Skevaki, C.; et al. Review of trials currently testing treatment and prevention of COVID-19. Clin. Microbiol. Infect. 2020, 26, 988–998. [Google Scholar] [CrossRef]
- Everts, M.; Cihlar, T.; Bostwick, J.R.; Whitley, R.J. Accelerating Drug Development: Antiviral Therapies for Emerging Viruses as a Model. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Langlais, D.; Cencic, R.; Moradin, N.; Kennedy, J.M.; Ayi, K.; Brown, L.E.; Crandall, I.; Tarry, M.J.; Schmeing, M.; Kain, K.C.; et al. Rocaglates as dual-targeting agents for experimental cerebral malaria. Proc. Natl. Acad. Sci. USA 2018, 115, E2366–E2375. [Google Scholar] [CrossRef] [Green Version]
- Iyer, K.R.; Whitesell, L.; Porco, J.A.; Henkel, T.; Brown, L.E.; Robbins, N.; Cowen, L.E. Translation Inhibition by Rocaglates Activates a Species-Specific Cell Death Program in the Emerging Fungal Pathogen Candida auris. MBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Years | Disease | Causative Agent | Epidemic/Pandemic | Taxonomic Family |
|---|---|---|---|---|
| 2019–present | COVID-19 | SARS-CoV-2 | Pandemic | Coronaviridae |
| 2015–present | Zika virus disease | ZIKV | Epidemic | Flaviviridae |
| 2015/2016 | Chikungunya fever | CHIKV | Epidemic | Togaviridae |
| 2014–2016 | Ebola hemorrhagic fever | EBOV | Epidemic | Filoviridae |
| 2012–present | Middle East respiratory syndrome | MERS-CoV | Epidemic | Coronaviridae |
| 2009/2010 | Influenza | IAV/H1N1 | Pandemic | Orthomyxoviridae |
| 2002/2003 | Severe acute respiratory syndrome | SARS-CoV | Epidemic | Coronaviridae |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taroncher-Oldenburg, G.; Müller, C.; Obermann, W.; Ziebuhr, J.; Hartmann, R.K.; Grünweller, A. Targeting the DEAD-Box RNA Helicase eIF4A with Rocaglates—A Pan-Antiviral Strategy for Minimizing the Impact of Future RNA Virus Pandemics. Microorganisms 2021, 9, 540. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030540
Taroncher-Oldenburg G, Müller C, Obermann W, Ziebuhr J, Hartmann RK, Grünweller A. Targeting the DEAD-Box RNA Helicase eIF4A with Rocaglates—A Pan-Antiviral Strategy for Minimizing the Impact of Future RNA Virus Pandemics. Microorganisms. 2021; 9(3):540. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030540
Chicago/Turabian StyleTaroncher-Oldenburg, Gaspar, Christin Müller, Wiebke Obermann, John Ziebuhr, Roland K. Hartmann, and Arnold Grünweller. 2021. "Targeting the DEAD-Box RNA Helicase eIF4A with Rocaglates—A Pan-Antiviral Strategy for Minimizing the Impact of Future RNA Virus Pandemics" Microorganisms 9, no. 3: 540. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030540