
The Janus Kinase Inhibitor Ruxolitinib Prevents Terminal Shock in a Mouse Model of Arenavirus Hemorrhagic Fever



Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Ethics Statement
2.2. Virus Titration and Infection
2.3. Treatment with Ruxolitinib, Anti-TNF and Anti-IFN-γ
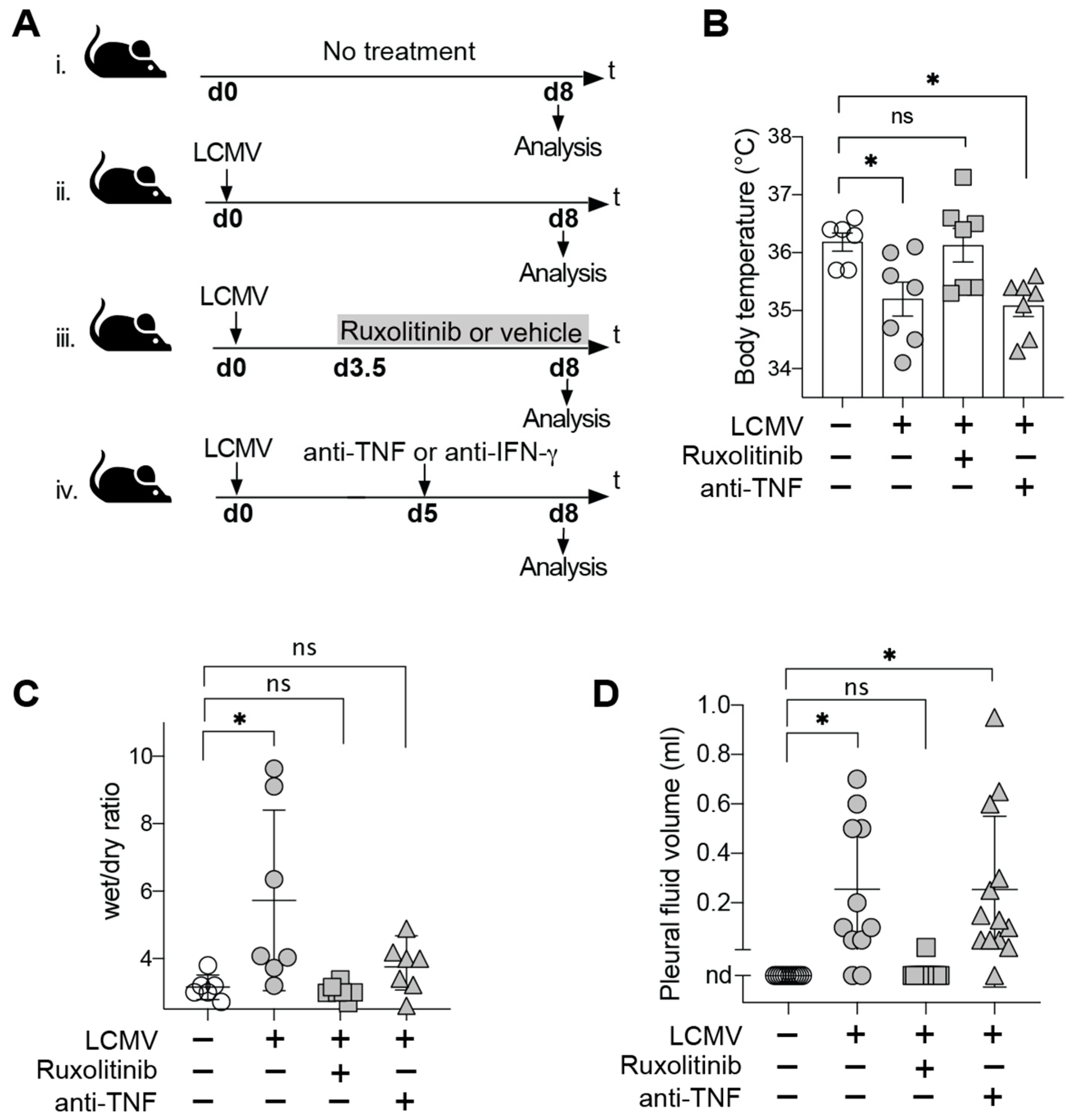
2.4. Determination of Body Temperature, Pleural Effusions and Skin Edema
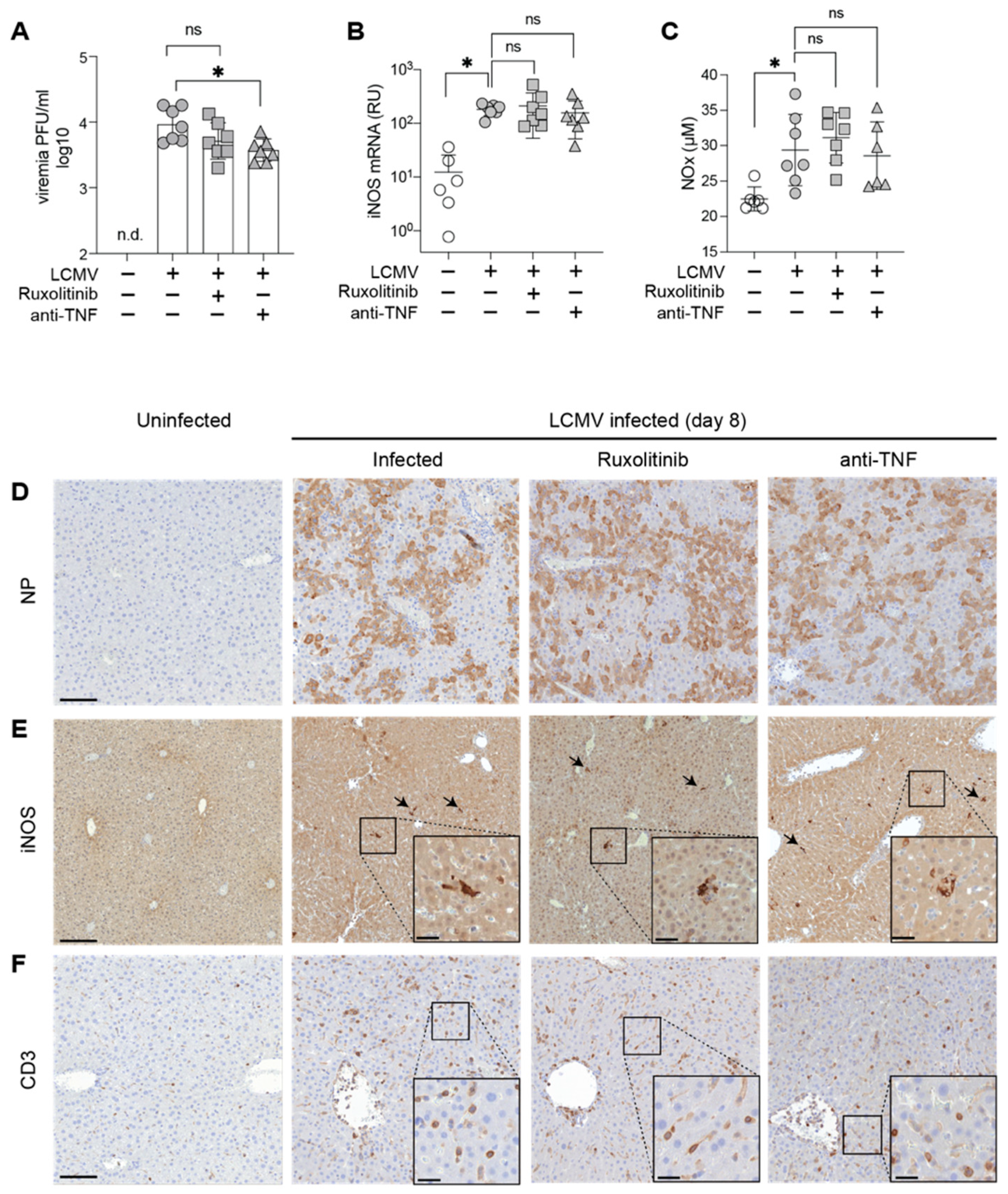
2.5. NOx Determination
2.6. iNOS mRNA Expression Analysis
2.7. Histology and Immunohistochemistry
2.8. Statistical Analysis
3. Results
3.1. JAK Inhibitor Ruxolitinib but Not TNF Blockade Prevents Terminal Disease in LCMV-Infected HHD Mice
3.2. Ruxolitinib Protection in AVHF Is Unrelated to Viral Loads, Systemic NO or Hepatic iNOS Expression Levels
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geisbert, T.W.; Jahrling, P.B. Exotic emerging viral diseases: Progress and challenges. Nat. Med. 2004, 10, S110. [Google Scholar] [CrossRef]
- Edington, G.; White, H. The pathology of Lassa fever: A tribute to the late Dr. JM Troup. Trans. R. Soc. Trop. Med. Hyg. 1972, 66, 381–389. [Google Scholar] [CrossRef]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Johnson, K.M.; O’Sullivan, R.; Smith, E.S.; Trippel, S.; Tong, T.C. A case-control study of the clinical diagnosis and course of Lassa fever. J. Infect. Dis. 1987, 155, 445–455. [Google Scholar] [CrossRef]
- Schmitz, H.; Köhler, B.; Laue, T.; Drosten, C.; Veldkamp, P.J.; Günther, S.; Emmerich, P.; Geisen, H.P.; Fleischer, K.; Beersma, M.F. Monitoring of clinical and laboratory data in two cases of imported Lassa fever. Microbes Infect. 2002, 4, 43–50. [Google Scholar] [CrossRef]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W. T cell-dependence of Lassa fever pathogenesis. PLoS Pathog. 2010, 6, e1000836. [Google Scholar] [CrossRef] [Green Version]
- Remy, M.M.; Sahin, M.; Flatz, L.; Regen, T.; Xu, L.; Kreutzfeldt, M.; Fallet, B.; Doras, C.; Rieger, T.; Bestmann, L. Interferon-γ-Driven iNOS: A Molecular Pathway to Terminal Shock in Arenavirus Hemorrhagic Fever. Cell Host Microbe 2017, 22, 354–365.e355. [Google Scholar] [CrossRef] [Green Version]
- Lukashevich, I.S.; Djavani, M.; Rodas, J.D.; Zapata, J.C.; Usborne, A.; Emerson, C.; Mitchen, J.; Jahrling, P.B.; Salvato, M.S. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J. Med. Virol. 2002, 67, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hommes, D.W.; Mikhajlova, T.L.; Stoinov, S.; Štimac, D.; Vucelic, B.; Lonovics, J.; Zákuciová, M.; D’Haens, G.; Van Assche, G.; Ba, S. Fontolizumab, a humanised anti-interferon γ antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 2006, 55, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Lounder, D.T.; Bin, Q.; de Min, C.; Jordan, M.B. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. 2019, 3, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajayi, S.; Becker, H.; Reinhardt, H.; Engelhardt, M.; Zeiser, R.; von Bubnoff, N.; Wäsch, R. Ruxolitinib. In Small Molecules in Hematology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 119–132. [Google Scholar]
- Wang, J.; Wang, Y.; Wu, L.; Wang, X.; Jin, Z.; Gao, Z.; Wang, Z. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica 2020, 105, e210–e212. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, A.; Del Poggio, P.; Bracchi, F.; Cesana, G.; Sertori, N.; Di Mauro, D.; Fargnoli, A.; Motta, M.; Giussani, C.; Moro, P.; et al. Low-dose ruxolitinib plus steroid in severe SARS-CoV-2 pneumonia. Leukemia 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Heise, M.T.; Virgin, H. The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J. Virol. 1995, 69, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atrasheuskaya, A.; Petzelbauer, P.; Fredeking, T.M.; Ignatyev, G. Anti-TNF antibody treatment reduces mortality in experimental dengue virus infection. FEMS Immunol. Med. Microbiol. 2003, 35, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Branche, E.; Tang, W.W.; Viramontes, K.M.; Young, M.P.; Sheets, N.; Joo, Y.; Nguyen, A.-V.T.; Shresta, S. Synergism between the tyrosine kinase inhibitor sunitinib and anti-TNF antibody protects against lethal dengue infection. Antivir. Res. 2018, 158, 1–7. [Google Scholar] [CrossRef]
- Pascolo, S.; Bervas, N.; Ure, J.M.; Smith, A.G.; Lemonnier, F.A.; Pérarnau, B. HLA-A2. 1–restricted education and cytolytic activity of CD8+ T lymphocytes from β2 microglobulin (β2m) HLA-A2. 1 monochain transgenic H-2Db β2m double knockout mice. J. Exp. Med. 1997, 185, 2043–2051. [Google Scholar] [CrossRef] [Green Version]
- Battegay, M.; Cooper, S.; Althage, A.; Bänziger, J.; Hengartner, H.; Zinkernagel, R.M. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24-or 96-well plates. J. Virol. Methods 1991, 33, 191–198. [Google Scholar] [CrossRef]
- Flatz, L.; Hegazy, A.N.; Bergthaler, A.; Verschoor, A.; Claus, C.; Fernandez, M.; Gattinoni, L.; Johnson, S.; Kreppel, F.; Kochanek, S. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 2010, 16, 339. [Google Scholar] [CrossRef]
- Lander, H.M.; Grant, A.M.; Albrecht, T.; Hill, T.; Peters, C.J. Endothelial cell permeability and adherens junction disruption induced by junin virus infection. Am. J. Trop. Med. Hyg. 2014, 90, 993–1002. [Google Scholar] [CrossRef] [Green Version]
- Rathore, A.P.; Mantri, C.K.; Aman, S.A.; Syenina, A.; Ooi, J.; Jagaraj, C.J.; Goh, C.C.; Tissera, H.; Wilder-Smith, A.; Ng, L.G. Dengue virus–elicited tryptase induces endothelial permeability and shock. J. Clin. Investig. 2019, 129, 4180–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematologica 2013, 98, 1865. [Google Scholar] [CrossRef] [PubMed]
- Dell’Albani, P.; Santangelo, R.; Torrisi, L.; Nicoletti, V.d.; De Vellis, J.; Giuffrida Stella, A. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J. Neurosci. Res. 2001, 65, 417–424. [Google Scholar] [CrossRef]
- Marrero, M.B.; Venema, V.J.; He, H.; Caldwell, R.B.; Venema, R.C. Inhibition by the JAK/STAT pathway of IFNγ-and LPS-stimulated nitric oxide synthase induction in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998, 252, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Chao, Y.; Chang, Y.H.; Hsu, M.J.; Lin, W.W. Inhibition of cytokine-induced JAK-STAT signalling pathways by an endonuclease inhibitor aurintricarboxylic acid. Br. J. Pharmacol. 2002, 137, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.J.; Levy, D.E.; Johnstone, R.W.; Clarke, C.J. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008, 19, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.; Verbist, K.C.; Tedrick, P.E.; Tillman, H.; Picarsic, J.; Bassett, R.; Nichols, K.E. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood J. Am.Soc. Hematol. 2019, 134, 147–159. [Google Scholar] [CrossRef]
- Jhan, M.K.; HuangFu, W.C.; Chen, Y.F.; Kao, J.C.; Tsai, T.T.; Ho, M.R.; Shen, T.J.; Tseng, P.C.; Wang, Y.T.; Lin, C.F. Anti-TNF-α restricts dengue virus-induced neuropathy. J. Leukoc. Biol. 2018, 104, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.; Isaza, D.; Salazar, C.; Ramírez, R.; Ospina, M.; Alvarez, L. Serum levels of interleukin-6, tumor necrosis factor-alpha and interferon-gamma in infants with and without dengue. Rev. Soc. Bras. Med. Trop. 2008, 41, 6–10. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Yeleswaram, S.; Smith, P.; Burn, T.; Covington, M.; Juvekar, A.; Li, Y.; Squier, P.; Langmuir, P. Inhibition of cytokine signaling by ruxolitinib and implications for COVID-19 treatment. Clin. Immunol. 2020, 218, 108517. [Google Scholar] [CrossRef] [PubMed]
- La Rosée, F.; Bremer, H.; Gehrke, I.; Kehr, A.; Hochhaus, A.; Birndt, S.; Fellhauer, M.; Henkes, M.; Kumle, B.; Russo, S. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia 2020, 34, 1805–1815. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef] [PubMed]
- Lo Caputo, S.; Corso, G.; Clerici, M.; Santantonio, T.A. Baricitinib: A chance to treat COVID-19? J. Med. Virol. 2020, 92, 2343–2344. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Experiment a | Treatment b | Diseased Animals/Tested Animals c | Day of Termination d | Diseased Mice % e |
|---|---|---|---|---|
| 1 | None Vehicle Ruxolitinib | 5/8 6/8 0/8 | 8/8/10/12/12 8/8/10/10/11/11 - | 62.5 75 0 |
| 2 | None Ruxolitinib anti-TNF | 1/8 0/8 2/8 | 8 - 8/8 | 12.5 0 25 |
| 3 | None anti-TNF anti-IFN-γ | 3/7 4/7 0/7 | 7/8/8 7/7/8/8 - | 42.9 57.1 0 |
| 1–3 combined | None Ruxolitinib anti-TNF anti-IFN-γ | 9/23 0/16 6/15 0/7 | 7/8/8/8/8/8/10/12/12 - 7/7/8/8/8/8 - | 39.1 0 40 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahin, M.; Remy, M.M.; Merkler, D.; Pinschewer, D.D. The Janus Kinase Inhibitor Ruxolitinib Prevents Terminal Shock in a Mouse Model of Arenavirus Hemorrhagic Fever. Microorganisms 2021, 9, 564. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030564
Sahin M, Remy MM, Merkler D, Pinschewer DD. The Janus Kinase Inhibitor Ruxolitinib Prevents Terminal Shock in a Mouse Model of Arenavirus Hemorrhagic Fever. Microorganisms. 2021; 9(3):564. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030564
Chicago/Turabian StyleSahin, Mehmet, Melissa M. Remy, Doron Merkler, and Daniel D. Pinschewer. 2021. "The Janus Kinase Inhibitor Ruxolitinib Prevents Terminal Shock in a Mouse Model of Arenavirus Hemorrhagic Fever" Microorganisms 9, no. 3: 564. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030564