
Transcriptomic Analysis Reveals Host miRNAs Correlated with Immune Gene Dysregulation during Fatal Disease Progression in the Ebola Virus Cynomolgus Macaque Disease Model

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
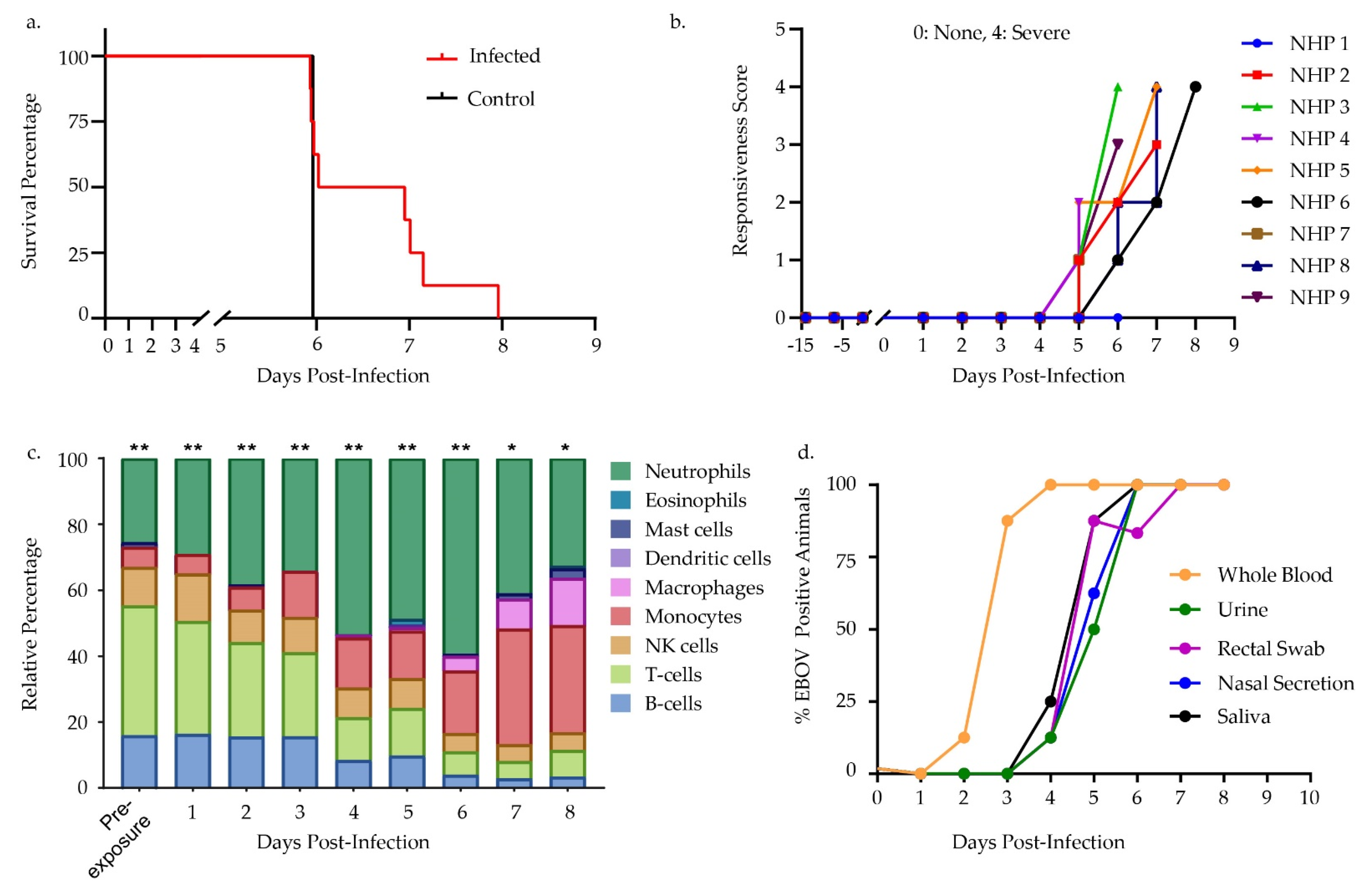
2.2. Animals and Study Design
2.3. Challenge Virus Stock
2.4. Blood Chemistries and Sample Collection
2.5. EBOV Antibody and Antigen Detection
2.6. RNA Extraction and Quantitation
2.7. Determination of Viremia in NHP Clinical Specimens by RT-qPCR
2.8. NanoString Processing and Analysis
2.9. miRNA Library Preparation, Sequencing and Analysis
2.10. Pathway and Correlation Analysis
3. Results
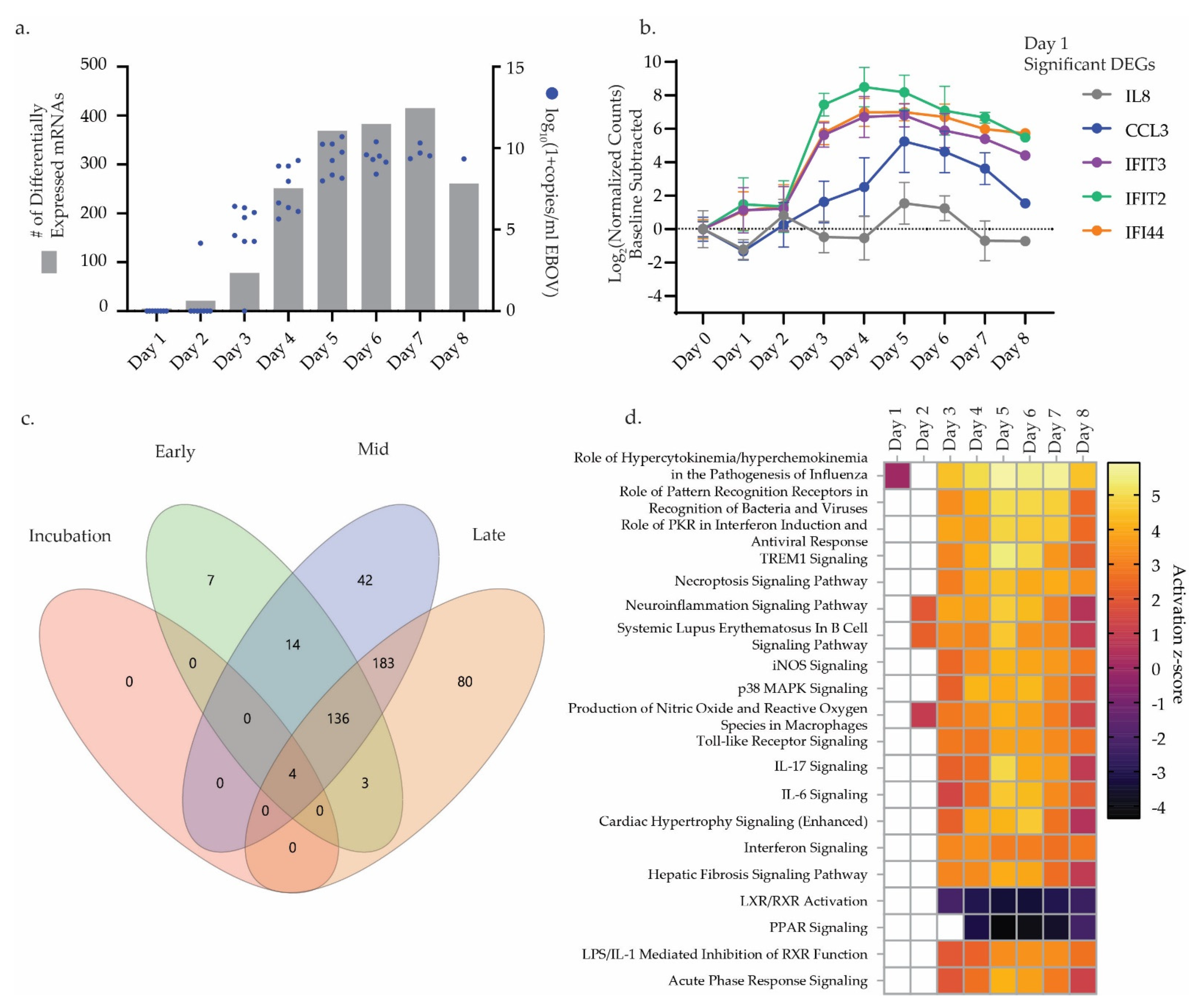
3.1. Challenge Design and Sample Characterization
3.2. Differential Gene Expression Trends
3.3. Differential miRNA Expression Trends
3.4. Correlative Analysis of Significant miRNA and mRNA Signatures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The Pathogenesis of Ebola Virus Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 387–418. [Google Scholar] [CrossRef]
- Malvy, D.; McElroy, A.K.; de Clerck, H.; Günther, S.; van Griensven, J. Ebola virus disease. Lancet 2019, 393, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Jacob, S.T.; Crozier, I.; Wahl, V.; Griffiths, A.; Bollinger, L.; Kuhn, J.H.; Fischer, W.A., II; Hewlett, A.; Kraft, C.S.; De La Vega, M.-A.; et al. Ebola virus disease. Nat. Rev. Dis. Prim. 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamontagne, F.; A Fowler, R.; Adhikari, N.K.; Murthy, S.; Brett-Major, D.M.; Jacobs, M.; Uyeki, T.M.; Vallenas, C.; Norris, S.L.; A Fischer, W.; et al. Evidence-based guidelines for supportive care of patients with Ebola virus disease. Lancet 2018, 391, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Colavita, F.; Biava, M.; Castilletti, C.; Lanini, S.; Miccio, R.; Portella, G.; Vairo, F.; Ippolito, G.; Capobianchi, M.R.; Di Caro, A.; et al. Inflammatory and Humoral Immune Response during Ebola Virus Infection in Survivor and Fatal Cases Occurred in Sierra Leone during the 2014–2016 Outbreak in West Africa. Viruses 2019, 11, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, J.B.; Marzi, A.; Safronetz, D.; Robertson, S.J.; Feldmann, H.; Best, S.M. Immunobiology of Ebola and Lassa virus infections. Nat. Rev. Immunol. 2017, 17, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Geisbert, T.W. Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005, 37, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Wahl-Jensen, V.; Kurz, S.; Feldmann, F.; Buehler, L.K.; Kindrachuk, J.; DeFilippis, V.; Correia, J.D.S.; Früh, K.; Kuhn, J.H.; Burton, D.R.; et al. Ebola Virion Attachment and Entry into Human Macrophages Profoundly Effects Early Cellular Gene Expression. PLOS Negl. Trop. Dis. 2011, 5, e1359. [Google Scholar] [CrossRef]
- Reynard, S.; Journeaux, A.; Gloaguen, E.; Schaeffer, J.; Varet, H.; Pietrosemoli, N.; Mateo, M.; Baillet, N.; Laouenan, C.; Raoul, H.; et al. Immune parameters and outcomes during Ebola virus disease. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Jankeel, A.; Menicucci, A.R.; Woolsey, C.; Fenton, K.A.; Mendoza, N.; Versteeg, K.; Cross, R.W.; Geisbert, T.W.; Messaoudi, I. Early Transcriptional Changes within Liver, Adrenal Gland, and Lymphoid Tissues Significantly Contribute to Ebola Virus Pathogenesis in Cynomolgus Macaques. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef]
- Younan, P.; Iampietro, M.; Bukreyev, A. Disabling of lymphocyte immune response by Ebola virus. PLoS Pathog. 2018, 14, e1006932. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; Castilletti, C.; Casetti, R.; Sacchi, A.; Falasca, L.; Turchi, F.; Tumino, N.; Bordoni, V.; Cimini, E.; Viola, D.; et al. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. 2016, 7, e2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohue, D.E.; Gautam, A.; Miller, S.-A.; Srinivasan, S.; Abu-Amara, D.; Campbell, R.; Marmar, C.R.; Hammamieh, R.; Jett, M. Gene expression profiling of whole blood: A comparative assessment of RNA-stabilizing collection methods. PLoS ONE 2019, 14, e0223065. [Google Scholar] [CrossRef]
- Rubins, K.H.; Hensley, L.E.; Wahl-Jensen, V.; Daddario DiCaprio, K.M.; Young, H.A.; Reed, D.S.; Jahrling, P.B.; Brown, P.O.; Relman, D.A.; Geisbert, T.W. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 2007, 8, R174. [Google Scholar] [CrossRef] [Green Version]
- Speranza, E.; Connor, J.H. Host Transcriptional Response to Ebola Virus Infection. Vaccines 2017, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speranza, E.; Altamura, L.A.; Kulcsar, K.; Bixler, S.L.; Rossi, C.A.; Schoepp, R.J.; Nagle, E.; Aguilar, W.; Douglas, C.E.; Delp, K.L.; et al. Comparison of Transcriptomic Platforms for Analysis of Whole Blood from Ebola-Infected Cynomolgus Macaques. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Speranza, E.; Bixler, S.L.; Altamura, L.A.; Arnold, C.E.; Pratt, W.D.; Taylor-Howell, C.; Burrows, C.; Aguilar, W.; Rossi, F.; Shamblin, J.D.; et al. A conserved transcriptional response to intranasal Ebola virus exposure in nonhuman primates prior to onset of fever. Sci. Transl. Med. 2018, 10, eaaq1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Fan, J.; Belasco, J.G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Kumar, A.; Ingle, H.; Kumar, H. The Interplay Between Viral-Derived miRNAs and Host Immunity During Infection. Front. Immunol. 2020, 10, 3079. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sun, J.; Zhang, H.; Wang, M.; Gao, G.F.; Li, X. Ebola virus encodes a miR-155 analog to regulate importin-α5 expression. Cell. Mol. Life Sci. 2016, 73, 3733–3744. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, J.; Sun, C.; Jin, G.; Xin, L.; Jin, Z.; Wang, D.; Li, Z.; Wang, L.; Wang, N. MicroRNA expression levels as diagnostic biomarkers for intraductal papillary mucinous neoplasm. Oncotarget 2017, 8, 58765–58770. [Google Scholar] [CrossRef] [Green Version]
- Szilágyi, B.; Fejes, Z.; Pócsi, M.; Kappelmayer, J.; Nagy, B., Jr. Role of sepsis modulated circulating microRNAs. EJIFCC 2019, 30, 128–145. [Google Scholar]
- Girardi, E.; López, P.; Pfeffer, S. On the Importance of Host MicroRNAs During Viral Infection. Front. Genet. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Claire, M.C.; Ragland, D.R.; Bollinger, L.; Jahrling, P.B. Animal Models of Ebolavirus Infection. Comp. Med. 2017, 67, 253–262. [Google Scholar]
- Shurtleff, A.; Biggins, J.; Keeney, A.; Zumbrun, E.; Bloomfield, H.; Kuehne, A.; Audet, J.; Alfson, K.; Griffiths, A.; Olinger, G.; et al. Standardization of the Filovirus Plaque Assay for Use in Preclinical Studies. Viruses 2012, 4, 3511–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trombley, A.R.; Wachter, L.; Garrison, J.; Buckley-Beason, V.A.; Jahrling, J.; Hensley, L.E.; Schoepp, R.J.; Norwood, D.A.; Goba, A.; Fair, J.N.; et al. Comprehensive Panel of Real-Time TaqMan™ Polymerase Chain Reaction Assays for Detection and Absolute Quantification of Filoviruses, Arenaviruses, and New World Hantaviruses. Am. J. Trop. Med. Hyg. 2010, 82, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Zhang, Y.; Zhang, S.J.; Ma, Y.N.; He, Y. Comprehensive analysis of key genes and microRNAs in radioresistant nasopharyngeal carcinoma. BMC Med Genom. 2019, 12, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzi, A.; Feldmann, F.; Hanley, P.W.; Scott, D.P.; Günther, S.; Feldmann, H. Delayed Disease Progression in Cynomolgus Macaques Infected with Ebola Virus Makona Strain. Emerg. Infect. Dis. 2015, 21, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisler, R.B.; Yu, C.; Donofrio, M.J.; Warren, T.K.; Wells, J.B.; Stuthman, K.S.; Garza, N.L.; VanTongeren, S.A.; Donnelly, G.C.; Kane, C.D.; et al. Clinical Laboratory Values as Early Indicators of Ebola Virus Infection in Nonhuman Primates. Emerg. Infect. Dis. 2017, 23, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanini, S.; Portella, G.; Vairo, F.; Kobinger, G.P.; Pesenti, A.; Langer, M.; Kabia, S.; Brogiato, G.; Amone, J.; Castilletti, C.; et al. Relationship Between Viremia and Specific Organ Damage in Ebola Patients: A Cohort Study. Clin. Infect. Dis. 2017, 66, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Havelka, A.; Sejersen, K.; Venge, P.; Pauksens, K.; Larsson, A. Calprotectin, a new biomarker for diagnosis of acute respiratory infections. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tisserand, J.; Khetchoumian, K.; Thibault, C.; Dembélé, D.; Chambon, P.; Losson, R. Tripartite Motif 24 (Trim24/Tif1α) Tumor Suppressor Protein Is a Novel Negative Regulator of Interferon (IFN)/Signal Transducers and Activators of Transcription (STAT) Signaling Pathway Acting through Retinoic Acid Receptor α (Rarα) Inhibition. J. Biol. Chem. 2011, 286, 33369–33379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Duy, J.; Koehler, J.W.; Honko, A.N.; Schoepp, R.J.; Wauquier, N.; Gonzalez, J.-P.; Pitt, M.L.; Mucker, E.M.; Johnson, J.C.; O’Hearn, A.; et al. Circulating microRNA profiles of Ebola virus infection. Sci. Rep. 2016, 6, 24496. [Google Scholar] [CrossRef]
- Caballero, I.S.; Honko, A.N.; Gire, S.K.; Winnicki, S.M.; Melé, M.; Gerhardinger, C.; Lin, A.E.; Rinn, J.L.; Sabeti, P.C.; Hensley, L.E.; et al. In vivo Ebola virus infection leads to a strong innate response in circulating immune cells. BMC Genom. 2016, 17, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Huang, Y.; Xu, K.; Liu, Y.; Li, X.; Xu, Y.; Zhong, W.; Hao, P. Differential responses of innate immunity triggered by different subtypes of influenza a viruses in human and avian hosts. BMC Med. Genom. 2017, 10, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Zapata, J.C.; Salvato, M.S. Genomic profiling of host responses to Lassa virus: Therapeutic potential from primate to man. Futur. Virol. 2015, 10, 233–256. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W. Interferon-stimulated genes: Roles in viral pathogenesis. Curr. Opin. Virol. 2014, 6, 40–46. [Google Scholar] [CrossRef]
- Olejnik, J.; Forero, A.; Deflubé, L.R.; Hume, A.J.; Manhart, W.A.; Nishida, A.; Marzi, A.; Katze, M.G.; Ebihara, H.; Rasmussen, A.L.; et al. Ebolaviruses Associated with Differential Pathogenicity Induce Distinct Host Responses in Human Macrophages. J. Virol. 2017, 91, e00179-17. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 MAP Kinase Family as Regulators of Proinflammatory Cytokine Production in Degenerative Diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar]
- Herlaar, E.; Brown, Z. p38 MAPK signalling cascades in inflammatory disease. Mol. Med. Today 1999, 5, 439–447. [Google Scholar] [CrossRef]
- Johnson, J.C.; Martinez, O.; Honko, A.N.; Hensley, L.E.; Olinger, G.G.; Basler, C.F. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire ebolavirus in human dendritic cells. Antivir. Res. 2014, 107, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banadyga, L.; Siragam, V.; Zhu, W.; He, S.; Cheng, K.; Qiu, X. The Cytokine Response Profile of Ebola Virus Disease in a Large Cohort of Rhesus Macaques Treated With Monoclonal Antibodies. Open Forum Infect. Dis. 2019, 6, ofz046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, A.K.; Mühlberger, E.; Muñoz-Fontela, C. Immune barriers of Ebola virus infection. Curr. Opin. Virol. 2018, 28, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, O.; Moroso, M.; Siracusa, J.; Jarjaval, F.; Goriot, M.-E.; Peyrefitte, C.N.; Banzet, S. Circulating microRNA profile in a mouse model of Crimean-Congo haemorrhagic fever. Virus Res. 2019, 263, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Seppälä, I.; Lyytikäinen, L.-P.; Viikari, J.; Ala-Korpela, M.; Soininen, P.; Kangas, A.J.; Waldenberger, M.; Klopp, N.; Illig, T.; et al. Blood hsa-miR-122-5p and hsa-miR-885-5p levels associate with fatty liver and related lipoprotein metabolism—The Young Finns Study. Sci. Rep. 2016, 6, 38262. [Google Scholar] [CrossRef]
- Zhou, X.-J.; Wang, J.; Ye, H.-H.; Fa, Y.-Z. Signature MicroRNA expression profile is associated with lipid metabolism in African green monkey. Lipids Health Dis. 2019, 18, 55. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zang, Y.; Zheng, C.; Li, Z.; Gu, X.; Zhou, M.; Wang, Z.; Xiang, J.; Chen, Z.; Zhou, Y. CD3D is associated with immune checkpoints and predicts favorable clinical outcome in colon cancer. Immunotherapy 2020, 12, 25–35. [Google Scholar] [CrossRef]
- Wiedemann, A.; POSTEBOGUI Study Group; Foucat, E.; Hocini, H.; Lefebvre, C.; Hejblum, B.P.; Durand, M.; Krüger, M.; Keita, A.K.; Ayouba, A.; et al. Long-lasting severe immune dysfunction in Ebola virus disease survivors. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Kolokoltsov, A.A.; Saeed, M.F.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Identification of novel cellular targets for therapeutic intervention against Ebola virus infection by siRNA screening. Drug Dev. Res. 2009, 70, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Tani-Ichi, S.; Shimba, A.; Wagatsuma, K.; Miyachi, H.; Kitano, S.; Imai, K.; Hara, T.; Ikuta, K. Interleukin-7 receptor controls development and maturation of late stages of thymocyte subpopulations. Proc. Natl. Acad. Sci. USA 2012, 110, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Cruikshank, W.; Little, F. lnterleukin-16: The ins and outs of regulating T-cell activation. Crit. Rev. Immunol. 2008, 28, 467–483. [Google Scholar] [CrossRef]
- Skundric, D.S.; Cruikshank, W.W.; Drulovic, J. Role of IL-16 in CD4+ T cell-mediated regulation of relapsing multiple sclerosis. J. Neuroinflamm. 2015, 12, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, A.Y.-L.; Zhao, J.L.; Baltimore, D. The Yin and Yang of microRNAs: Leukemia and immunity. Immunol. Rev. 2013, 253, 129–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat. Rev. Immunol. 2004, 4, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Mohamadzadeh, M.; Chen, L.; Schmaljohn, A.L. How Ebola and Marburg viruses battle the immune system. Nat. Rev. Immunol. 2007, 7, 556–567. [Google Scholar] [CrossRef]
- Reed, D.S.; Hensley, L.E.; Geisbert, J.B.; Jahrling, P.B.; Geisbertl, T.W. Depletion of Peripheral Blood T Lymphocytes and NK Cells During the Course of Ebola Hemorrhagic Fever in Cynomolgus Macaques. Viral Immunol. 2004, 17, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Aid, M.; Ram, D.R.; Bosinger, S.E.; Barouch, D.H.; Reeves, R.K. Delineation and Modulation of the Natural Killer Cell Transcriptome in Rhesus Macaques During ZIKV and SIV Infections. Front. Cell. Infect. Microbiol. 2020, 10, 194. [Google Scholar] [CrossRef]
- Hanna, J.; Bechtel, P.; Zhai, Y.; Youssef, F.; McLachlan, K.; Mandelboim, O. Novel Insights on Human NK Cells’ Immunological Modalities Revealed by Gene Expression Profiling. J. Immunol. 2004, 173, 6547–6563. [Google Scholar] [CrossRef]
- Bongen, E.; Vallania, F.; Utz, P.J.; Khatri, P. KLRD1-expressing natural killer cells predict influenza susceptibility. Genome Med. 2018, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.-M.; Lin, K.-Y.; Chen, Y.-Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, H.; Li, C.; Adereth, Y.; Hsu, T.; Watson, D.K.; Li, R. Caspase-1 Is a Direct Target Gene of ETS1 and Plays a Role in ETS1-Induced Apoptosis. Cancer Res. 2005, 65, 7205–7213. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhan, M.; Chen, T.; Chen, W.; Zhang, Y.; Xu, S.; Yan, J.; Huang, Q.; Wang, J. miR-125b-5p enhances chemotherapy sensitivity to cisplatin by down-regulating Bcl2 in gallbladder cancer. Sci. Rep. 2017, 7, srep43109. [Google Scholar] [CrossRef] [PubMed]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human Fatal Zaire Ebola Virus Infection Is Associated with an Aberrant Innate Immunity and with Massive Lymphocyte Apoptosis. PLoS Negl. Trop. Dis. 2010, 4, e837. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.-C.; Capron, M.; Lansoud-Soukate, J.; Debré, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef]
- Garamszegi, S.; Yen, J.Y.; Honko, A.N.; Geisbert, J.B.; Rubins, K.H.; Geisbert, T.W.; Xia, Y.; Hensley, L.E.; Connor, J.H. Transcriptional Correlates of Disease Outcome in Anticoagulant-Treated Non-Human Primates Infected with Ebolavirus. PLoS Negl. Trop. Dis. 2014, 8, e3061. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.R.; Ho, A.; Pius, R.; Buchan, I.; Carson, G.; Drake, T.M.; Dunning, J.; Fairfield, C.J.; Gamble, C.; A Green, C.; et al. Risk stratification of patients admitted to hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Development and validation of the 4C Mortality Score. BMJ 2020, 370, m3339. [Google Scholar] [CrossRef] [PubMed]
- Vastrad, B.; Vastrad, C.; Tengli, A. Identification of potential mRNA panels for severe acute respiratory syndrome coronavirus 2 (COVID-19) diagnosis and treatment using microarray dataset and bioinformatics methods. 3 Biotech 2020, 10, 1–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | mRNA | Spearman Rho | Prob>|Rho| | miRNA | mRNA | Spearman Rho | Prob>|Rho| |
|---|---|---|---|---|---|---|---|
| hsa-miR-125b-5p * | IL16 | −0.767 | <0.0001 | hsa-miR-423-5p | LTF | −0.5655 | <0.0001 |
| hsa-miR-424-5p | AKT3 | −0.7083 | <0.0001 | hsa-miR-143-3p | GZMK | −0.5647 | <0.0001 |
| hsa-miR-125b-5p * | ETS1 | −0.7004 | <0.0001 | hsa-miR-424-5p | ICOS | −0.5634 | <0.0001 |
| hsa-miR-424-5p | IL7R | −0.6876 | <0.0001 | hsa-miR-423-5p | CHIT1 | −0.5619 | <0.0001 |
| hsa-miR-21-5p | MALT1 | −0.6873 | <0.0001 | hsa-miR-199b-5p | ETS1 | −0.5581 | <0.0001 |
| hsa-miR-143-3p | CD3D | −0.6799 | <0.0001 | hsa-miR-122-5p | CD3E | −0.5548 | <0.0001 |
| hsa-miR-143-3p | MAP4K1 | −0.6787 | <0.0001 | hsa-miR-423-5p | TNFRSF13C | −0.5541 | <0.0001 |
| hsa-miR-223-5p | KLRC3 | −0.6655 | <0.0001 | hsa-miR-338-3p * | ETS1 | −0.5505 | <0.0001 |
| hsa-miR-122-5p | LILRB1 | −0.6615 | <0.0001 | hsa-miR-19b-3p | RPS6KA5 | −0.549 | <0.0001 |
| hsa-miR-125b-5p * | BCL2 | −0.6473 | <0.0001 | hsa-miR-34a-5p | KLRK1 | −0.549 | <0.0001 |
| hsa-miR-122-5p | CCL5 | −0.6421 | <0.0001 | hsa-miR-424-5p | HLA-DQA1 | −0.5477 | <0.0001 |
| hsa-miR-199a-3p | PLCB1 | −0.6324 | <0.0001 | hsa-miR-199b-5p | GZMB | −0.5455 | <0.0001 |
| hsa-miR-27a-3p | IRF4 | −0.6323 | <0.0001 | hsa-miR-223-5p | HLA-DPB1 | −0.545 | <0.0001 |
| hsa-miR-423-5p | MAFG | −0.6271 | <0.0001 | hsa-miR-19b-3p | MAF | −0.5431 | <0.0001 |
| hsa-miR-424-5p | BCL2 | −0.6242 | <0.0001 | hsa-miR-1260b | TNFRSF13C | −0.5386 | <0.0001 |
| hsa-miR-1260b | MAFG | −0.6224 | <0.0001 | hsa-miR-423-3p | MAFG | −0.5381 | <0.0001 |
| hsa-miR-370-3p | SMAD3 | −0.622 | <0.0001 | hsa-miR-671-5p | KLRC3 | −0.5373 | <0.0001 |
| hsa-miR-424-5p | CD3E | −0.6184 | <0.0001 | hsa-miR-423-5p | ATG7 | −0.5363 | <0.0001 |
| hsa-miR-27a-3p | HLA-DRA | −0.6113 | <0.0001 | hsa-miR-27a-3p | MEF2C | −0.535 | <0.0001 |
| hsa-miR-1260b | TNFSF14 | −0.6082 | <0.0001 | hsa-miR-19b-3p | GZMK | −0.5329 | <0.0001 |
| hsa-miR-296-5p | IKBKE | −0.6064 | <0.0001 | hsa-miR-143-3p | MAF | −0.5326 | <0.0001 |
| hsa-miR-199b-5p | KLRF1 | −0.6012 | <0.0001 | hsa-miR-125b-5p * | CASP2 | −0.5308 | <0.0001 |
| hsa-miR-144-3p * | ETS1 | −0.5972 | <0.0001 | hsa-miR-29c-3p | DPP4 | −0.529 | <0.0001 |
| hsa-miR-34a-5p | HLA-DQA1 | −0.5925 | <0.0001 | hsa-miR-432-5p | RELA | −0.5284 | <0.0001 |
| hsa-miR-23a-3p | KLRF1 | −0.5905 | <0.0001 | hsa-miR-125b-5p | CD244 | −0.5235 | <0.0001 |
| hsa-miR-125b-5p * | PPP1R12B | −0.5882 | <0.0001 | hsa-miR-1260b | CHIT1 | −0.5228 | <0.0001 |
| hsa-miR-10b-5p | CD3G | −0.5852 | <0.0001 | hsa-miR-676-3p | CD3G | −0.5212 | <0.0001 |
| hsa-miR-143-3p | HLA-DPB1 | −0.5805 | <0.0001 | hsa-miR-1260b | DAXX | −0.5208 | <0.0001 |
| hsa-miR-127-3p | PLAUR | −0.5763 | <0.0001 | hsa-miR-143-3p | BCL2 | −0.5199 | <0.0001 |
| hsa-miR-27a-3p | ICOS | −0.5752 | <0.0001 | hsa-miR-1262 * | ANP32B | −0.5167 | <0.0001 |
| hsa-miR-6865-5p | THY1 | −0.5751 | <0.0001 | hsa-miR-423-5p | TNFRSF14 | −0.5164 | <0.0001 |
| hsa-miR-34a-5p | KLRD1 | −0.5749 | <0.0001 | hsa-miR-423-5p | PLA2G6 | −0.5154 | <0.0001 |
| hsa-miR-23a-3p | BCL2 | −0.5747 | <0.0001 | hsa-miR-296-5p | CD22 | −0.514 | <0.0001 |
| hsa-miR-21-5p | TGFBI | −0.5714 | <0.0001 | hsa-miR-423-5p | CSF3 | −0.5125 | <0.0001 |
| hsa-miR-125b-5p * | IRF4 | −0.5683 | <0.0001 | hsa-miR-29c-3p | TRAF5 | −0.5113 | <0.0001 |
| hsa-miR-223-5p | DPP4 | −0.5675 | <0.0001 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefan, C.P.; Arnold, C.E.; Shoemaker, C.J.; Zumbrun, E.E.; Altamura, L.A.; Douglas, C.E.; Taylor-Howell, C.L.; Graham, A.S.; Delp, K.L.; Blancett, C.D.; et al. Transcriptomic Analysis Reveals Host miRNAs Correlated with Immune Gene Dysregulation during Fatal Disease Progression in the Ebola Virus Cynomolgus Macaque Disease Model. Microorganisms 2021, 9, 665. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030665
Stefan CP, Arnold CE, Shoemaker CJ, Zumbrun EE, Altamura LA, Douglas CE, Taylor-Howell CL, Graham AS, Delp KL, Blancett CD, et al. Transcriptomic Analysis Reveals Host miRNAs Correlated with Immune Gene Dysregulation during Fatal Disease Progression in the Ebola Virus Cynomolgus Macaque Disease Model. Microorganisms. 2021; 9(3):665. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030665
Chicago/Turabian StyleStefan, Christopher P., Catherine E. Arnold, Charles J. Shoemaker, Elizabeth E. Zumbrun, Louis A. Altamura, Christina E. Douglas, Cheryl L. Taylor-Howell, Amanda S. Graham, Korey L. Delp, Candace D. Blancett, and et al. 2021. "Transcriptomic Analysis Reveals Host miRNAs Correlated with Immune Gene Dysregulation during Fatal Disease Progression in the Ebola Virus Cynomolgus Macaque Disease Model" Microorganisms 9, no. 3: 665. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9030665