
Wide Genetic Diversity of Blastocystis in White-Tailed Deer (Odocoileus virginianus) from Maryland, USA


Abstract
:1. Introduction
2. Materials and Methods
2.1. Source and Collection of Specimens
2.2. Parasite Concentration from Feces and DNA Extraction
2.3. Molecular Detection and Subtype Identification Using Next Generation Amplicon Sequencing
2.4. PCR Amplification and Sequencing of the Full-Length SSU rRNA Gene
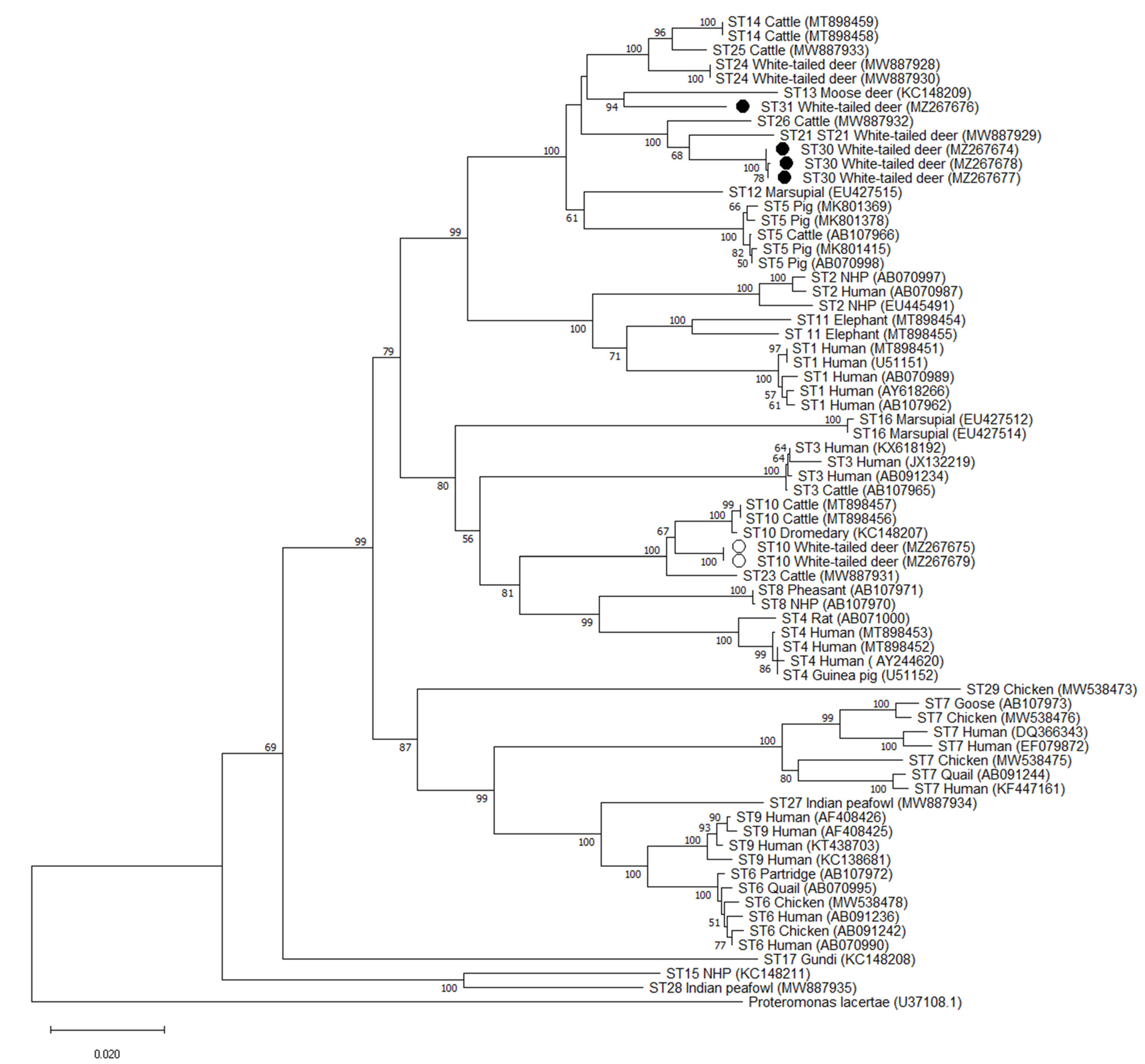
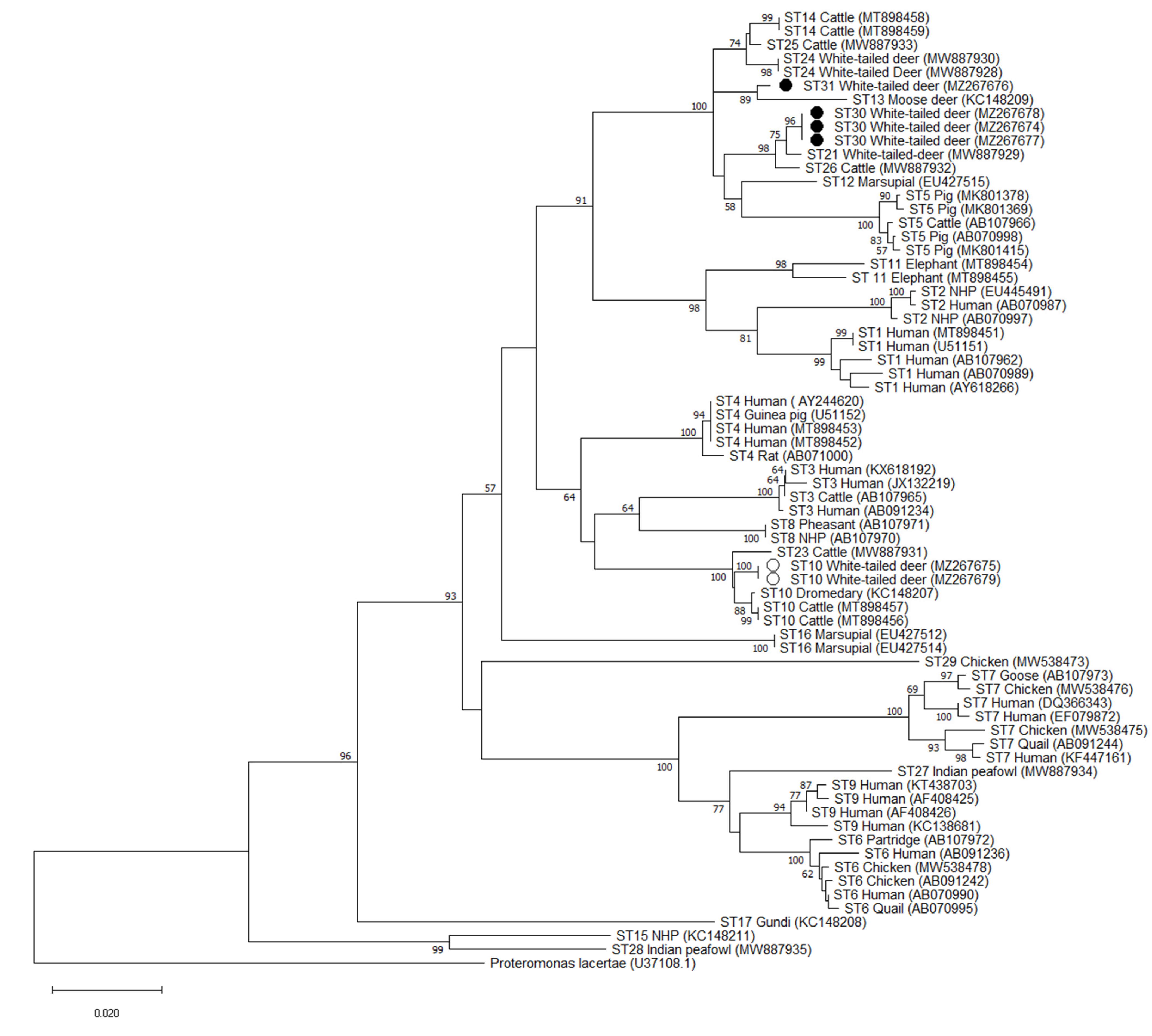
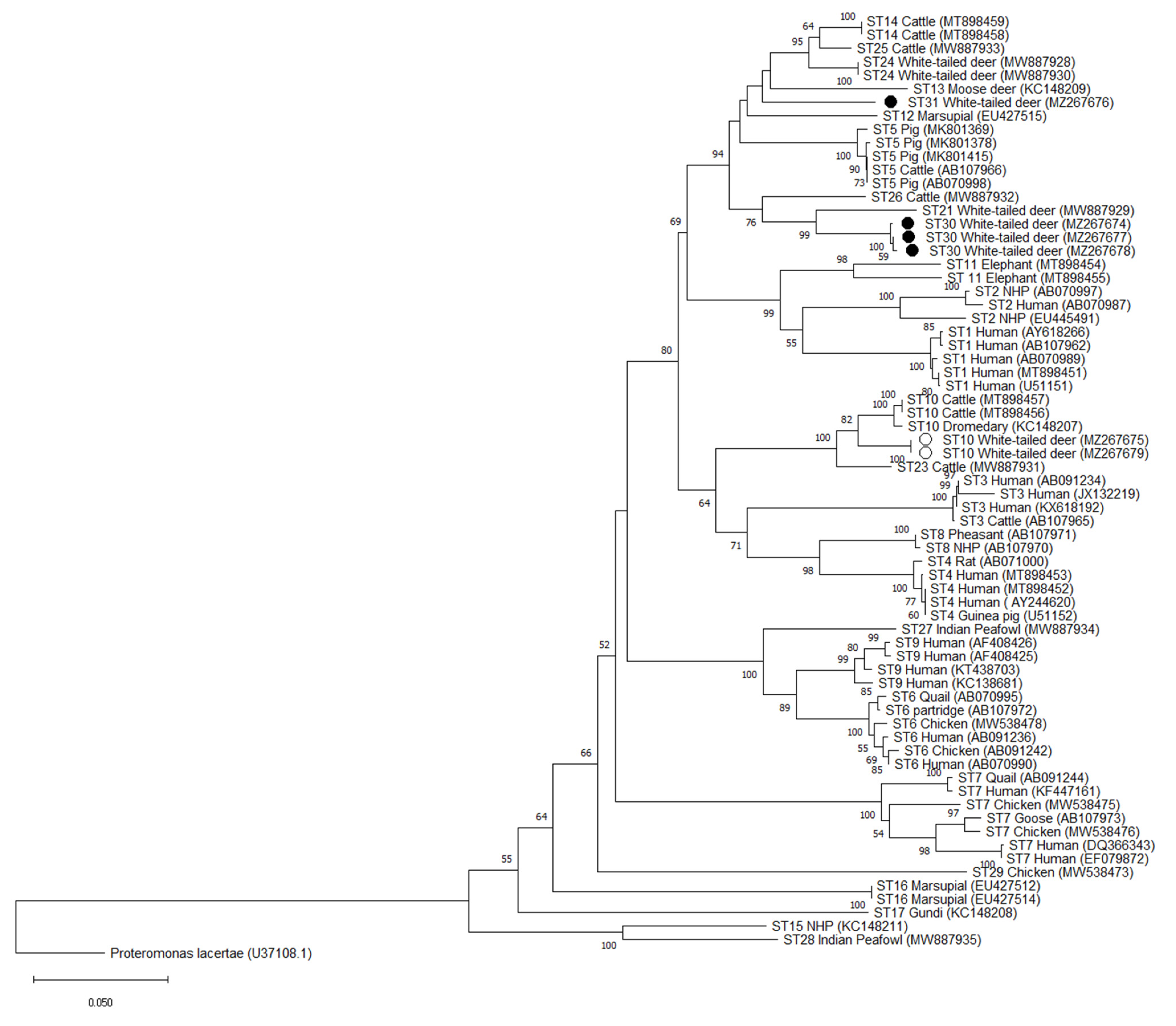
2.5. Phylogenetic and Pairwise Distance Analysis
3. Results
3.1. Prevalence of Blastocystis in White-Tailed Deer
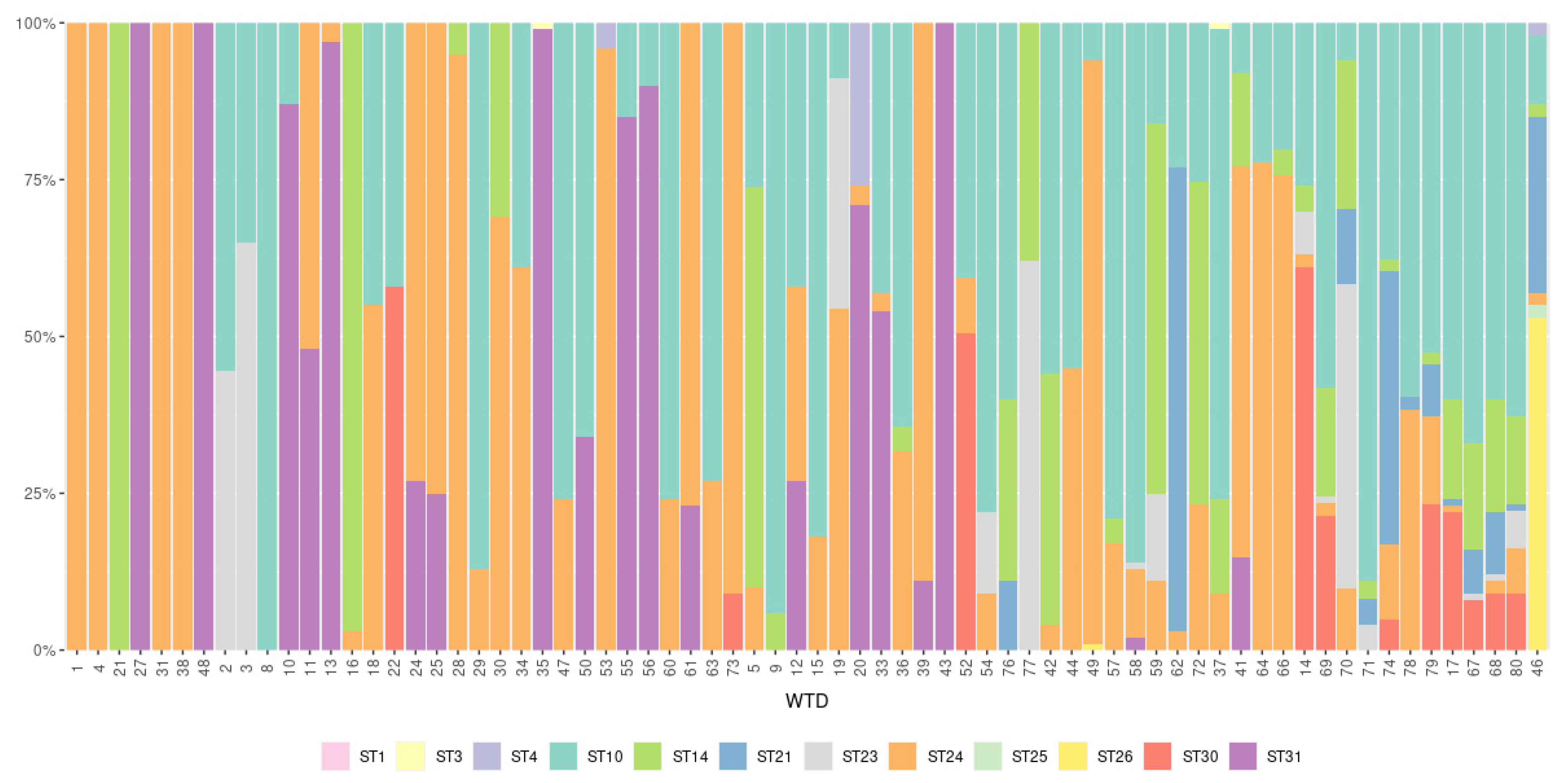
3.2. Subtypes of Blastocystis in White-Tailed Deer
3.3. Blastocystis Intra-Subtype Variation
3.4. Validation of Novel Subtypes ST30 and ST31
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Safadi, D.; Cian, A.; Nourrisson, C.; Pereira, B.; Morelle, C.; Bastien, P.; Bellanger, A.P.; Botterel, F.; Candolfi, E.; Desoubeaux, G.; et al. Prevalence, risk factors for infection and subtype distribution of the intestinal parasite Blastocystis sp. from a large-scale multi-center study in France. BMC Infect. Dis. 2016, 16, 451. [Google Scholar] [CrossRef]
- Tito, R.Y.; Chaffron, S.; Caenepeel, C.; Lima-Mendez, G.; Wang, J.; Vieira-Silva, S.; Falony, G.; Hildebrand, F.; Darzi, Y.; Rymenans, L.; et al. Population-level analysis of Blastocystis subtype prevalence and variation in the human gut microbiota. Gut 2019, 68, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Hublin, J.S.Y.; Maloney, J.G.; Santin, M. Blastocystis in domesticated and wild mammals and birds. Res. Vet. Sci. 2021, 135, 260–282. [Google Scholar] [CrossRef]
- Deng, L.; Wojciech, L.; Gascoigne, N.R.J.; Peng, G.; Tan, K.S.W. New insights into the interactions between Blastocystis, the gut microbiota, and host immunity. PLoS Pathog. 2021, 17, e1009253. [Google Scholar] [CrossRef]
- Ajjampur, S.S.R.; Tan, K.S.W. Pathogenic mechanisms in Blastocystis SPP—Interpreting results from in vitro and in vivo studies. Parasitol. Int. 2016, 65, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.D.; Mongi, F.; Sánchez, A.; Ramírez, J.D. Blastocystis and urticaria: Examination of subtypes and morphotypes in an unusual clinical manifestation. Acta Trop. 2015, 148, 156–161. [Google Scholar] [CrossRef]
- Rojas-Velázquez, L.; Maloney, J.G.; Molokin, A.; Morán, P.; Serrano-Vázquez, A.; González, E.; Pérez-Juárez, H.; Ximénez, C.; Santin, M. Use of next-generation amplicon sequencing to study Blastocystis genetic diversity in a rural human population from Mexico. Parasit. Vectors 2019, 12, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Chye, T.; Karmacharya, B.; Govind, S. Blastocystis SP: Waterborne zoonotic organism, a possibility? Parasit. Vectors 2012, 5, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caradonna, T.; Marangi, M.; Del Chierico, F.; Ferrari, N.; Reddel, S.; Bracaglia, G.; Normanno, G.; Putignani, L.; Giangaspero, A. Detection and prevalence of protozoan parasites in ready-to-eat packaged salads on sale in Italy. Food Microbiol. 2017, 67, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Angelici, M.C.; Nardis, C.; Scarpelli, R.; Ade, P. Blastocystis hominis transmission by non-potable water: A case report in Italy. New Microbiol. 2018, 41, 173–177. [Google Scholar] [PubMed]
- Rodrigues, A.C.; da Silva, M.D.C.; Pereira, R.Â.S.; Pinto, L.C. Prevalence of contamination by intestinal parasites in vegetables (Lactuca sativa L. and Coriandrum sativum L.) sold in markets in Belém, Northern Brazil. J. Sci. Food Agric. 2020, 100, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Suresh, G.K.; Tan, K.S.; Thompson, R.C.; Traub, R.J.; Viscogliosi, E.; Yoshikawa, H.; Clark, C.G. Terminology for Blastocystis subtypes—A consensus. Trends Parasitol. 2007, 23, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Clark, C.G. Pre-empting Pandora’s Box: Blastocystis subtypes revisited. Trends Parasitol. 2020, 36, 229–232. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; da Cunha, M.J.R.; Cury, M.C.; Santin, M. Blastocystis subtype distribution in domestic and captive wild bird species from Brazil using next generation amplicon sequencing. Parasite Epidemiol. Control 2020, 9, e00138. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.G.; da Cunha, M.J.R.; Molokin, A.; Cuty, M.C.; Santin, M. Next generation sequencing reveals widespread presence of human-pathogenic Blastocystis subtypes in chickens. Parasitol. Res. 2021, 21, 2219–2231. [Google Scholar] [CrossRef]
- Maloney, J.G.; Santin, M. Mind the gap: New full-length sequences of Blastocystis subtypes generated via Oxford Nanopore Minion sequencing allow for comparisons between full-length and partial sequences of the small subunit of the ribosomal RNA gene. Microorganisms 2021, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Tan, K.S.W.; Clark, C.G. Blastocystis. Trends Parasitol. 2020, 36, 315–316. [Google Scholar] [CrossRef]
- Yan, Y.; Su, S.; Ye, J.; Lai, X.; Lai, R.; Liao, H.; Chen, G.; Zhang, R.; Hou, Z.; Luo, X. Blastocystis sp. subtype 5: A possibly zoonotic genotype. Parasitol. Res. 2007, 101, 1527–1532. [Google Scholar] [CrossRef]
- Parkar, U.; Traub, R.J.; Vitali, S.; Elliot, A.; Levecke, B.; Robertson, I.; Geurden, T.; Steele, J.; Drake, B.; Thompson, R.C. Molecular characterization of Blastocystis isolates from zoo animals and their animal-keepers. Vet. Parasitol. 2010, 169, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Owen, H.; Traub, R.J.; Cuttell, L.; Inpankaew, T.; Bielefeldt-Ohmann, H. Molecular epidemiology of Blastocystis in pigs and their in-contact humans in Southeast Queensland, Australia, and Cambodia. Vet. Parasitol. 2014, 203, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Greige, S.; El Safadi, D.; Bécu, N.; Gantois, N.; Pereira, B.; Chabé, M.; Benamrouz-Vanneste, S.; Certad, G.; El Hage, R.; Chemaly, M.; et al. Prevalence and subtype distribution of Blastocystis sp. isolates from poultry in Lebanon and evidence of zoonotic potential. Parasit. Vectors 2018, 11, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köster, P.C.; Dashti, A.; Bailo, B.; Muadica, A.S.; Maloney, J.G.; Santín, M.; Chicharro, C.; Migueláñez, S.; Nieto, F.J.; Cano-Terriza, D.; et al. Occurrence and genetic diversity of protist parasites in captive non-human primates, zookeepers, and free-living sympatric rats in the Córdoba Zoo Conservation Centre, Southern Spain. Animals 2021, 11, 700. [Google Scholar] [CrossRef]
- Wang, J.; Gong, B.; Liu, X.; Zhao, W.; Bu, T.; Zhang, W.; Liu, A.; Yang, F. Distribution and genetic diversity of Blastocystis subtypes in various mammal and bird species in northeastern China. Parasit. Vectors 2018, 11, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.H.; Hu, X.F.; Liu, T.L.; Hu, R.S.; Yu, Z.Q.; Yang, W.B.; Wu, Y.L.; Yu, S.K.; Song, J.K. Molecular characterization of Blastocystis SP. in captive wild animals in Qinling Mountains. Parasitol. Res. 2017, 116, 2327–2333. [Google Scholar] [CrossRef] [PubMed]
- Alfellani, M.A.; Taner-Mulla, D.; Jacob, A.S.; Imeede, C.A.; Yoshikawa, H.; Stensvold, C.R.; Clark, C.G. Genetic diversity of Blastocystis in livestock and zoo animals. Protist 2013, 164, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Arbex, A.P.; David, É.B.; Tenório, M.D.S.; Cicchi, P.J.P.; Patti, M.; Coradi, S.T.; Lucheis, S.B.; Jim, J.; Guimarães, S. Diversity of Blastocystis subtypes in wild mammals from a zoo and two conservation units in southeastern Brazil. Infect. Genet. Evol. 2020, 78, 104053. [Google Scholar] [CrossRef] [PubMed]
- Betts, E.L.; Gentekaki, E.; Thomasz, A.; Breakell, V.; Carpenter, A.I.; Tsaousis, A.D. Genetic diversity of Blastocystis in non-primate animals. Parasitology 2018, 145, 1228–1234. [Google Scholar] [CrossRef] [Green Version]
- Betts, E.L.; Gentekaki, E.; Tsaousis, A.D. Exploring micro-eukaryotic diversity in the gut: Co-occurrence of Blastocystis subtypes and other protists in zoo animals. Front. Microbiol. 2020, 11, 288. [Google Scholar] [CrossRef]
- Kim, K.T.; Noh, G.; Lee, H.; Kim, S.H.; Jeong, H.; Kim, Y.; Jheong, W.H.; Oem, J.K.; Kim, T.H.; Kwon, O.D.; et al. Genetic diversity and zoonotic potential of Blastocystis in Korean water deer, Hydropotes inermis argyropus. Pathogens 2020, 9, 955. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.; Stark, D.; Harkness, J.; Ellis, J. Subtype distribution of Blastocystis isolates from a variety of animals from New South Wales, Australia. Vet. Parasitol. 2013, 196, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Zou, Y.; Pan, J.; Liang, Q.L.; Zeng, Z.; Meng, Y.M.; Wang, X.L.; Wang, H.N.; Zhu, X.Q. Prevalence and subtypes of Blastocystis sp. infection in zoo animals in three cities in China. Parasitol. Res. 2020, 119, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Alfellani, M.A.; Nørskov-Lauritsen, S.; Prip, K.; Victory, E.L.; Maddox, C.; Nielsen, H.V.; Clark, C.G. Subtype distribution of Blastocystis isolates from synanthropic and zoo animals and identification of a new subtype. Int. J. Parasitol. 2009, 39, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, H.B.; Gong, Q.L.; Zhang, N.Z.; Zhao, Q.; Tao, W.F.; Qiu, H.Y.; Fei, Y.C.; Zhang, X.X. Molecular detection of Blastocystis in black bears and sika deer in northern China. Parasitol. Res. 2021, 120, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Karim, M.R.; Li, D.; Rahaman Sumon, S.M.M.; Siddiki, S.H.M.F.; Rume, F.I.; Sun, R.; Jia, Y.; Zhang, L. Molecular characterization of Blastocystis sp. in captive wildlife in Bangladesh National Zoo: Non-human primates with high prevalence and zoonotic significance. Int. J. Parasitol. Parasites Wildl. 2019, 10, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Santín, M.; Trout, J.M.; Xiao, L.; Zhou, L.; Greiner, E.; Fayer, R. Prevalence and age-related variation of Cryptosporidium species and genotypes in dairy calves. Vet. Parasitol. 2004, 122, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.G.; Molokin, A.; Santin, M. Next generation amplicon sequencing improves detection of Blastocystis mixed subtype infections. Infect. Genet. Evol. 2019, 73, 119–125. [Google Scholar] [CrossRef]
- Santín, M.; Gómez-Muñoz, M.T.; Solano-Aguilar, G.; Fayer, R. Development of a new PCR protocol to detect and subtype Blastocystis SPP. from humans and animals. Parasitol. Res. 2011, 109, 205–212. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Download. SourceForge.net. 2014. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 27 April 2020).
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PEER J. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Maloney, J.G.; Molokin, A.; Santin, M. Use of Oxford Nanopore MinION to generate full-length sequences of the Blastocystis small subunit (SSU) rRNA gene. Parasit. Vectors 2020, 13, 595. [Google Scholar] [CrossRef]
- Xiao, L.; Escalante, L.; Yang, C.; Sulaiman, I.; Escalante, A.A.; Montali, R.J.; Fayer, R.; Lal, A.A. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl. Environ. Microbiol. 1999, 65, 1578–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Scicluna, S.M.; Tawari, B.; Clark, C.G. DNA barcoding of Blastocystis. Protist 2006, 157, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Stensvold, C.R.; Clark, C.G. Current status of Blastocystis: A personal view. Parasit. Int. 2016, 65, 763–771. [Google Scholar] [CrossRef]
- Maloney, J.G.; Lombard, J.E.; Urie, N.J.; Shively, C.B.; Santin, M. Zoonotic and genetically diverse subtypes of Blastocystis in US pre-weaned dairy heifer calves. Parasitol. Res. 2019, 118, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Song, J.K.; Yin, Y.L.; Yuan, Y.J.; Tang, H.; Ren, G.J.; Zhang, H.J.; Li, Z.X.; Zhang, Y.M.; Zhao, G.H. First genotyping of Blastocystis sp. in dairy, meat, and cashmere goats in northwestern China. Acta Trop. 2017, 176, 277–282. [Google Scholar] [CrossRef]
- Ren, M.; Song, J.K.; Yang, F.; Zou, M.; Wang, P.X.; Wang, D.; Zhang, H.J.; Zhao, G.H.; Lin, Q. First genotyping of Blastocystis in yaks from Qinghai Province, Northwestern China. Parasit. Vectors 2019, 12, 171. [Google Scholar] [CrossRef]
- Calero-Bernal, R.; Santín, M.; Maloney, J.G.; Martín-Pérez, M.; Habela, M.A.; Fernández-García, J.L.; Figueiredo, A.; Nájera, F.; Palacios, M.J.; Mateo, M.; et al. Blastocystis sp. subtype diversity in wild carnivore species from Spain. J. Eukaryot. Microbiol. 2020, 67, 273–278. [Google Scholar] [CrossRef]
- Russini, V.; Di Filippo, M.M.; Fanelli, R.; Polidori, M.; Berrilli, F.; Di Cave, D.; Novelletto, A.; Calderini, P. Characterization of prevalence and genetic subtypes of Blastocystis sp. in wild and domestic Suidae of central Italy aided by amplicon NGS. Vet. Parasitol. Reg. Stud. Rep. 2020, 22, 100472. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host (Scientific Name) | Country | Source of Deer | No. of Samples Examined/ No. of Positives (%) | Subtype(s) | References |
|---|---|---|---|---|---|
| Eurasia elk (Alces alces) | United Kingdom | Zoo | 2/1 (50%) a | ST4(1), ST10(1), ST14(1), ND(1) | [27] |
| United Kingdom | Zoo | 3/1 (33.3%) a | ST4(1), ST10(1), ST14(4) | [28] | |
| Fallow deer (Dama dama) | China | Captive | 2/1 (50%) | ST10(1) | [24] |
| Mauritius | Zoo | 2/2 (100%) | ST10(2) | [25] | |
| Gray brocket (Mazama gouazoubira) | Brazil | Captive | 1/1 (100%) | ST5(1) | [26] |
| Korean water deer (Hydropotes inermis argyropus) | South Korea | Wild | 125/51 (40.8%) | ST4(1), ST14(25) b | [29] |
| Marsh deer (Blastocerus dichotomus) | Brazil | Captive | 1/1 (100%) | ND(1) b | [26] |
| Muntjac deer (Muntiacus reevesi) | United Kingdom | Zoo | 1/1(100%) | ST14(1) | [27] |
| United Kingdom | Zoo | 1/1(100%) | ST13(1) | [28] | |
| Red deer (Cervus elaphus) | Australia | Wild | 50/1 (2%) | ST4(1) | [30] |
| China | Captive | 3/1 (33.3%) | ST10(1) | [24] | |
| China | Zoo | 5/2 (40%) | ST10(2) | [31] | |
| United Kingdom | Zoo | 1/1(100%) a | ST4(2), ST10(6) | [27] | |
| United Kingdom | Zoo | 3/1(33.3%) a | ST4(3), ST10(5) | [28] | |
| Reindeer (Rangifer tarandus) | China | Farm | 104/7 (6.7%) | ST10(3), ST13(4) | [23] |
| United Kingdom | Zoo | 1/1(100%) | ST10(1) | [28] | |
| Roe deer (Capreolus capreolus) | Denmark | Zoo | 1 c | ST10(1) | [32] |
| United Kingdom | Zoo | 2/1 (50%) | ST5(1) | [25] | |
| Sika deer (Cervus nippon) | China | Farm | 6/760 (0.8%) | ST10(5), ST14(1) | [33] |
| China | Captive | 8/3 (37.5%) | ST10(3) | [24] | |
| China | Farm | 82/12 (14.6%) | ST10(10), ST14(2) | [23] | |
| China | Zoo | 11/1 (9.1%) | ST1(1) | [4] | |
| Spotted deer (Axis axis) | Bangladesh | Zoo | 30/1 (3.3%) | ST14(1) | [34] |
| White-lipped deer (Cervus albirostris) | China | Captive | 1/1 (100%) | ST10(1) | [24] |
| White tailed-deer (Odocoileus virginianus) | United States | Wild | 80/71 (88.8) | ST1(1), ST3(2), ST4(3), ST10(51), ST14(30), ST21(14), ST23(14), ST24(55), ST25(1), ST26(2), ST30(11), ST31(19) d | This study |
| No. of WTD | No. Blastocystis Positives (%) | Subtypes Identified | Subtypes Combinations Observed in Individual Samples | ||
|---|---|---|---|---|---|
| Age group a | Fawn | 3 | 3 (100) | ST10, ST14, ST21, ST23, ST24, ST30 | ST10/ST14/ST21(1); ST14/ST21/ST23(1); ST10/ST14/ST21/ST23/ST24/ST30(1) |
| Yearling | 22 | 19 (86.4) | ST3, ST10, ST14, ST23, ST24, ST26, ST30, ST31 | ST3/ST31(1); ST3/ST10/ST14/ST24(1); ST10/ST14/ST24(2); ST10/ST24(4); ST10/ST24/ST26(1); ST10/ST23/ST24(1); ST10/ST30(1); ST10/ST31(2); ST10/ST24/ST31(2); ST14/ST24(2); ST24(1); ST24/ST31(1) | |
| Adult | 54 | 48 (88.9) | ST1, ST4, ST10, ST14, ST21, ST23, ST24, ST25, ST26, ST30, ST31 | ST1/ST10/ST21/ST24(1); ST4/ST24(1); ST4/ST24/ST31(1); ST4/ST10/ST14/ST21/ST24/ST25/ST26(1); ST10/ST14(2); ST10/ST14/ST23/ST24(1); ST10/ST14/ST24(5); ST10/ST14/ST31 (1); ST10/ST14/ST21/ST24/ST30(3); ST10/ST14/ST21/ST23/ST24(2); ST10/ST14/ST21/ST23/ST24/ST30(2); ST10/ST14/ST23/ST24/ST30(2); ST10/ST14/ST24/ST31(1); ST10/ST31(2); ST10/ST21/ST24(2); ST10/ST23(2); ST10/ST23/ST24(1); ST10/ST24(4); ST10/ST24/ST30(1); ST10/ST24/ST31(1); ST14(1); ST14/ST24(1); ST24(3); ST24/ST30(1); ST24/ST31(4); ST31(2) | |
| Gender a | Males | 36 | 31 (86.1) | ST3, ST4, ST10, ST14, ST21, ST23, ST24, ST25, ST26, ST30, ST31 | ST3/ST31(1); ST3/ST10/ST14/ST24(1); ST4/ST24(1); ST4/ST24/ST31(1); ST4/ST10/ST14/ST21/ST24/ST25/ST26(1); ST10/ST14/ST23/ST24(2); ST10/ST14/ST24(4); ST10/ST14/ST21/ST24/ST30(1); ST10/ST14/ST21/ST23/ST24(1); ST10/ST14/ST23/ST24/ST30(1); ST10/ST14/ST24/ST31(1); ST14/ST21/ST23(1); ST10/ST30(1); ST10/ST31(1); ST10/ST23(1); ST10/ST24(3); ST10/ST24/ST26(1); ST10/ST24/ST31(2); ST14/ST24(2); ST24(2); ST24/ST30(1); ST24/ST31(2) |
| Females | 43 | 39 (90.7) | ST1, ST10, ST14, ST21, ST23, ST24, ST30, ST31 | ST1/ST10/ST21/ST24(1); ST10/ST14(2); ST10/ST14/ST21(1); ST10/ST14/ST24(3); ST10/ST14/ST31 (1); ST10/ST14/ST21/ST24/ST30(2); ST10/ST14/ST21/ST23/ST24(1); ST10/ST14/ST21/ST23/ST24/ST30(3); ST10/ST14/ST23/ST24/ST30(1); ST10/ST31(3); ST10/ST21/ST24(2); ST10/ST23(1); ST10/ST23/ST24(2); ST10/ST24(5); ST10/ST24/ST30(1); ST10/ST24/ST31(1); ST14(1); ST14/ST24(1); ST24(2); ST24/ST31(3); ST31(2) | |
| Total | 80 | 71 (88.8) | ST1, ST3, ST4, ST10, ST14, ST21, ST23, ST24, ST25, ST26, ST30, and ST31 | ST1/ST10/ST21/ST24(1); ST3/ST10/ST14/ST24(1); ST3/ST31(1); ST4/ST24(1); ST4/ST24/ST31(1); ST4/ST10/ST14/ST21/ST24/ST25/ST26(1); ST10/ST14(2); ST10/ST14/ST23/ST24(1); ST10/ST14/ST24(7); ST10/ST14/ST31 (1); ST10/ST23/ST24/ST31(1); ST10/ST14/ST21(1); ST10/ST14/ST21/ST23/ST24(2); ST10/ST14/ST21/ST24/ST30(3); ST10/ST14/ST21/ST23/ST24/ST30(3); ST10/ST14/ST23/ST24/ST30(2); ST10/ST14/ST24/ST31(1); ST10/ST30(1); ST10/ST31(4); ST14/ST21/ST23(1);ST10/ST21/ST24(2); ST10/ST23(2); ST10/ST23/ST24(2); ST10/ST24(8); ST10/ST24/ST26(1); ST10/ST24/ST30(1); ST10/ST24/ST31(3); ST14(1); ST14/ST24(3); ST24(4); ST24/ST30(1); ST24/ST31(5); ST31(2) | |
| Blastocystis Subtype | No. of Blastocystis-Positive Samples | Percentage of Positive WTD | No. of Unique Sequences within Subtype |
|---|---|---|---|
| ST1 | 1 | 1.4 | 2 |
| ST3 | 2 | 2.8 | 2 |
| ST4 | 3 | 4.2 | 2 |
| ST10 | 51 | 71.8 | 14 |
| ST14 | 30 | 42.3 | 3 |
| ST21 | 14 | 19.7 | 3 |
| ST23 | 14 | 19.7 | 1 |
| ST24 | 55 | 77.5 | 3 |
| ST25 | 1 | 1.4 | 1 |
| ST26 | 2 | 2.8 | 3 |
| ST30 | 11 | 15.5 | 3 |
| ST31 | 19 | 26.8 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, J.G.; Jang, Y.; Molokin, A.; George, N.S.; Santin, M. Wide Genetic Diversity of Blastocystis in White-Tailed Deer (Odocoileus virginianus) from Maryland, USA. Microorganisms 2021, 9, 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061343
Maloney JG, Jang Y, Molokin A, George NS, Santin M. Wide Genetic Diversity of Blastocystis in White-Tailed Deer (Odocoileus virginianus) from Maryland, USA. Microorganisms. 2021; 9(6):1343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061343
Chicago/Turabian StyleMaloney, Jenny G., Yunah Jang, Aleksey Molokin, Nadja S. George, and Monica Santin. 2021. "Wide Genetic Diversity of Blastocystis in White-Tailed Deer (Odocoileus virginianus) from Maryland, USA" Microorganisms 9, no. 6: 1343. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9061343