
Genomic Analysis of Prophages Recovered from Listeria monocytogenes Lysogens Found in Seafood and Seafood-Related Environment




Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Representative L. monocytogenes Lysogens for Genome Sequencing
2.2. DNA Extraction and Library Preparation for Sequencing
2.3. Genome Assembly and Annotation of L. monocytogenes Lysogens
2.4. Identification and Annotation of Prophages and Prophage-Related Regions in the Newly Sequenced Lysogenic L. monocytogenes Genomes
2.5. Variation of Prophages Inserted in Multiple Loci of L. monocytogenes Genomes
2.6. Comparison of Prophages Present in the Lysogenic Genomes and the Corresponding Induced Phages
2.7. Comparative Genomic Analysis of the Newly Sequenced Listeria Phages and the Previously Sequenced Temperate Listeria Phages
2.8. Nucleotide Sequence Accession Numbers
3. Results
3.1. Draft Genomes of Lysogenic L. monocytogenes
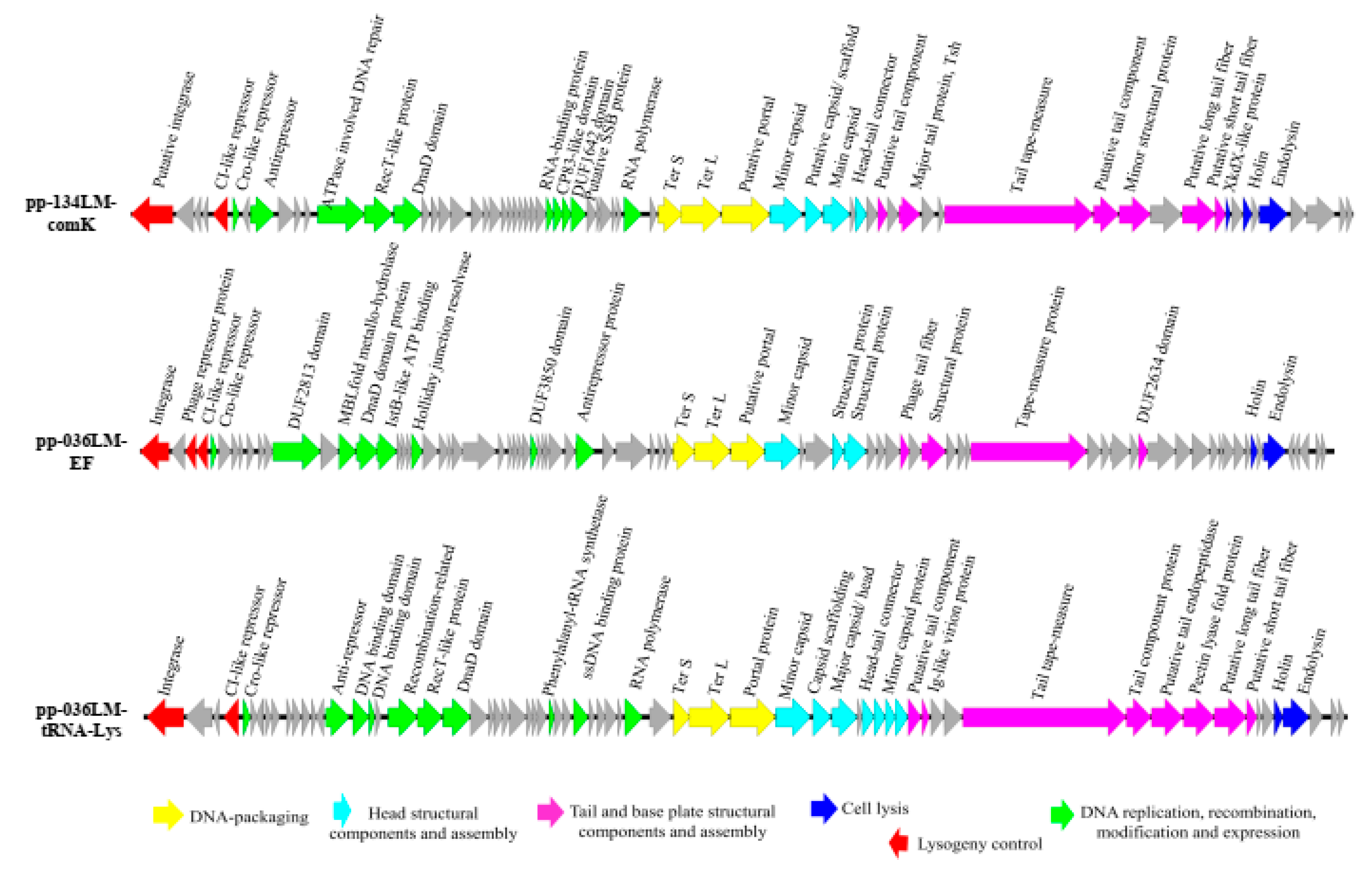
3.2. Identification and Annotation of Prophages and Prophage-Related Regions in the Genomes of Lysogenic L. monocytogenes Newly Sequenced
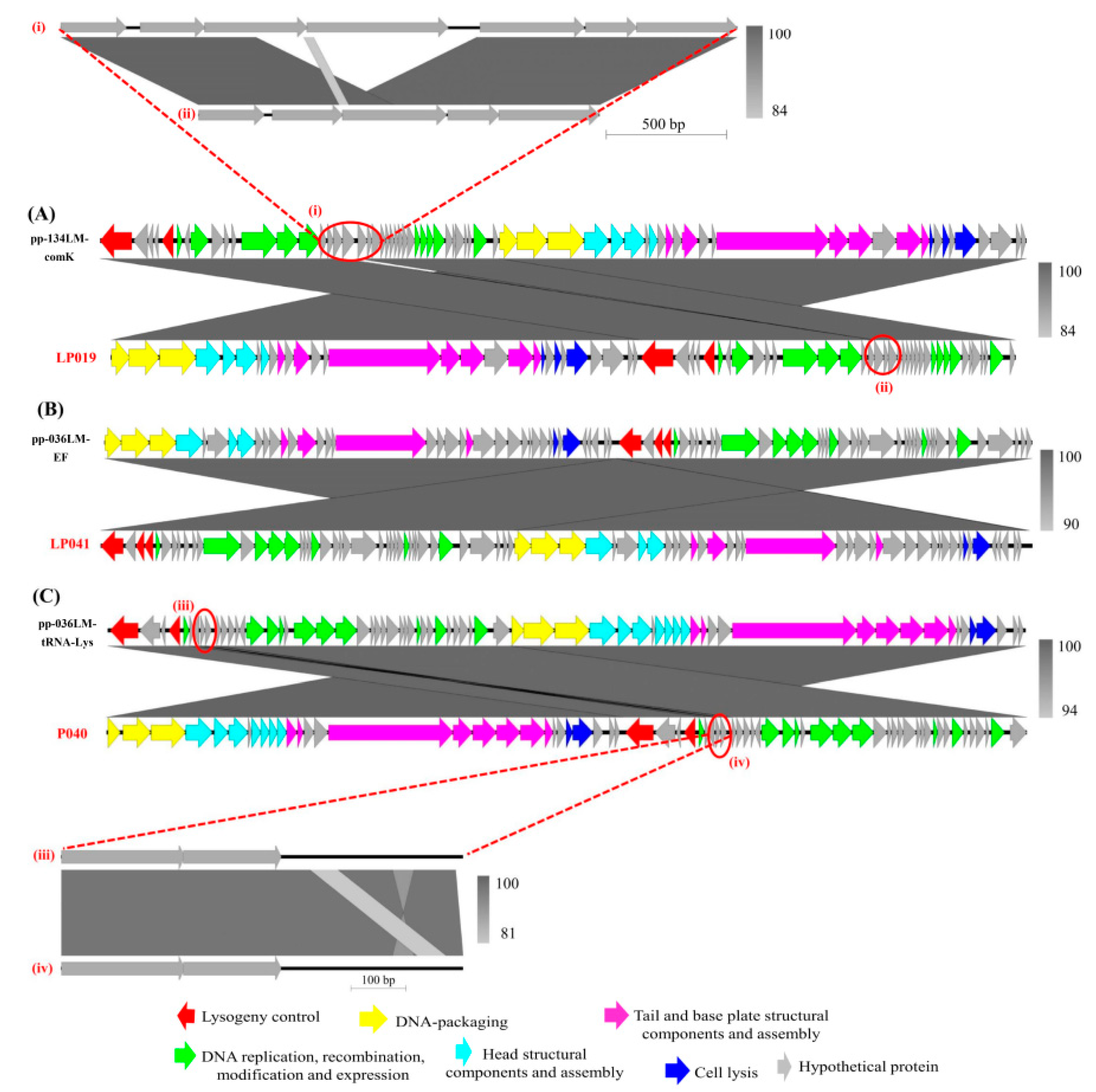
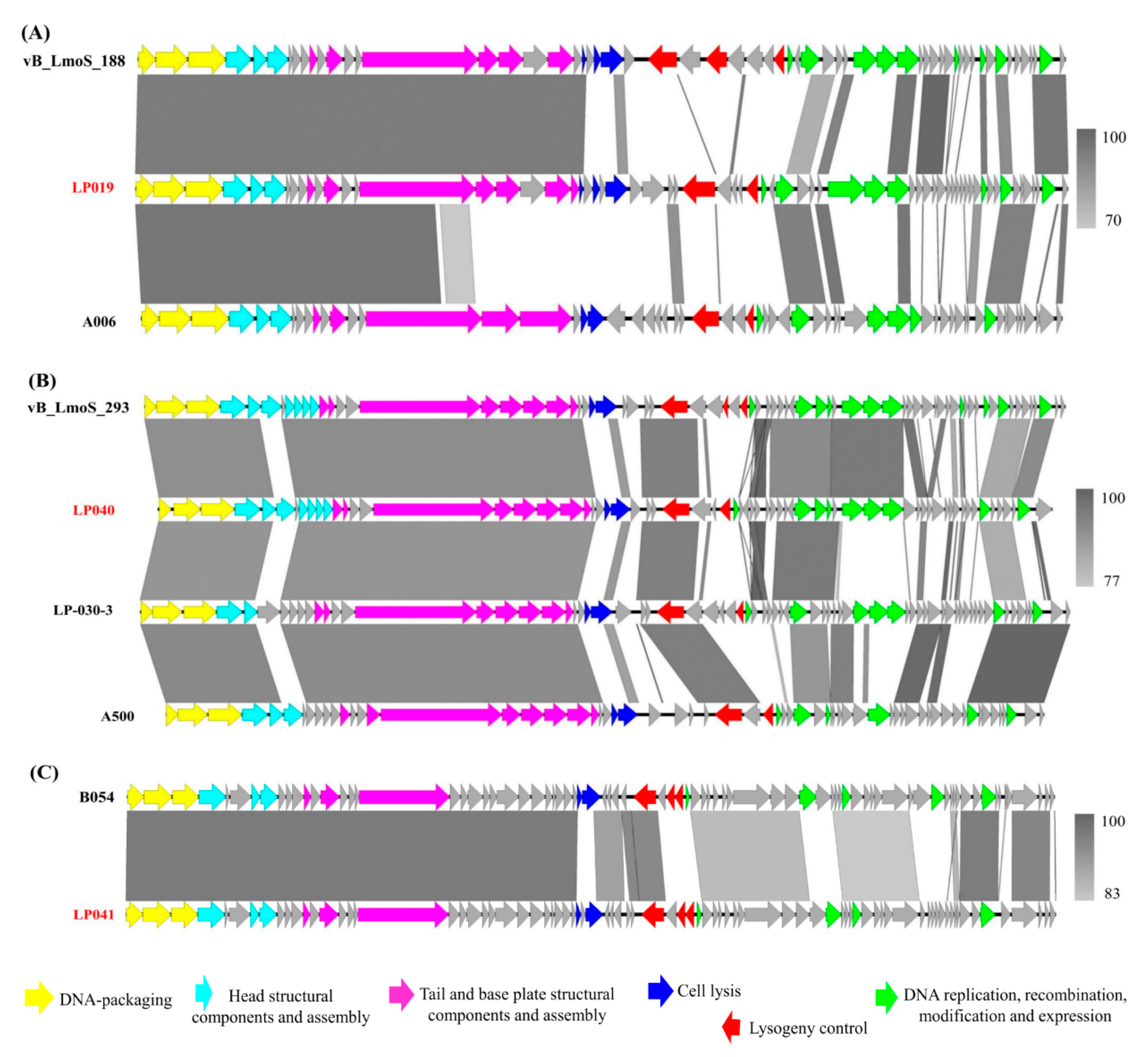
3.3. Sequence Comparison of Prophages and Their Corresponding Induced Phages
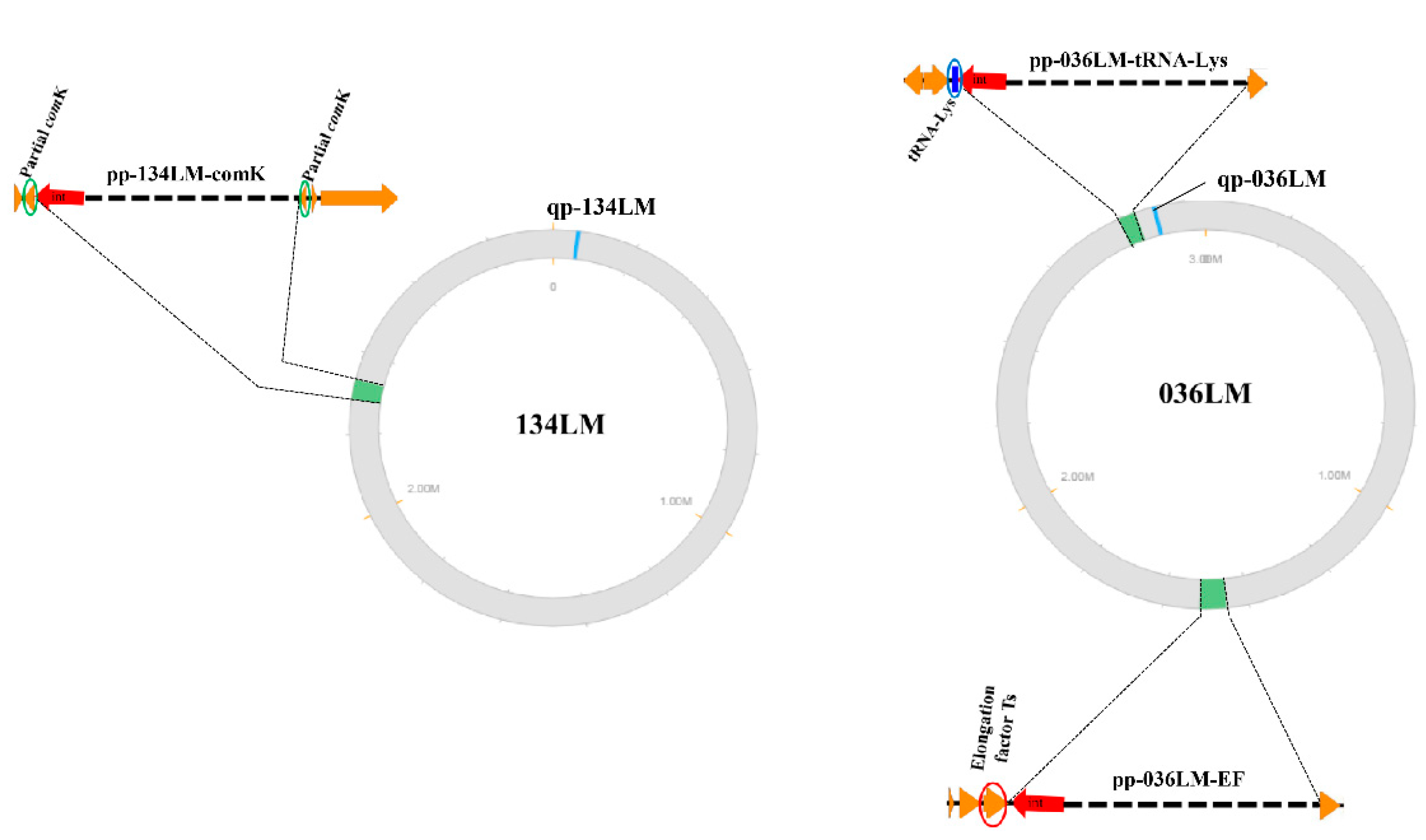
3.4. Prophages Inserted in Multiple Loci of L. monocytogenes Host Genomes
3.5. Variation of Prophage Sequences Found in L. monocytogenes Genomes
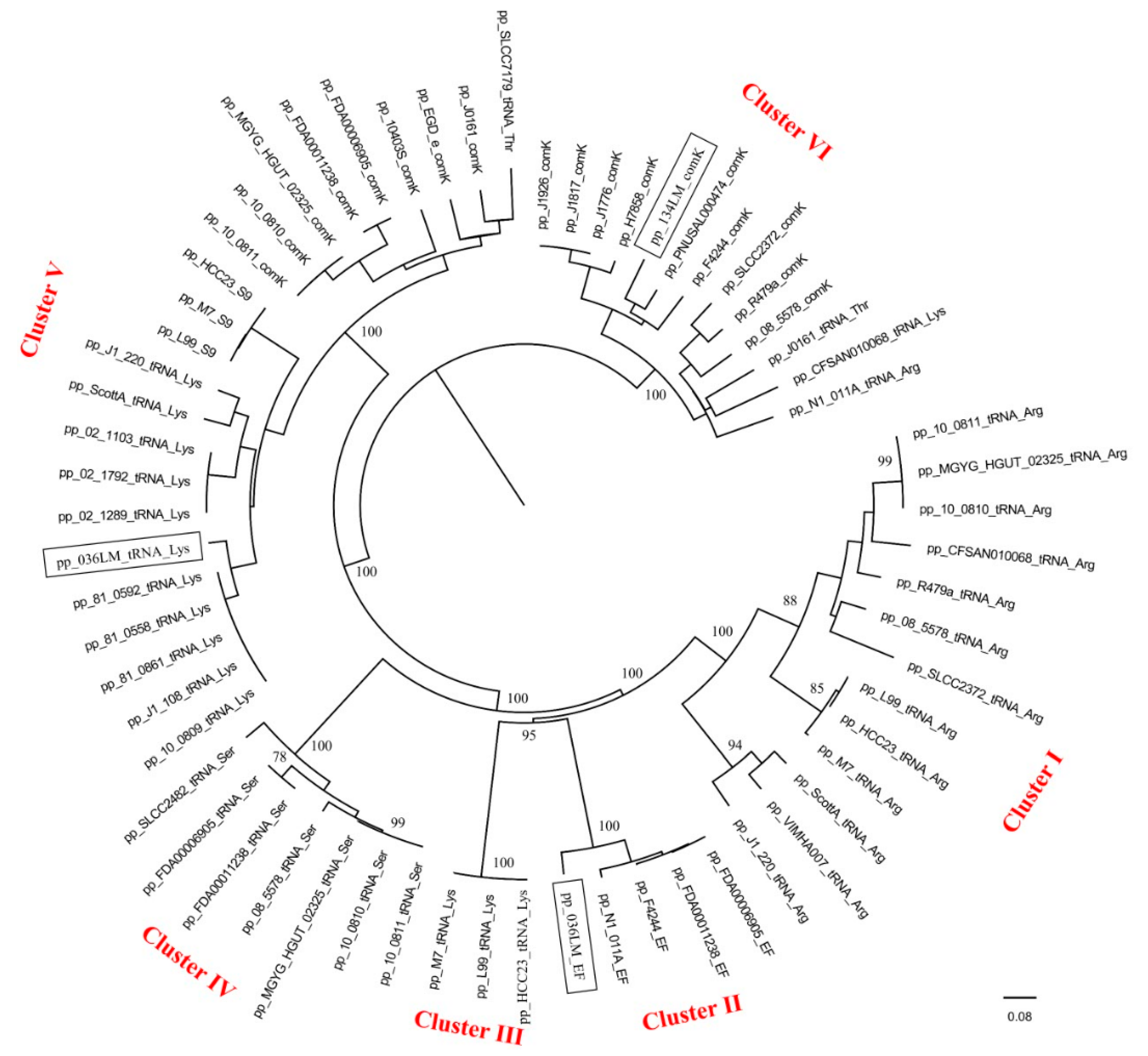
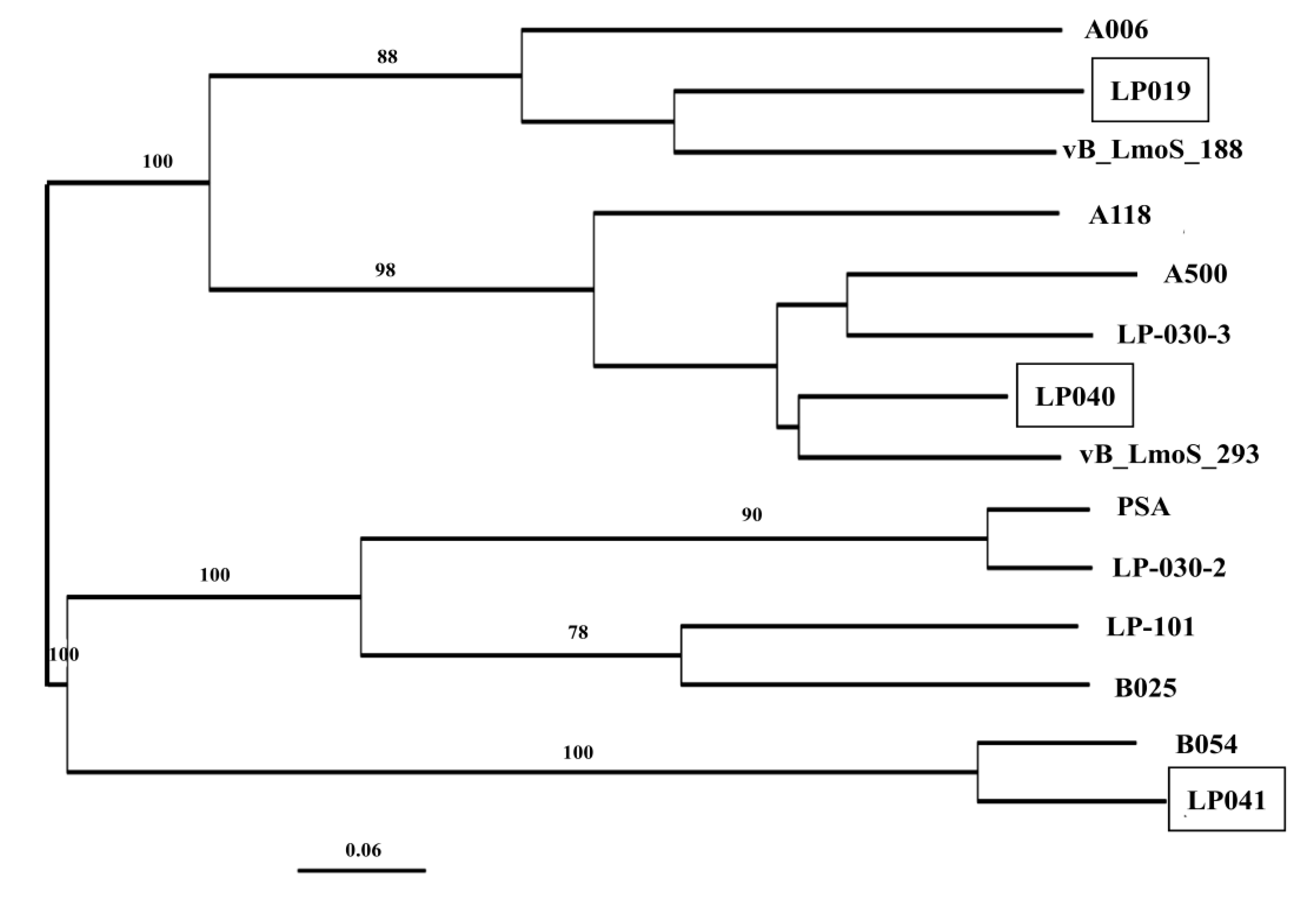
3.6. Comparative Analysis of the Newly Sequenced Induced Phages and the Temperate Listeria Phages from Other Regions
4. Discussion
4.1. Prophages and Prophage-Related Regions Contribute to the Diversity of L. monocytogenes Genomes
4.2. Change in the Genome Organization between Prophage and the Corresponding Induced Listeria Phage
4.3. Uniqueness of Induced Phages and Temperate Listeria Phages from Various Sources and Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Swaminathan, B.; Gerner-Smidt, P. The epidemiology of human listeriosis. Microbes Infect. 2007, 9, 1236–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human Listeriosis. Available online: http://www.who.int/iris/handle/10665/62689 (accessed on 10 June 2016).
- CDC Foodborne Outbreaks. Available online: http://www.cdc.gov/foodsafety/outbreaks/index.html (accessed on 12 December 2014).
- CDC Foodborne Outbreaks. Available online: http://www.cdc.gov/listeria/outbreaks/index.html (accessed on 20 December 2015).
- Tolvanen, R.; Hellström, S.; Elsser, D.; Morgenstern, H.; Björkroth, J.; Korkeala, H. Survival of Listeria monocytogenes strains in a dry sausage model. J. Food Prot. 2008, 71, 1550–1555. [Google Scholar] [CrossRef]
- Burgess, C.M.; Gianotti, A.; Gruzdev, N.; Holah, J.; Knøchel, S.; Lehner, A.; Margas, E.; Esser, S.S.; Sela (Saldinger), S.; Tresse, O. The response of foodborne pathogens to osmotic and desiccation stresses in the food chain. Int. J. Food Microbiol. 2016, 221, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.A.; Williams, A.C. Resistance of Listeria monocytogenes to freezing in foods. Food Microbiol. 1991, 8, 63–68. [Google Scholar] [CrossRef]
- Tompkin, R.B. Control of Listeria monocytogenes in the food-processing environment. J. Food Prot. 2002, 65, 709–725. [Google Scholar] [CrossRef]
- den Bakker, H.C.; Desjardins, C.A.; Griggs, A.D.; Peters, J.E.; Zeng, Q.; Young, S.K.; Kodira, C.D.; Yandava, C.; Hepburn, T.A.; Haas, B.J.; et al. Evolutionary dynamics of the accessory genome of Listeria monocytogenes. PLoS ONE 2013, 8, e67511. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, V.; Barbosa, J.; Stasiewicz, M.; Vongkamjan, K.; Moreno Switt, A.; Hogg, T.; Gibbs, P.; Teixeira, P.; Wiedmann, M. Diverse geno- and phenotypes of persistent Listeria monocytogenes isolates from fermented meat sausage production facilities in Portugal. Appl. Environ. Microbiol. 2011, 77, 2701–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verghese, B.; Lok, M.; Wen, J.; Alessandria, V.; Chen, Y.; Kathariou, S.; Knabel, S. comK prophage junction fragments as markers for Listeria monocytogenes genotypes unique to individual meat and poultry processing plants and a model for rapid niche-specific adaptation, biofilm formation, and persistence. Appl. Environ. Microbiol. 2011, 77, 3279–3292. [Google Scholar] [CrossRef] [Green Version]
- Orsi, R.H.; Borowsky, M.L.; Lauer, P.; Young, S.K.; Nusbaum, C.; Galagan, J.E.; Birren, B.W.; Ivy, R.A.; Sun, Q.; Graves, L.M.; et al. Short-term genome evolution of Listeria monocytogenes in a non-controlled environment. BMC Genom. 2008, 9, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genom. 2013, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.E.; Fouts, D.E.; Mongodin, E.F.; Ravel, J.; DeBoy, R.T.; Kolonay, J.F.; Rasko, D.A.; Angiuoli, S.V.; Gill, S.R.; Paulsen, I.T.; et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004, 32, 2386–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Luo, Y.; Curry, P.; Timme, R.; Melka, D.; Doyle, M.; Parish, M.; Hammack, T.S.; Allard, M.W.; Brown, E.W.; et al. Assessing the genome level diversity of Listeria monocytogenes from contaminated ice cream and environmental samples linked to a listeriosis outbreak in the United States. PLoS ONE 2017, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.; Bru, H. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, M.; Sattelberger, E.; Inman, R.B.; Calendar, R.; Loessner, M.J. Genome and proteome of Listeria monocytogenes phage PSA: An unusual case for programmed + 1 translational frameshifting in structural protein synthesis. Mol. Microbiol. 2003, 50, 303–317. [Google Scholar] [CrossRef]
- Loessner, M.J.; Inman, R.B.; Lauer, P.; Calendar, R. Complete nucleotide sequence, molecular analysis and genome structure of bacteriophage A118 of Listeria monocytogenes: Implications for phage evolution. Mol. Microbiol. 2000, 35, 324–340. [Google Scholar] [CrossRef]
- Dorscht, J.; Klumpp, J.; Bielmann, R.; Schmelcher, M.; Born, Y.; Zimmer, M.; Calendar, R.; Loessner, M.J. Comparative genome analysis of Listeria bacteriophages reveals extensive mosaicism, programmed translational frameshifting, and a novel prophage insertion site. J. Bacteriol. 2009, 191, 7206–7215. [Google Scholar] [CrossRef] [Green Version]
- Denes, T.; Vongkamjan, K.; Ackermann, H.W.; Moreno Switt, A.I.; Wiedmann, M.; den Bakker, H.C. Comparative genomic and morphological analyses of Listeria phages isolated from farm environments. Appl. Environ. Microbiol. 2014, 80, 4616–4625. [Google Scholar] [CrossRef] [Green Version]
- Casey, A.; Jordan, K.; Coffey, A.; McAuliffe, O. Complete genome sequences of vB_LmoS_188 and vB_LmoS_293, two bacteriophages with specificity for Listeria monocytogenes strains of serotypes 4b and 4e. Genome Announc. 2015, 3, e00040-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, H.T.K.; Stasiewicz, M.J.; Benjakul, S.; Vongkamjan, K. Genome sequences of Listeria phages induced from lysogenic isolates of Listeria monocytogenes from seafood and a seafood processing environment in Thailand. Genome Announc. 2018, 6, e00546-18. [Google Scholar] [CrossRef] [Green Version]
- Vongkamjan, K.; Fuangpaiboon, J.; Turner, M.P.; Vuddhakul, V. Various ready-to-eat products from retail stores linked to occurrence of diverse Listeria monocytogenes and Listeria spp. isolates. J. Food Prot. 2016, 79, 239–245. [Google Scholar] [CrossRef]
- Vongkamjan, K.; Fuangpaiboon, J.; Jirachotrapee, S.; Turner, M.P. Occurrence and diversity of Listeria spp. in seafood processing plant environments. Food Control 2015, 50, 265–272. [Google Scholar] [CrossRef]
- Vu, H.T.K.; Benjakul, S.; Vongkamjan, K. Characterization of Listeria prophages in lysogenic isolates from foods and food processing environments. PLoS ONE 2019, 14, e0214641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Jolley, K.A.; Maiden, M.C.J. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [Green Version]
- Pightling, A.W.; Petronella, N.; Pagotto, F. The Listeria monocytogenes Core-Genome Sequence Typer (LmCGST): A bioinformatic pipeline for molecular characterization with next-generation sequence data. BMC Microbiol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Hyden, P.; Pietzka, A.; Lennkh, A.; Murer, A.; Springer, B.; Blaschitz, M.; Indra, A.; Huhulescu, S.; Allerberger, F.; Ruppitsch, W.; et al. Whole genome sequence-based serogrouping of Listeria monocytogenes isolates. J. Biotechnol. 2016, 235, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Monnier, A.L.; Brisse, S. A new perspective on Listeria monocytogenes evolution. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef] [Green Version]
- Chenal-francisque, V.; Lopez, J.; Cantinelli, T.; Caro, V.; Tran, C.; Leclercq, A.; Lecuit, M.; Brisse, S. Worldwide distribution of major clones of Listeria monocytogenes. Emerg. Infect. Dis. 2011, 17, 1110–1112. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, A.; Zhu, R.; Lan, R.; Jin, D.; Cui, Z.; Wang, Y.; Li, Z.; Wang, Y.; Xu, J.; et al. Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 2012, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Esser, S.; Müller, A.; Stessl, B.; Wagner, M. Genomes of sequence type 121 Listeria monocytogenes strains harbor highly conserved plasmids and prophages. Front. Microbiol. 2015, 6, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FigTree 1.4.3—A Graphical Viewer of Phylogenetic Trees and a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 June 2018).
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2014, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, W.; Knabel, S.J. Multi-virulence-locus sequence typing identifies single nucleotide polymorphisms which differentiate epidemic clones and outbreak strains of Listeria monocytogenes. J. Clin. Microbiol. 2007, 45, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Kruczkiewicz, P.A. Comparative Genomic Framework for the In Silico Design and Assessment of Molecular Typing Methods Using Whole-Genome Sequence Data with Application to Listeria Monocytogenes. Master’s of Science Thesis, University of Lethbridge, Lethbridge, AB, Canada, 2013. [Google Scholar]
- Reimer, A.; Weedmark, K.; Petkau, A.; Peterson, C.-L.; Walker, M.; Knox, N.; Kent, H.; Mabon, P.; Berry, C.; Tyler, S.; et al. Shared genome analyses of notable listeriosis outbreaks, highlighting the critical importance of epidemiological evidence, input datasets and interpretation criteria. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J. The distribution, diversity and functional characterization of the Listeria genomic island 1. Master’s of Science Thesis, University of Manitoba, Winnipeg, MB, Canada, 2011. [Google Scholar]
- Briers, Y.; Klumpp, J.; Schuppler, M.; Loessner, M.J. Genome sequence of Listeria monocytogenes Scott A, a clinical isolate from a food-borne listeriosis outbreak. J. Bacteriol. 2011, 193, 4284–4285. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Luo, Y.; Carleton, H.; Timme, R.; Melka, D.; Muruvanda, T.; Wang, C.; Kastanis, G.; Katz, L.S.; Turner, L.; et al. Whole genome and core genome multilocus sequence typing and single nucleotide polymorphism analyses of Listeria monocytogenes associated with an outbreak linked to cheese, United States, 2013. Appl. Environ. Microbiol. 2017, 83, e00633-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherifi, T.; Carrillo, C.; Lambert, D.; Miniaï, I.; Quessy, S.; Larivière-Gauthier, G.; Blais, B.; Fravalo, P. Genomic characterization of Listeria monocytogenes isolates reveals that their persistence in a pig slaughterhouse is linked to the presence of benzalkonium chloride resistance genes. BMC Microbiol. 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Matle, I.; Pierneef, R.; Mbatha, K.R.; Magwedere, K.; Madoroba, E. Genomic diversity of common sequence types of Listeria monocytogenes isolated from ready-to-eat products of animal origin in South Africa. Genes (Basel) 2019, 10, 1007. [Google Scholar] [CrossRef] [Green Version]
- Burall, L.S.; Grim, C.J.; Mammel, M.K.; Datta, A.R. Whole genome sequence analysis using JSpecies tool establishes clonal relationships between Listeria monocytogenes strains from epidemiologically unrelated listeriosis outbreaks. PLoS ONE 2016, 11, e0150797. [Google Scholar] [CrossRef]
- Schmitz-Esser, S.; Gram, L.; Wagner, M. Complete genome sequence of the persistent Listeria monocytogenes strain R479a. Genome Announc. 2015, 3, e00150-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, P.; Frangeul, L.; Buchrieser, C.; Rusniok, C.; Amend, A.; Baquero, F.; Berche, P.; Bloecker, H.; Brandt, P.; Chakraborty, T.; et al. Comparative genomics of Listeria species. Science 2001, 294, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Hain, T.; Ghai, R.; Billion, A.; Kuenne, C.; Steinweg, C.; Izar, B.; Mohamed, W.; Mraheil, M.; Domann, E.; Schaffrath, S.; et al. Comparative genomics and transcriptomics of lineages I, II, and III strains of Listeria monocytogenes. BMC Genom. 2012, 13, 144. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.L.; Donaldson, J.R.; Paul, D.; Banes, M.M.; Arick, T.; Bridges, S.M.; Lawrence, M.L. Genome sequence of lineage III Listeria monocytogenes strain HCC23. J. Bacteriol. 2011, 193, 3679–3680. [Google Scholar] [CrossRef]
- Chen, J.; Xia, Y.; Cheng, C.; Fang, C.; Shan, Y.; Jin, G.; Fang, W. Genome sequence of the nonpathogenic Listeria monocytogenes serovar 4a strain M7. J. Bacteriol. 2011, 193, 5019–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.W.; do Nascimento, N.C.; Bhunia, A.K. Genome sequence of Listeria monocytogenes strain F4244, a 4b serotype. Genome Announc. 2017, 5, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmour, M.W.; Graham, M.; Van Domselaar, G.; Tyler, S.; Kent, H.; Trout-Yakel, K.M.; Larios, O.; Allen, V.; Lee, B.; Nadon, C. High-throughput genome sequencing of two Listreia monocytogenes clinical isolates during a large foodborne outbreak. BMC Genom. 2010, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Zink, R.; Loessner, M.J.; Scherer, S. Characterization of cryptic prophages (monocins) in Listeria and sequence analysis of a holin/endolysin gene. Microbiology 1995, 141, 2577–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychli, K.; Wagner, E.M.; Ciolacu, L.; Zaiser, A.; Tasara, T.; Wagner, M.; Schmitz-Esser, S. Comparative genomics of human and non-human Listeria monocytogenes sequence type 121 strains. PLoS ONE 2017, 12, e0176857. [Google Scholar] [CrossRef]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Fox, E.M.; Allnutt, T.; Bradbury, M.I.; Fanning, S.; Chandry, P.S. Comparative genomics of the Listeria monocytogenes ST204 subgroup. Front. Microbiol. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, B. Characterization of the integrase gene and attachment site for the Myxococcus xanthus Bacteriophage Mx9. J. Bacteriol. 2003, 185, 6325–6330. [Google Scholar] [CrossRef] [Green Version]
- Fineran, P.C.; Petty, N.K.; Salmond, G.P.C. Transduction: Host DNA transfer by bacteriophages. In The Encyclopedia of microbiology; Schaechter, M., Ed.; Academic Press: London, UK, 2009; pp. 666–679. ISBN 9780123739445. [Google Scholar]
- Eggers, C.H.; Gray, C.M.; Preisig, A.M.; Glenn, D.M.; Pereira, J.; Ayers, R.W.; Alshahrani, M.; Acabbo, C.; Becker, M.R.; Bruenn, K.N.; et al. Phage-mediated horizontal gene transfer of both prophage and heterologous DNA by ϕBB-1, a bacteriophage of Borrelia burgdorferi. Pathog. Dis. 2016, 74, ftw107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth, A.C.; Calos, M.P. Phage integrases: Biology and applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Gandon, S. Why be temperate: Lessons from bacteriophage λ. Trends Microbiol. 2016, 24, 356–365. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Information of the Lysogen | Length (bp) of Prophage Identified in Each Insertion Site | Reference ** | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain/Isolates ID | Sequence Type | Serogroup | Lineage | CC | Source | Year | Country | Accession No. | comK | tRNA-Lys | tRNA-Arg | tRNA-Ser | tRNA-Thr | EF-Ts | Ribosomal Protein S9 | |
| 134LM | ST1 | IVb * | I | CC1 | Food | 2013 | Thailand | QSWG00000000.1 | 39,532 | This study *** | ||||||
| PNUSAL000474 | ST1 | IVb * | I | CC1 | Unknown | - | USA | JSWS01000001.1 | 38,978 | Unpublished | ||||||
| J1-108 | ST1 | 4b | I | CC1 | Food | 1981 | Canada | CP006596.1 | 39,322 | [49] | ||||||
| 81-0558 | ST1 | 4b | I | CC1 | Human | 1981 | Canada | CP007525.1 | 39,322 | [50] | ||||||
| 81-0861 | ST1 | 4b | I | CC1 | Human | 1981 | Canada | CP006874.1 | 39,322 | [51] | ||||||
| 81-0592 | ST1 | 4b | I | CC1 | Human | 1981 | Canada | CP007526.1 | 39,322 | [52] | ||||||
| 10-0809 | ST1 | 4b | I | CC1 | Human | 1981 | Canada | CP007167.1 | 39,322 | [50] | ||||||
| 02-1792 | ST1 | 4b | I | CC1 | Food | 2002 | Canada | CP007461.1 | 41,248 | [50] | ||||||
| 02-1289 | ST1 | 4b | I | CC1 | Human | 2002 | Canada | CP007460.1 | 41,248 | [50] | ||||||
| 02-1103 | ST1 | 4b | I | CC1 | Human | 2002 | Canada | CP007459.1 | 41,248 | [50] | ||||||
| Scott A | ST1 | 4b | I | CC2 | Human | 1983 | USA | CM001159.1 | 42,003 | 37,502 | [53] | |||||
| 036LM | ST1 | IVb * | I | CC1 | Environment | 2013 | Thailand | QOUY00000000.1 | 39,488 | 48,684 | This study | |||||
| VIMHA007 | ST2 | 4b | I | CC2 | Human | - | Russia | CP018149.1 | 36,576 | Unpublished | ||||||
| J1-220 | ST2 | 4b | I | CC2 | Human | 1979 | USA | CP006046.3 | 42,348 | 38,700 | [49] | |||||
| SLCC2482 | ST3 | 7 | I | CC3 | Human | 1966 | - | FR720325 | 33,409 | [13] | ||||||
| N1-001A | ST3 | IIb * | I | CC3 | Environment | - | USA | CP006597.1 | 38,108 | 47,525 | Unpublished | |||||
| CFSAN010068 | ST5 | IIb * | I | CC5 | Food | 2014 | USA | CP014250.1 | 41,864 | 44,935 | [54] | |||||
| 10-0810 | ST5 | 1/2b | I | CC5 | Human | 1996 | Canada | CP007168.1 | 41,583 | 45,827 | 33,225 | [55] | ||||
| 10-0811 | ST5 | 1/2b | I | CC5 | Human | 1996 | Canada | CP007169.1 | 41,583 | 45,827 | 33,225 | [55] | ||||
| MGYG-HGUT-02325 | ST5 | IIb * | I | CC5 | Human | - | UK | LR698978.1 | 41,583 | 45,827 | 33,225 | Unpublished | ||||
| FDA00006905 | ST5 | IIb * | I | CC5 | Environment | 2011 | USA | CP023052.1 | 40,883 | 32,205 | 47,272 | Unpublished | ||||
| FDA00011238 | ST5 | IIb * | I | CC5 | Environment | 2017 | USA | CP023050.1 | 40,883 | 32,203 | 47,272 | Unpublished | ||||
| J1776 | ST6 | 4b | I | CC6 | Human | 2002 | USA | CP006598 | 39,825 | [56] | ||||||
| J1926 | ST6 | 4b | I | CC6 | Food | 2002 | USA | CP006600 | 39,825 | [57] | ||||||
| J1817 | ST6 | 4b | I | CC6 | Environment | 2002 | USA | CP006599 | 39,810 | [56] | ||||||
| H7858 | ST6 | 4b | I | CC6 | Food | 1998 | USA | AADR00000000 | 40,484 | [14] | ||||||
| R479a | ST8 | 1/2a | II | CC8 | Food | 1996 | Denmark | HG813247.1 | 38,927 | 42,048 | [58] | |||||
| J0161 | ST11 | 1/2a | II | CC11 | Human | 2000 | USA | NC_017545.1 | 41,530 | 38,519 | [12] | |||||
| EGD-e | ST35 | 1/2a | II | CC9 | Animal | 1926 | UK | NC_003210.1 | 41,459 | [59] | ||||||
| 10403S | ST85 | 1/2a | II | CC7 | Human | 1968 | USA | CP002002 | 37,474 | [9] | ||||||
| SLCC7179 | ST91 | 3a | II | CC14 | Food | 1986 | Austria | FR733650 | 38,696 | [13] | ||||||
| SLCC2372 | ST122 | 1/2c | II | CC9 | Human | 1935 | UK | FR733648 | 38,319 | 42,317 | [13] | |||||
| L99 | ST201 | 4a | III | CC69 | Food | 1950 | Netherlands | FM211688.1 | 39,372 | 41,757 | 41,172 | [60] | ||||
| HCC23 | ST201 | 4a | III | CC69 | Animal | 2000 | USA | CP001175.1 | 39,369 | 41,757 | 41,661 | [61] | ||||
| M7 | ST201 | 4a | III | CC69 | Human | 2009 | China | NC_017537 | 39,426 | 43,788 | 41,172 | [62] | ||||
| F4244 | ST1347 | 4b | I | CC6 | Human | 1991 | USA | CP015508.1 | 40,067 | 47,934 | [63] | |||||
| 08-5578 | ST292 | 1/2a | II | CC8 | Human | 2008 | Canada | CP001602 | 39,425 | 44,365 | 36,088 | [64] | ||||
| Temperate Listeria Phage | Genome Size (bp) | NCBI Accession No. | Reference |
|---|---|---|---|
| LP019 | 38,601 | MH341451 | [22] |
| LP040 | 39,585 | MH341452 | [22] |
| LP041 | 48,286 | MH341453 | [22] |
| A118 | 40,834 | NC_003216.1 | [18] |
| A006 | 38,124 | NC_009815.1 | [19] |
| B025 | 42,653 | NC_009812.1 | [19] |
| B054 | 48,172 | NC_009813.1 | [19] |
| A500 | 38,867 | NC_009810.1 | [19] |
| PSA | 37,618 | AJ312240.2 | [17] |
| LP-101 | 43,767 | NC_024387.1 | [20] |
| LP-030-2 | 38,275 | JX120799.2 | [20] |
| LP-030-3 | 41,156 | NC_024384.1 | [20] |
| vB_LmoS_188 | 38,392 | KP399677 | [21] |
| vB_LmoS_293 | 40,759 | KP399678 | [21] |
| Description | PSU-KV-134LM (134LM) | PSU-KV-036LM (036LM) |
|---|---|---|
| No. of reads (pre-filtered) | 27,525,582 | 24,257,104 |
| No. of reads (post filtered) | 21,137,912 | 19,286,526 |
| Assembly size (bp) | 2,953,877 | 3,000,399 |
| Sequencing coverage | 715× | 643× |
| GC content (%) | 37.80 | 37.83 |
| Number of contigs | 11 | 11 |
| Shortest contig size (bp) | 1778 | 3570 |
| Longest contig size (bp) | 766,006 | 1,291,185 |
| N50 value | 476,941 | 556,523 |
| No. of predicted CDSs | 2955 | 3027 |
| No. of unique rRNAs (5S, 16S, 23S) | 2, 1, 1 | 1, 1, 1 |
| No. of tRNAs | 54 | 52 |
| No. of prophage regions | 2 | 3 |
| Length of intact prophage (bp) | 39,532 | 48,684 and 39,488 |
| Length of questionable prophage (bp) | 10,729 | 10,729 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, H.T.K.; Stasiewicz, M.J.; Benjakul, S.; Vongkamjan, K. Genomic Analysis of Prophages Recovered from Listeria monocytogenes Lysogens Found in Seafood and Seafood-Related Environment. Microorganisms 2021, 9, 1354. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071354
Vu HTK, Stasiewicz MJ, Benjakul S, Vongkamjan K. Genomic Analysis of Prophages Recovered from Listeria monocytogenes Lysogens Found in Seafood and Seafood-Related Environment. Microorganisms. 2021; 9(7):1354. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071354
Chicago/Turabian StyleVu, Hue Thi Kim, Matthew J. Stasiewicz, Soottawat Benjakul, and Kitiya Vongkamjan. 2021. "Genomic Analysis of Prophages Recovered from Listeria monocytogenes Lysogens Found in Seafood and Seafood-Related Environment" Microorganisms 9, no. 7: 1354. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071354