
Insights into Autotrophic Activities and Carbon Flow in Iron-Rich Pelagic Aggregates (Iron Snow)

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lake Characteristics and Iron Snow Sampling
2.2. Geochemical Analysis
2.3. Nucleic Acid Extraction of Iron Snow Microbiome
2.4. 16S rRNA Gene Amplicon Sequencing
2.5. Amplicon Sequencing Data Analysis
2.6. Metatranscriptome Sequencing
2.7. Taxonomic Annotation of rRNA-Derived Sequences
2.8. Taxonomic and Functional Profiling of mRNA Sequences
2.9. Taxonomic Annotation of Functional Subsets
2.10. Metatranscriptome Read Recruitment Analysis of Dominant Fe-Cycling Microbes
2.11. Oxic and Anoxic Microcosm Incubation Experiments
2.12. Nucleic Acid and Protein Extraction of Microcosms
2.13. Metaproteomics Analysis: Identification, Taxonomy, Function and 13C Quantification of Peptides from Proteins
2.14. Statistics
2.15. Figure Generation
3. Results
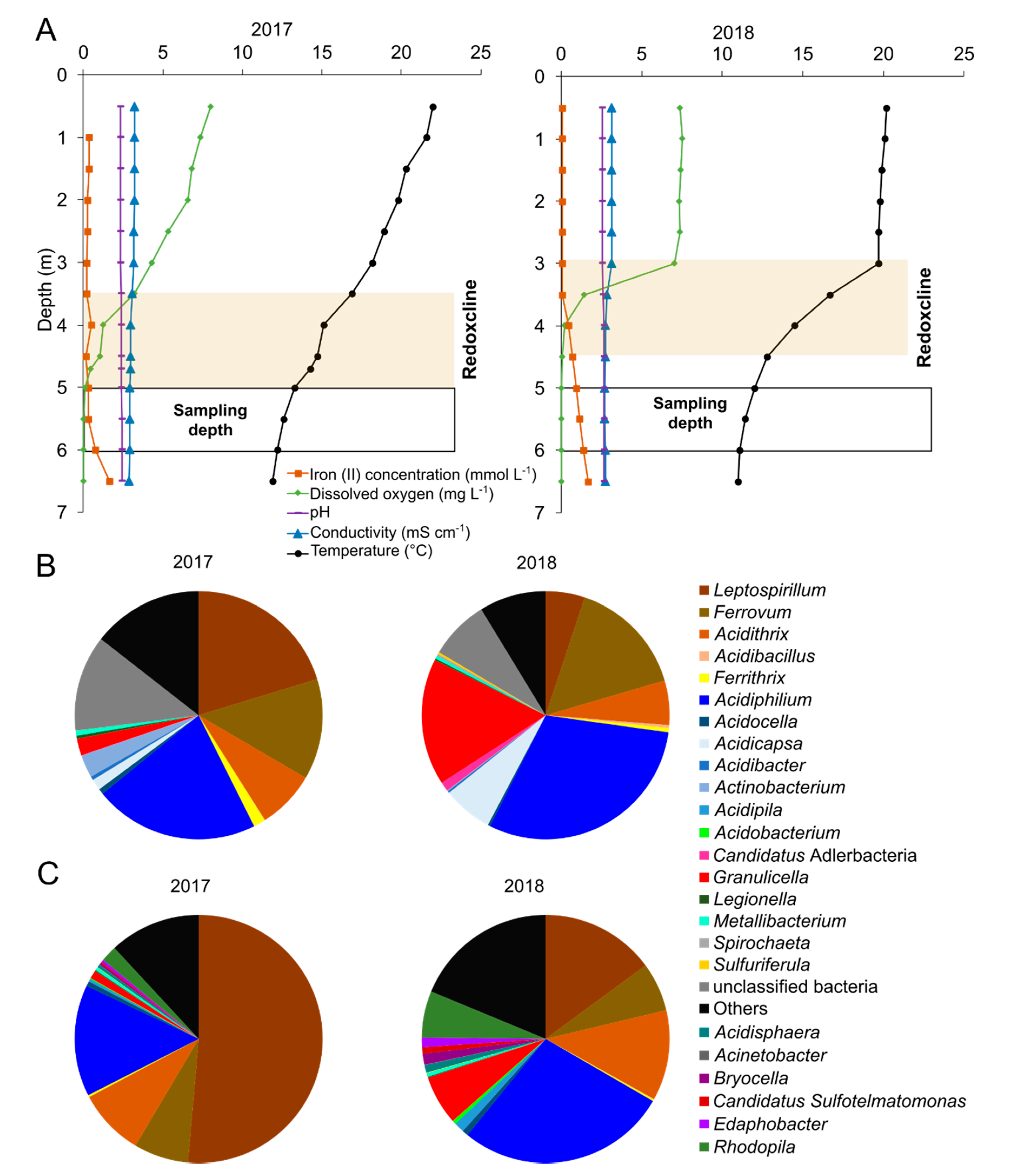
3.1. Physicochemical Characterization and Microbial Community Composition and Diversity of Lake 77
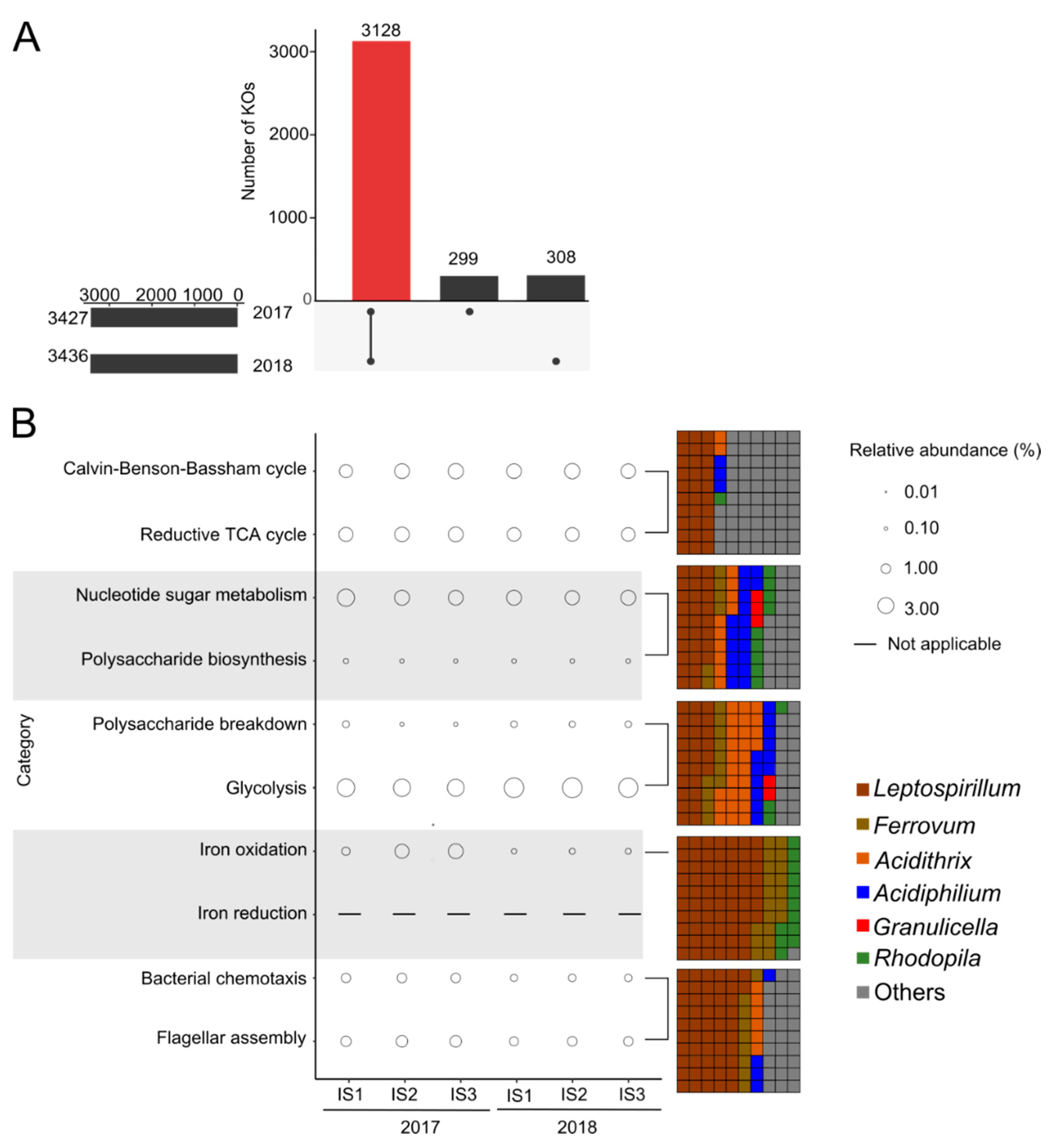
3.2. Gene Expression Analysis of Iron Snow Microbial Communities
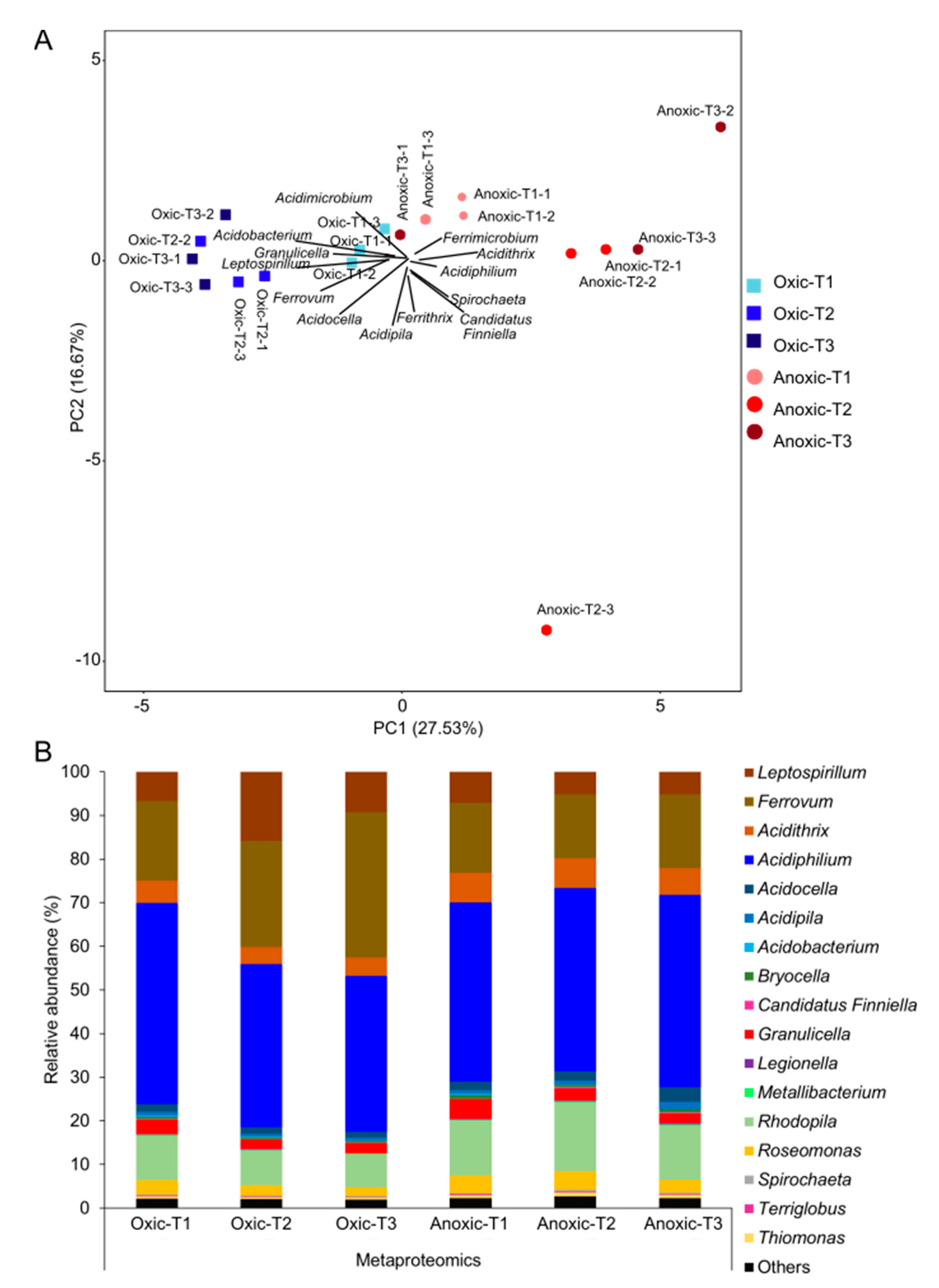
3.3. Taxonomic Profiles of Active FeOB and FeRB Taxa in Oxic and Anoxic Iron Snow Microcosm Incubations
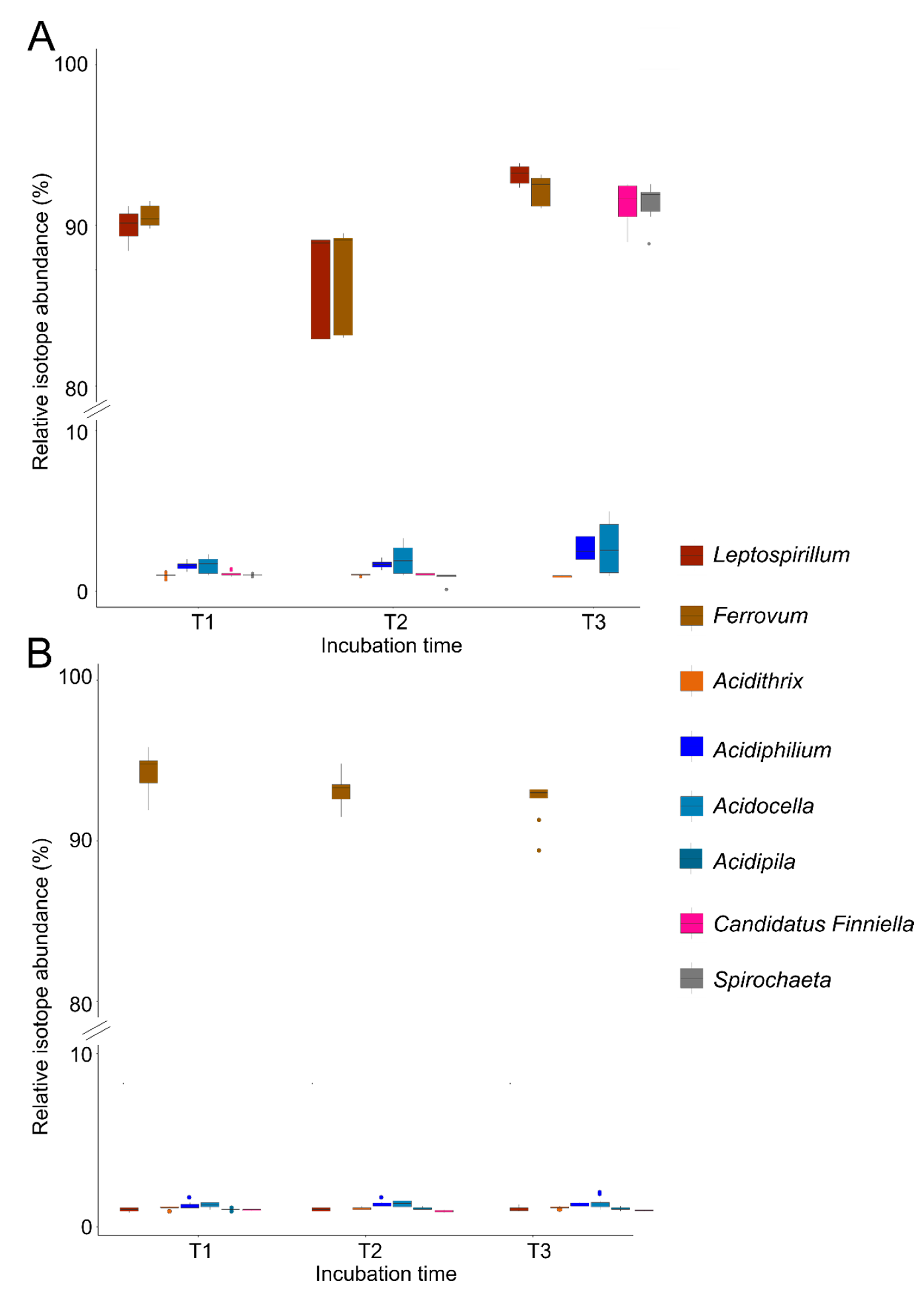
3.4. Carbon Flow of Dominant Iron Cycling Bacteria in Iron Snow Microcosms
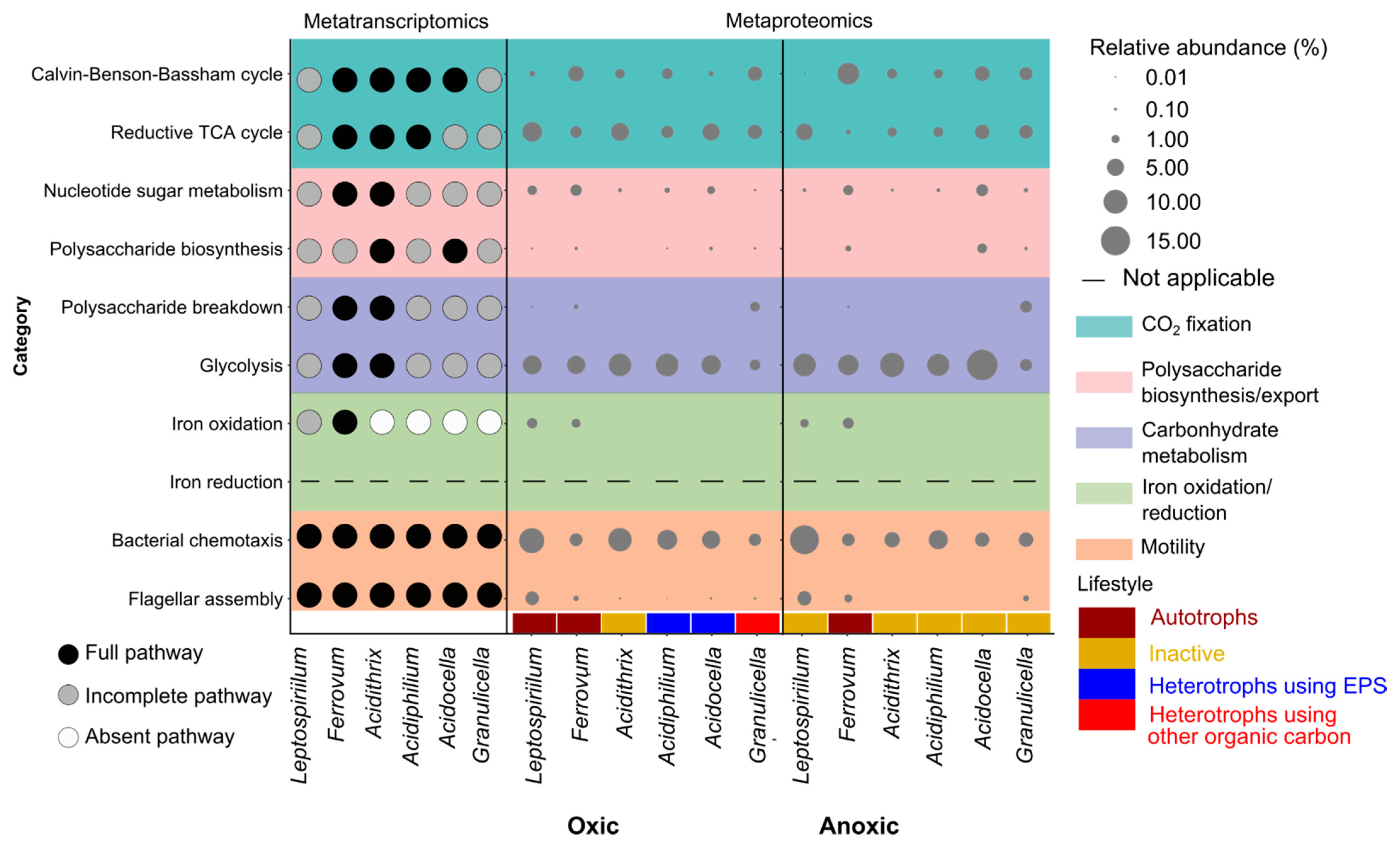
3.5. Metatranscriptomic and Metaproteomic Functional Profile of Dominant Microbes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alldredge, A.L.; Silver, M.W. Characteristics, dynamics and significance of marine snow. Prog. Oceanogr. 1988, 20, 41–82. [Google Scholar] [CrossRef]
- Thornton, D. Diatom aggregation in the sea: Mechanisms and ecological implications. Eur. J. Phycol. 2002, 37, 149–161. [Google Scholar] [CrossRef]
- Simon, M.; Grossart, H.-P.; Schweitzer, B.; Ploug, H. Microbial ecology of organic aggregates in aquatic ecosystems. Aquat. Microb. Ecol. 2002, 28, 175–211. [Google Scholar] [CrossRef] [Green Version]
- Hmelo, L.R.; Mincer, T.J.; Van Mooy, B.A.S. Possible influence of bacterial quorum sensing on the hydrolysis of sinking particulate organic carbon in marine environments. Environ. Microbiol. Rep. 2011, 3, 682–688. [Google Scholar] [CrossRef]
- Passow, U.; Carlson, C. The biological pump in a high CO2 world. Mar. Ecol. Prog. Ser. 2012, 470, 249–271. [Google Scholar] [CrossRef] [Green Version]
- Ploug, H.; Grossart, H.-P.; Azam, F.; Jørgensen, B.B. Photosynthesis, respiration, and carbon turnover in sinking marine snow from surface waters of Southern California Bight: Implications for the carbon cycle in the ocean. Mar. Ecol. Prog. Ser. 1999, 179, 1–11. [Google Scholar] [CrossRef]
- Turner, J.T. Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump. Prog. Oceanogr. 2015, 130, 205–248. [Google Scholar] [CrossRef]
- Reiche, M.; Lu, S.; Ciobotǎ, V.; Neu, T.R.; Nietzsche, S.; Rösch, P.; Popp, J.; Küsel, K. Pelagic boundary conditions affect the biological formation of iron-rich particles (iron snow) and their microbial communities. Limnol. Oceanogr. 2011, 56, 1386–1398. [Google Scholar] [CrossRef]
- Crowe, S.A.; Katsev, S.; Leslie, K.; Sturm, A.; Magen, C.; Nomosatryo, S.; Pack, M.A.; Kessler, J.D.; Reeburgh, W.S.; Roberts, J.A.; et al. The methane cycle in ferruginous Lake Matano. Geobiology 2011, 9, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Posth, N.R.; Huelin, S.; Konhauser, K.O.; Kappler, A. Size, density and composition of cell-mineral aggregates formed during anoxygenic phototrophic Fe(II) oxidation: Impact on modern and ancient environments. Geochim. Cosmochim. Acta 2010, 74, 3476–3493. [Google Scholar] [CrossRef]
- Crowe, S.A.; Jones, C.A.; Katsev, S.; Magen, C.; O’Neill, A.H.; Sturm, A.; Canfield, D.E.; Haffner, G.D.; Mucci, A.; Sundby, B.; et al. Photoferrotrophs thrive in an Archean Ocean analogue. Proc. Natl. Acad. Sci. USA 2008, 105, 15938–15943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, X.A.; Picazo, A.; Miracle, M.R.; Vicente, E.; Camacho, A.; Aragno, M.; Zopfi, J. Phototrophic Fe(II)-oxidation in the chemocline of a ferruginous meromictic lake. Front. Microbiol. 2014, 5, 713. [Google Scholar] [CrossRef] [Green Version]
- Camacho, A.; Walter, X.A.; Picazo, A.; Zopfi, J. Photoferrotrophy: Remains of an ancient photosynthesis in modern environments. Front. Microbiol. 2017, 8, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshaies, M. Metamorphosis of Mining Landscapes in the Lower Lusatian Lignite Basin (Germany): New uses and new image of a mining region. Cah. la Rech. Archit. Urbaine Paysagère 2020, 7, 1–24. [Google Scholar] [CrossRef]
- Geller, W.; Klapper, H.; Schultze, M. Natural and Anthropogenic Sulfuric Acidification of Lakes. In Acidic Mining Lakes: Acid Mine Drainage, Limnology and Reclamation; Geller, W., Klapper, H., Salomons, W., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1998; pp. 3–14. ISBN 978-3-642-71954-7. [Google Scholar]
- Johnson, D.B.; Kanao, T.; Hedrich, S. Redox Transformations of Iron at Extremely Low pH: Fundamental and Applied Aspects. Front. Microbiol. 2012, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixdorf, B.; Krumbeck, H.; Jander, J.; Beulker, C. Comparison of bacterial and phytoplankton productivity in extremely acidic mining lakes and eutrophic hard water lakes. Acta Oecologica 2003, 24, S281–S288. [Google Scholar] [CrossRef]
- Saeed, H.; Hartland, A.; Lehto, N.J.; Baalousha, M.; Sikder, M.; Sandwell, D.; Mucalo, M.; Hamilton, D.P. Regulation of phosphorus bioavailability by iron nanoparticles in a monomictic lake. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Chourey, K.; Reiche, M.; Nietzsche, S.; Shah, M.B.; Neu, T.R.; Hettich, R.L.; Küsel, K. Insights into the structure and metabolic function of microbes that shape pelagic iron-rich aggregates (“Iron snow”). Appl. Environ. Microbiol. 2013, 79, 4272–4281. [Google Scholar] [CrossRef] [Green Version]
- Mori, J.F.; Lu, S.; Händel, M.; Totsche, K.U.; Neu, T.R.; Iancu, V.V.; Tarcea, N.; Popp, J.; Küsel, K. Schwertmannite formation at cell junctions by a new filament-forming Fe(II)-oxidizing isolate affiliated with the novel genus Acidithrix. Microbiology 2016, 162, 62–71. [Google Scholar] [CrossRef]
- Mori, J.F.; Ueberschaar, N.; Lu, S.; Cooper, R.E.; Pohnert, G.; Küsel, K. Sticking together: Inter-species aggregation of bacteria isolated from iron snow is controlled by chemical signaling. ISME J. 2017, 11, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Hallberg, K.B.; Hedrich, S. Uncovering a Microbial Enigma: Isolation and Characterization of the Streamer-Generating, Iron-Oxidizing, Acidophilic Bacterium “Ferrovum myxofaciens”. Appl. Environ. Microbiol. 2014, 80, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, S.R.; Poehlein, A.; Tischler, J.S.; González, C.; Ossandon, F.J.; Daniel, R.; Holmes, D.S.; Schlömann, M.; Mühling, M. Genome analysis of the biotechnologically relevant acidophilic iron oxidising strain JA12 indicates phylogenetic and metabolic diversity within the novel genus “Ferrovum”. PLoS ONE 2016, 11, e0146832. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Goto, K. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III). Talanta 1974, 21, 314–318. [Google Scholar] [CrossRef]
- Tabatabai, M.A. A Rapid Method for Determination of Sulfate in Water Samples. Environ. Lett. 1974, 7, 237–243. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, 1–11. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Saeidipour, B.; Bakhshi, S. Cutadapt Removes Adapter Sequences from High-throughput sequenceing reads. Adv. Environ. Biol. 2013, 7, 2803–2809. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python Fabian. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Finn, R.D.; Bateman, A.; Eddy, S.R.; Petrov, A.I.; Kalvari, I.; Rivas, E. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2017, 46, D335–D342. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLOS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Poux, S.; Arighi, C.N.; Magrane, M.; Bateman, A.; Wei, C.H.; Lu, Z.; Boutet, E.; Bye-A-Jee, H.; Famiglietti, M.L.; Roechert, B.; et al. On expert curation and scalability: UniProtKB/Swiss-Prot as a case study. Bioinformatics 2017, 33, 3454–3460. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Eren, A.M.; Esen, Ö.C.; Sogin, M.L.; Quince, C.; Delmont, T.O.; Morrison, H.G.; Vineis, J.H. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2002, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Taubert, M.; Stöckel, S.; Geesink, P.; Girnus, S.; Jehmlich, N.; von Bergen, M.; Rösch, P.; Popp, J.; Küsel, K. Tracking active groundwater microbes with D2O labelling to understand their ecosystem function. Environ. Microbiol. 2018, 20, 369–384. [Google Scholar] [CrossRef]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Taubert, M.; Vogt, C.; Wubet, T.; Kleinsteuber, S.; Tarkka, M.T.; Harms, H.; Buscot, F.; Richnow, H.H.; Von Bergen, M.; Seifert, J. Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J. 2012, 6, 2291–2301. [Google Scholar] [CrossRef] [Green Version]
- Taubert, M.; Baumann, S.; Von Bergen, M.; Seifert, J. Exploring the limits of robust detection of incorporation of 13C by mass spectrometry in protein-based stable isotope probing (protein-SIP). Anal. Bioanal. Chem. 2011, 401, 1975–1982. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Itoh, S.; Iwaki, M.; Nagashima, K.V.P.; Matsuura, K.; Kobayashi, M.; Wakao, N. Reaction Center Complex Based on Zn-Bacteriochlorophyll from Acidiphilium Rubrum. In Photosynthesis: Mechanisms and Effects; Springer: Dordrecht, The Netherlands, 1998; pp. 909–912. [Google Scholar]
- Tomi, T.; Shibata, Y.; Ikeda, Y.; Taniguchi, S.; Haik, C.; Mataga, N.; Shimada, K.; Itoh, S. Energy and electron transfer in the photosynthetic reaction center complex of Acidiphilium rubrum containing Zn-bacteriochlorophyll a studied by femtosecond up-conversion spectroscopy. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, S.; Suthaus, A.; Melkonian, M. “Candidatus Finniella” (Rickettsiales, Alphaproteobacteria), novel endosymbionts of viridiraptorid amoeboflagellates (Cercozoa, Rhizaria). Appl. Environ. Microbiol. 2016, 82, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leschine, S.; Paster, B.J.; Canale-Parola, E. Free-Living Saccharolytic Spirochetes: The Genus Spirochaeta. In The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer New York: New York, NY, 2006; pp. 195–210. ISBN 978-0-387-30747-3. [Google Scholar]
- Bomberg, M.; Mäkinen, J.; Salo, M.; Kinnunen, P. High Diversity in Iron Cycling Microbial Communities in Acidic, Iron-Rich Water of the Pyhäsalmi Mine, Finland. Geofluids 2019, 2019, 7401304. [Google Scholar] [CrossRef] [Green Version]
- Santofimia, E.; González-Toril, E.; López-Pamo, E.; Gomariz, M.; Amils, R.; Aguilera, Á. Microbial Diversity and Its Relationship to Physicochemical Characteristics of the Water in Two Extreme Acidic Pit Lakes from the Iberian Pyrite Belt (SW Spain). PLoS ONE 2013, 8, e66746. [Google Scholar] [CrossRef] [Green Version]
- Kaltenbock, E.; Herndl, G.J. Ecology of amorphous aggregations (marine snow) in the northern Adriatic Sea. IV. Dissolved nutrients and the autotrophic community associated with marine snow. Mar. Ecol. Prog. Ser. 1992, 87, 147–159. [Google Scholar] [CrossRef]
- Simon, M.; Alldredge, A.; Azam, F. Bacterial carbon dynamics on marine snow. Mar. Ecol. Prog. Ser. 1990, 65, 205–211. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Simon, M.; Logan, B.E. Formation of macroscopic organic aggregates (lake snow) in a large lake: The significance of transparent exopolymer particles, phytoplankton, and Zooplankton. Limnol. Oceanogr. 1997, 42, 1651–1659. [Google Scholar] [CrossRef]
- Imhoff, J.F.; Rahn, T.; Künzel, S.; Neulinger, S.C. New insights into the metabolic potential of the phototrophic purple bacterium Rhodopila globiformis DSM 161T from its draft genome sequence and evidence for a vanadium-dependent nitrogenase. Arch. Microbiol. 2018, 200, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Grossart, H.-P.; Berman, T.; Simon, M.; Pohlmann, K. Occurrence and microbial dynamics of macroscopic organic aggregates (lake snow) in Lake Kinneret, Israel, in fall. Aquat. Microb. Ecol. 1998, 14, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, B.; Huber, I.; Amann, R.; Ludwig, W.; Simon, M. α- and β-Proteobacteria Control the Consumption and Release of Amino Acids on Lake Snow Aggregates. Appl. Environ. Microbiol. 2001, 67, 632–645. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, H. The Order Cytophagales. In The Prokaryotes; Springer New York: New York, NY, 2006; pp. 549–590. [Google Scholar]
- Grossart, H.-P.; Ploug, H. Microbial degradation of organic carbon and nitrogen on diatom aggregates. Limnol. Oceanogr. 2001, 46, 267–277. [Google Scholar] [CrossRef]
- Eigemann, F.; Vogts, A.; Voss, M.; Zoccarato, L.; Schulz-Vogt, H. Distinctive tasks of different cyanobacteria and associated bacteria in carbon as well as nitrogen fixation and cycling in a late stage Baltic Sea bloom. PLoS ONE 2019, 14, e0223294. [Google Scholar] [CrossRef] [Green Version]
- Shanks, A.L.; Reeder, M.L. Reducing microzones and sulfide production in marine snow. Mar. Ecol. Prog. Ser. 1993, 96, 43–47. [Google Scholar] [CrossRef]
- Dash, P.; Kashyap, D.; Mandal, S.C. Marine snow: Its formation and significance in fisheries and aquaculture. World Aquac. 2012, 6, 59–61. [Google Scholar]
- Kiørboe, T.; Hansen, J.L.S. Phytoplankton aggregate formation: Observations of patterns and mechanisms of cell sticking and the significance of exopolymeric material. J. Plankton Res. 1993, 15, 993–1018. [Google Scholar] [CrossRef]
- Stoderegger, K.E.; Herndl, G.J. Production and release of bacterial capsular material and its subsequent utilization by marine bacterioplankton. Limnol. Oceanogr. 1998, 43, 877–884. [Google Scholar] [CrossRef]
- Stoderegger, K.E.; Herndl, G.J. Production of exopolymer particles by marine bacterioplankton under contrasting turbulence conditions. Mar. Ecol. Prog. Ser. 1999, 189, 9–16. [Google Scholar] [CrossRef]
- Olson, J.B.; Steppe, T.F.; Litaker, R.W.; Paer, H.W. N2-Fixing Microbial Consortia Associated with the Ice Cover of Lake Bonney, Antarctica. Microb. Ecol. 1998, 36, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Klawonn, I.; Bonaglia, S.; Brüchert, V.; Ploug, H. Aerobic and anaerobic nitrogen transformation processes in N2-fixing cyanobacterial aggregates. ISME J. 2015, 9, 1456–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnelid, H.; Turk-Kubo, K.; Ploug, H.; Ossolinski, J.E.; Collins, J.R.; Van Mooy, B.A.S.; Zehr, J.P. Diverse diazotrophs are present on sinking particles in the North Pacific Subtropical Gyre. ISME J. 2019, 13, 170–182. [Google Scholar] [CrossRef]
- Tonolla, M.; Demarta, A.; Peduzzi, R. The chemistry of Lake Cadagno. Doc. Ist. Ital. Idrobiol. 1998, 63, 11–17. [Google Scholar]
- Peduzzi, S.; Tonolla, M.; Hahn, D. Isolation and characterization of aggregate-forming sulfate-reducing and purple sulfur bacteria from the chemocline of meromictic Lake Cadagno, Switzerland. FEMS Microbiol. Ecol. 2003, 45, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Tonolla, M.; Peduzzi, S.; Demarta, A.; Peduzzi, R.; Hahn, D. Phototropic sulfur and sulfate-reducing bacteria in the chemocline of meromictic Lake Cadagno, Switzerland. J. Limnol. 2004, 63, 161–170. [Google Scholar] [CrossRef]
- Küsel, K. Microbial cycling of iron and sulfur in acidic coal mining lake sediments. Water Air Soil Pollut. Focus 2003, 3, 67–90. [Google Scholar] [CrossRef]
- Meier, J.; Babenzien, H.-D.; Wendt-Potthoff, K. Microbial cycling of iron and sulfur in sediments of acidic and pH-neutral mining lakes in Lusatia (Brandenburg, Germany). Biogeochemistry 2004, 67, 135–156. [Google Scholar] [CrossRef]
- Church, C.D.; Wilkin, R.T.; Alpers, C.N.; Rye, R.O.; Blaine, R.B. Microbial sulfate reduction and metal attenuation in pH 4 acid mine water. Geochem. Trans. 2007, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschorreck, M. Microbial sulphate reduction at a low pH. FEMS Microbiol. Ecol. 2008, 64, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cooper, R.E.; Wegner, C.-E.; Küsel, K. Molecular Mechanisms Underpinning Aggregation in Acidiphilium sp. C61 Isolated from Iron-Rich Pelagic Aggregates. Microorganisms 2020, 8, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gram, L.; Grossart, H.-P.; Schlingloff, A.; Kiørboe, T. Possible quorum sensing in marine snow bacteria: Production of acylated homoserine lactones by Roseobacter strains isolated from marine snow. Appl. Environ. Microbiol. 2002, 68, 4111–4116. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Lovell, C. Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styp Von Rekowski, K.; Hempel, M.; Philipp, B. Quorum sensing by N-acylhomoserine lactones is not required for Aeromonas hydrophila during growth with organic particles in lake water microcosms. Arch. Microbiol. 2008, 189, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Méndez-García, C.; Peláez, A.I.; Mesa, V.; Sánchez, J.; Golyshina, O.V.; Ferrer, M. Microbial diversity and metabolic networks in acid mine drainage habitats. Front. Microbiol. 2015, 6, 475. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003, 44, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, S.R.; Poehlein, A.; Voget, S.; Hoppert, M.; Daniel, R.; Leimbach, A.; Tischler, J.S.; Schlömann, M.; Mühling, M. Permanent draft genome sequence of Acidiphilium sp. JA12-A1. Stand. Genomic Sci. 2015, 10, 1–10. [Google Scholar] [CrossRef]
- Küsel, K.; Dorsch, T.; Acker, G.; Stackebrandt, E. Microbial reduction of Fe(III) in acidic sediments: Isolation of Acidiphilium cryptum JF-5 capable of coupling the reduction of Fe(III) to the oxidation of glucose. Appl. Environ. Microbiol. 1999, 65, 3633–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küsel, K.; Roth, U.; Drake, H.L. Microbial reduction of Fe(III) in the presence of oxygen under low pH conditions. Environ. Microbiol. 2002, 4, 414–421. [Google Scholar] [CrossRef]
- Mühling, M.; Poehlein, A.; Stuhr, A.; Voitel, M.; Daniel, R.; Schlömann, M. Reconstruction of the metabolic potential of acidophilic Sideroxydans strains from the metagenome of an microaerophilic enrichment culture of acidophilic iron-oxidizing bacteria from a pilot plant for the treatment of acid mine drainage reveals met. Front. Microbiol. 2016, 7, 2082. [Google Scholar] [CrossRef] [Green Version]
- Winstanley, C.; Morgan, J.A.W. The bacterial flagellin gene as a biomarker for detection, population genetics and epidemiological analysis. Microbiology 1997, 143, 3071–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.G.; Pearson, L.; Dillon, S.; Kantalis, K. Natural assemblages of marine bacteria exhibiting high-speed motility and large accelerations. Appl. Environ. Microbiol. 1995, 61, 4436–4440. [Google Scholar] [CrossRef] [Green Version]
- Lambrecht, N.; Wittkop, C.; Katsev, S.; Fakhraee, M.; Swanner, E.D. Geochemical Characterization of Two Ferruginous Meromictic Lakes in the Upper Midwest, USA. J. Geophys. Res. Biogeosci. 2018, 123, 3403–3422. [Google Scholar] [CrossRef]
- Crowe, S.A.; O’Neill, A.H.; Katsev, S.; Hehanussa, P.; Haffner, G.D.; Sundby, B.; Mucci, A.; Fowle, D.A. The biogeochemistry of tropical lakes: A case study from Lake Matano, Indonesia. Limnol. Oceanogr. 2008, 53, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Oswald, K.; Jegge, C.; Tischer, J.; Berg, J.; Brand, A.; Miracle, M.R.; Soria, X.; Vicente, E.; Lehmann, M.F.; Zopfi, J.; et al. Methanotrophy under versatile conditions in the water column of the ferruginous meromictic Lake La Cruz (Spain). Front. Microbiol. 2016, 7, 1762. [Google Scholar] [CrossRef] [Green Version]
- Lu, S. Microbial Iron Cycling in Pelagic Aggregates (iron Snow) and Sediments of an Acidic Mine Lake. PhD’s Thesis, Friedrich-Schiller-Universität, Jena, Germany, 2012. [Google Scholar]
- Reinthaler, T.; van Aken, H.M.; Herndl, G.J. Major contribution of autotrophy to microbial carbon cycling in the deep North Atlantic’s interior. Deep. Res. Part II Top. Stud. Oceanogr. 2010, 57, 1572–1580. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Simon, M. Significance of limnetic organic aggregates (lake snow) for the sinking flux of particulate organic matter in a large lake. Aquat. Microb. Ecol. 1998, 15, 115–125. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Cooper, R.E.; Wegner, C.-E.; Taubert, M.; Jehmlich, N.; von Bergen, M.; Küsel, K. Insights into Autotrophic Activities and Carbon Flow in Iron-Rich Pelagic Aggregates (Iron Snow). Microorganisms 2021, 9, 1368. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071368
Li Q, Cooper RE, Wegner C-E, Taubert M, Jehmlich N, von Bergen M, Küsel K. Insights into Autotrophic Activities and Carbon Flow in Iron-Rich Pelagic Aggregates (Iron Snow). Microorganisms. 2021; 9(7):1368. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071368
Chicago/Turabian StyleLi, Qianqian, Rebecca E. Cooper, Carl-Eric Wegner, Martin Taubert, Nico Jehmlich, Martin von Bergen, and Kirsten Küsel. 2021. "Insights into Autotrophic Activities and Carbon Flow in Iron-Rich Pelagic Aggregates (Iron Snow)" Microorganisms 9, no. 7: 1368. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071368