
Impact of Landscape on Host–Parasite Genetic Diversity and Distribution Using the Puumala orthohantavirus–Bank Vole System


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Preparation
2.2. Microsatellite Genotyping of Voles
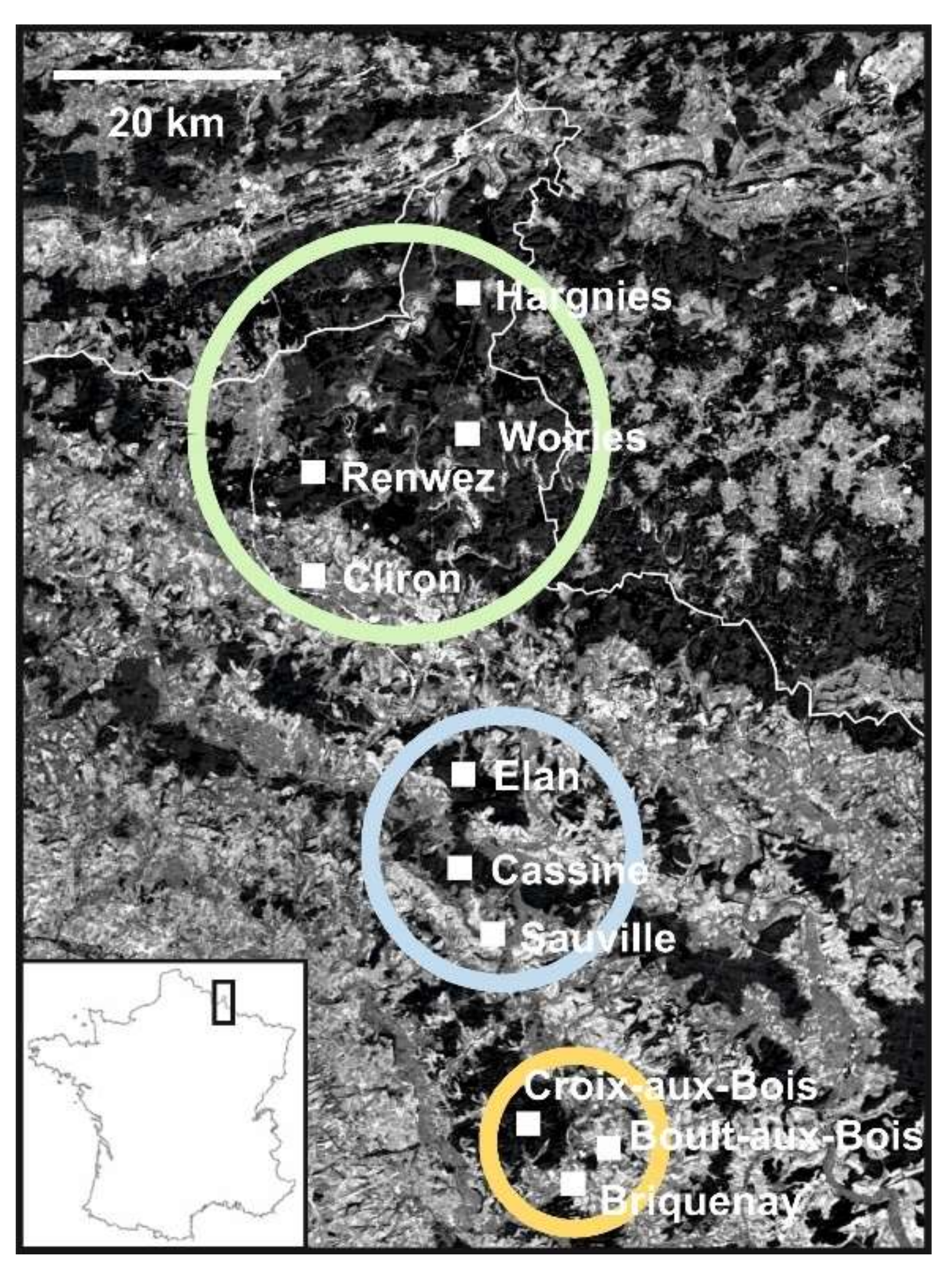
2.3. Landscape Characteristics of the Study Sites
2.4. Screening for PUUV
2.5. RT-PCR and Sequencing of the PUUV Genome
2.6. Molecular Signatures of Selection of PUUV Strains
2.7. Phylogenetic Analysis of PUUV Strains
2.8. Local Genetic Diversity
2.9. Large-Scale Genetic Structure
2.10. Landscape Analyses
3. Results
3.1. Detection and Genetic Diversity of PUUV and Bank Voles from the French Ardennes
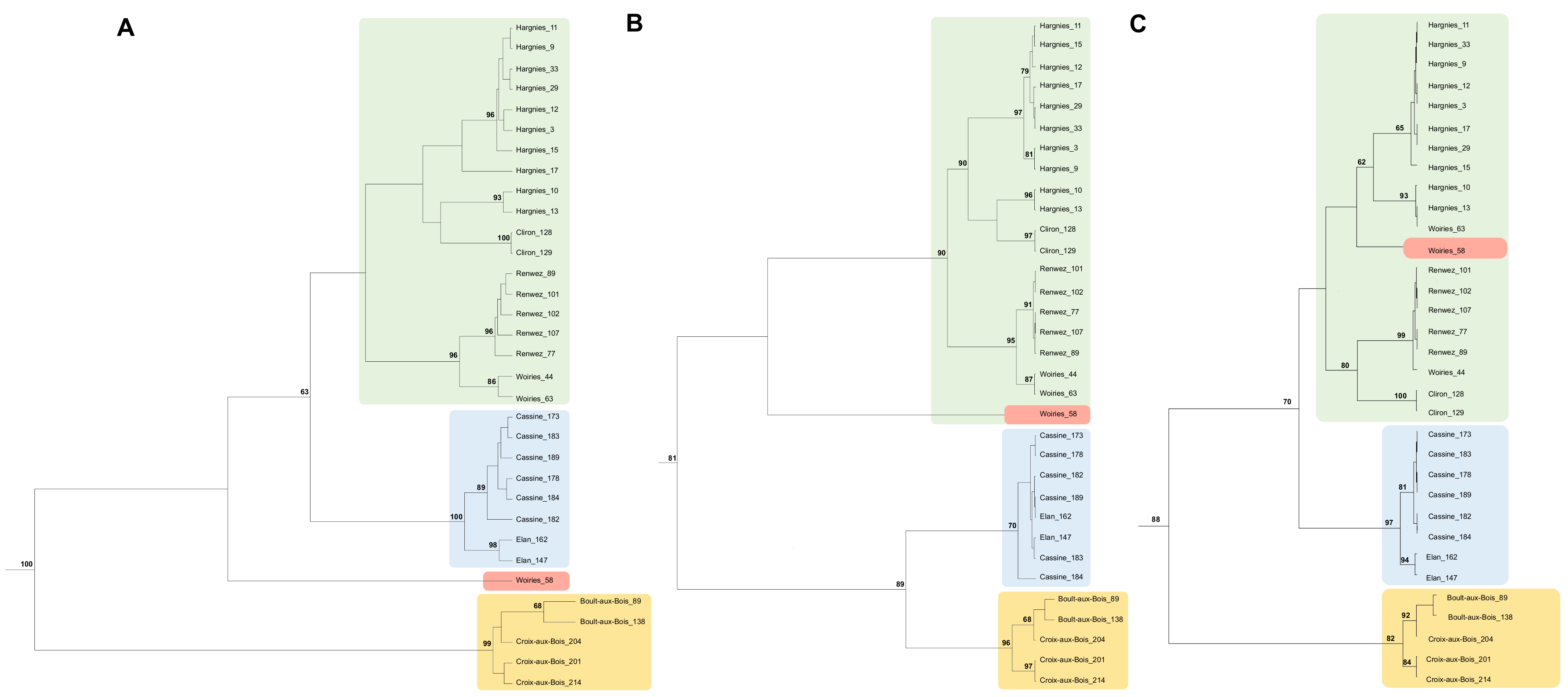
3.2. Phylogenetic Analysis of PUUV Strains from the French Ardennes
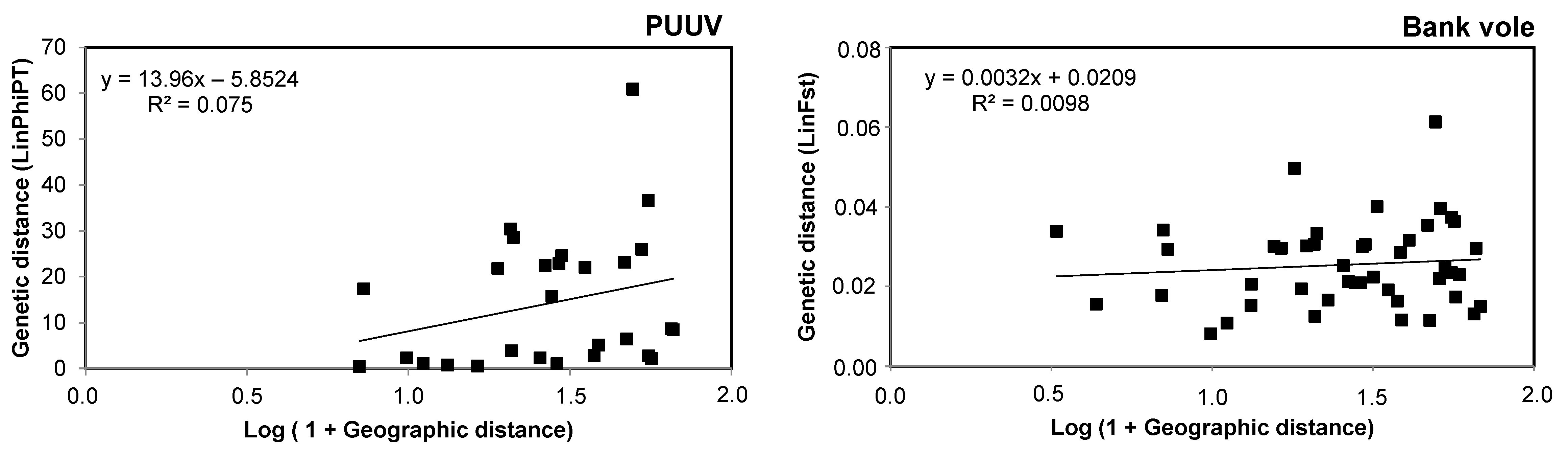
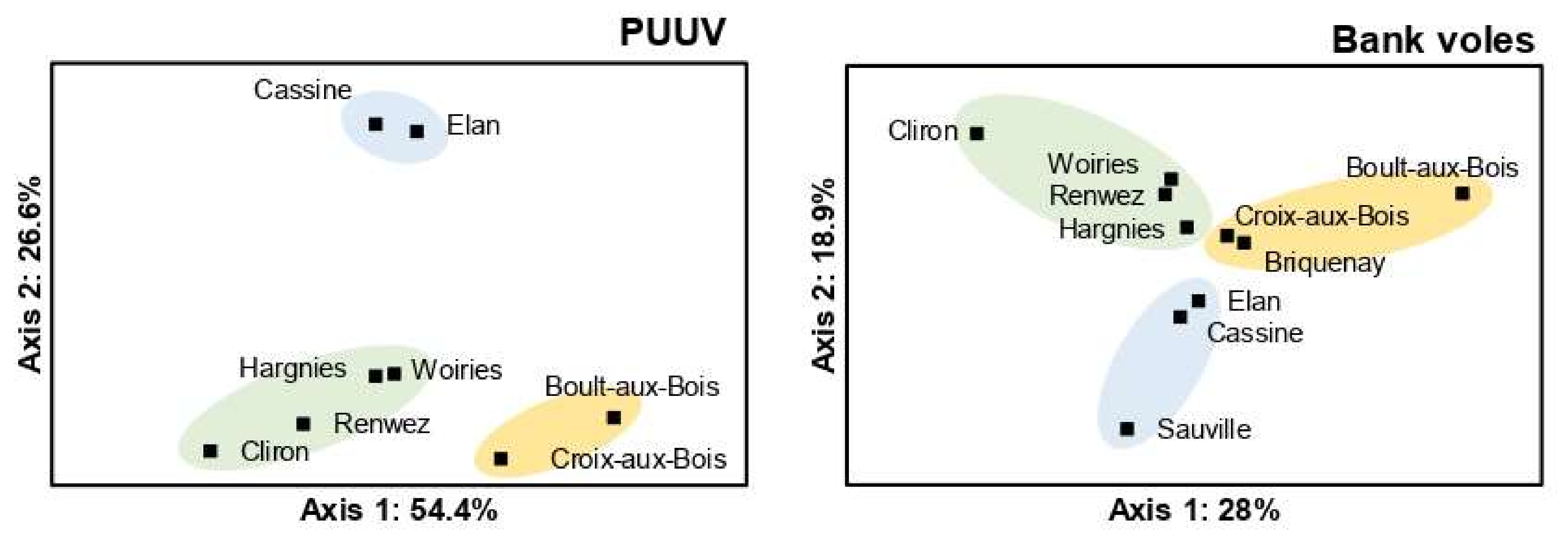
3.3. Spatial Genetic Structure of PUUV and Bank Voles from the French Ardennes
3.4. Effect of Landscape Features and Bank Vole Population Dynamics on PUUV Prevalence and Diversity
4. Discussion
4.1. Genetic Diversity of PUUV and Bank Voles from the French Ardennes
4.2. Phylogenetic Analysis of PUUV Strains from the French Ardennes
4.3. Spatial Genetic Structure of PUUV and Bank Voles from the French Ardennes
4.4. Effect of Landscape Features and Bank Vole Population Dynamics on PUUV Prevalence and Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Patz, J.A.; Graczyk, T.K.; Geller, N.; Vittor, A.Y. Effects of environmental change on emerging parasitic diseases. Int. J. Parasitol. 2000, 30, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.A.; McMichael, A.J. Social and environmental risk factors in the emergence of infectious diseases. Nat. Med. 2004, 10, S70–S76. [Google Scholar] [CrossRef]
- Patz, J.A.; Daszak, P.; Tabor, G.M.; Aguirre, A.A.; Pearl, M.; Epstein, J.; Wolfe, N.D.; Kilpatrick, A.M.; Foufopoulos, J.; Molyneux, D.; et al. Unhealthy landscapes: Policy recommendations on land use change and infectious disease emergence. Environ. Health Perspect. 2004, 112, 1092–1098. [Google Scholar] [CrossRef] [Green Version]
- Rouffaer, L.O.; Strubbe, D.; Teyssier, A.; Salleh Hudin, N.; van den Abeele, A.-M.; Cox, I.; Haesendonck, R.; Delmée, M.; Haesebrouck, F.; Pasmans, F.; et al. Effects of urbanization on host-pathogen interactions, using Yersinia in house sparrows as a model. PLoS ONE 2017, 12, e0189509. [Google Scholar] [CrossRef]
- Morse, S.S. Towards an evolutionary biology of viruses. In The Evolutionary Biology of Viruses; Morse, S.S., Ed.; Raven Press: New York, NY, USA, 1994; pp. 1–28. [Google Scholar]
- Gibbs, A.J.; Calisher, C.H.; Garcia-Arenal, F. Molecular Basis of Viral Evolution; Cambridge University Press: Cambridge, UK, 1995. [Google Scholar]
- Howard, C.R.; Fletcher, N.F. Emerging virus diseases: Can we ever expect the unexpected? Emerg. Microbes Infect. 2012, 1, e46. [Google Scholar] [CrossRef]
- Alexander, K.A.; Lewis, B.L.; Marathe, M.; Eubank, S.; Blackburn, J.K. Modeling of wildlife-associated zoonoses: Applications and caveats. Vector Borne Zoonotic Dis. 2012, 12, 1005–1018. [Google Scholar] [CrossRef]
- Cable, J.; Barber, I.; Boag, B.; Ellison, A.R.; Morgan, E.R.; Murray, K.; Pascoe, E.L.; Sait, S.M.; Wilson, A.J.; Booth, M. Global change, parasite transmission and disease control: Lessons from ecology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160088. [Google Scholar] [CrossRef] [PubMed]
- Martiny, J.B.H.; Bohannan, B.J.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krimins, J.J.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Manel, S.; Schwartz, M.K.; Luikart, G.; Taberlet, P. Landscape genetics: Combining landscape ecology and population genetics. Trends Ecol. Evol. 2003, 18, 189–197. [Google Scholar] [CrossRef]
- Manel, S.; Holderegger, R. Ten years of landscape genetics. Trends Ecol. Evol. 2013, 28, 614–621. [Google Scholar] [CrossRef]
- Kozakiewicz, C.P.; Burridge, C.; Funk, W.C.; Vandewoude, S.; Craft, M.E.; Crooks, K.R.; Ernest, H.B.; Fountain-Jones, N.M.; Carver, S. Pathogens in space: Advancing understanding of pathogen dynamics and disease ecology through landscape genetics. Evol. Appl. 2018, 11, 1763–1778. [Google Scholar] [CrossRef]
- Biek, R.; Real, L.A. The landscape genetics of infectious disease emergence and spread. Mol. Ecol. 2010, 19, 3515–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemming-Schroeder, E.; Lo, E.; Salazar, C.; Puente, S.; Yan, G. Landscape genetics: A toolbox for studying vector-borne diseases. Front. Ecol. Evol. 2018, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–752. [Google Scholar] [CrossRef]
- Whatson, D.C.; Sargianou, M.; Papa, A.; Chra, P.; Starakis, I.; Panos, G. Epidemiology of Hantavirus infections in humans: A comprehensive, global overview. Crit. Rev. Microbiol. 2014, 40, 261–272. [Google Scholar] [CrossRef]
- Heyman, P.; Vaheri, A.; ENIVD Members. Situation of hantavirus infections and haemorrhagic fever with renal syndrome in European countries as of December 2006. Eurosurveillance 2008, 13, 18925. [Google Scholar] [CrossRef] [PubMed]
- Heyman, P.; Ceianu, C.S.; Christova, I.; Tordo, N.; Beersma, M.; João Alves, M.; Lundvist, Å.; Hukic, M.; Papa, A.; Tenorio, A.; et al. A five-year perspective on the situation of haemorrhagic fever with renal syndrome and status of the hantavirus reservoirs in Europe, 2005–2010. Eurosurveillance 2011, 16, 2005–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laenen, L.; Vergote, V.; Calisher, C.H.; Klempa, B.; Klingström, J.; Kuhn, J.H.; Maes, P. Hantaviridae: Current classification and future perspectives. Viruses 2019, 11, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.P.; Lin, X.D.; Wang, W.; Tian, J.H.; Cong, M.L.; Zhang, H.L.; Wang, M.R.; Zhou, R.H.; Wang, J.B.; Li, M.H.; et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef] [Green Version]
- Botten, J.; Mirowsky, K.; Ye, C.; Gottlieb, K.; Saavedra, M.; Ponce, L.; Hjelle, B. Shedding and intracage transmission of Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus) model. J. Virol. 2002, 76, 7587–7594. [Google Scholar] [CrossRef] [Green Version]
- Kariwa, H.; Fujiki, M.; Yoshimatsu, K.; Arikawa, J.; Takashima, I.; Hashimoto, N. Urine-associated horizontal transmission of Seoul virus among rats. Arch. Virol. 1998, 143, 365–374. [Google Scholar] [CrossRef]
- Lee, H.W.; French, G.R.; Lee, P.W.; Baek, L.J.; Tsuchiya, K.; Foulke, R.S. Observations on natural and laboratory infection of rodents with the etiologic agent of Korean hemorrhagic fever. Am. J. Trop. Med. Hyg. 1981, 30, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [CrossRef]
- Ramsden, C.; Holmes, E.C.; Charleston, M.A. Hantavirus evolution in relation to its rodent and insectivore hosts: No evidence for co-divergence. Mol. Biol. Evolotution 2009, 26, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, F.; Drewes, S.; Imholt, C.; Saathoff, M.; Below, D.A.; Bendl, E.; Conraths, F.J.; Tenhaken, P.; Mylius, M.; Brockmann, S. Heterogeneous Puumala orthohantavirus situation in endemic regions in Germany in summer 2019. Transbound. Emerg. Dis. 2020, 67, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Essbauer, S.S.; Schmidt-Chanasit, J.; Madeja, E.L.; Wegener, W.; Friedrich, R.; Petraityte, R.; Sasnauskas, K.; Jacob, J.; Koch, J.; Dobler, G.; et al. Nephropathia epidemica in metropolitan area, Germany. Emerg. Infect. Dis. 2007, 13, 1271–1273. [Google Scholar] [CrossRef]
- Tersago, K.; Verhagen, R.; Vapalahti, O.; Heyman, P.; Ducoffre, G.; Leirs, H. Hantavirus outbreak in Western Europe: Reservoir host infection dynamics related to human disease patterns. Epidemiol. Infect. 2011, 139, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Vanwambeke, S.O.; Zeimes, C.B.; Drewes, S.; Ulrich, R.G.; Reil, D.; Jacob, J. Spatial dynamics of a zoonotic orthohantavirus disease through heterogenous data on rodents, rodent infections, and human disease. Sci. Rep. 2019, 9, 2329. [Google Scholar] [CrossRef]
- Cunze, S.; Kochmann, J.; Kuhn, T.; Frank, R.; Dörge, D.D.; Klimpel, S. Spatial and temporal patterns of human Puumala virus (PUUV) infections in Germany. PeerJ 2018, 6, e4255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linard, C.; Tersago, K.; Leirs, H.; Lambin, E.F. Environmental conditions and Puumala virus transmission in Belgium. Int. J. Health Geogr. 2007, 6, 55. [Google Scholar] [CrossRef]
- Monchatre-Leroy, E.; Crespin, L.; Boué, F.; Marianneau, P.; Calavas, D.; Hénaux, V. Spatial and temporal epidemiology of nephropathia epidemica incidence and hantavirus seroprevalence in rodent hosts: Identification of the main environmental factors in Europe. Transbound. Emerg. Dis. 2017, 64, 1210–1228. [Google Scholar] [CrossRef] [PubMed]
- Tersago, K.; Schreurs, A.; Linard, C.; Verhagen, R.; van Dongen, S.; Leirs, H. Population, environmental, and community effects on local bank vole (Myodes glareolus) Puumala virus infection in an area with low human incidence. Vector Borne Zoonotic Dis. 2008, 8, 235–244. [Google Scholar] [CrossRef]
- Wilson, D.E.; Reeder, D.M. Mammal Species of the World. A Taxonomic and Geographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; Volume 2, p. 142. [Google Scholar]
- Hutterer, R.; Kryštufek, B.; Yigit, N.; Mitsain, G.; Palomo, L.J.; Henttonen, H.; Vohralík, V.; Zagorodnyuk, I.; Juškaitis, R.; Meinig, H.; et al. Myodes Glareolus. Available online: www.iucnredlist.org (accessed on 12 July 2021). [CrossRef]
- Gliwicz, J.; Ims, R.A. Dispersal in the bank vole. Pol. J. Ecol. 2000, 48, 51–61. [Google Scholar]
- Kozakiewicz, M.; Apeldoorn, R.V.; Bergers, P.; Gortat, T.; Kozakiewicz, A. Landscape approach to bank vole ecology. Pol. J. Ecol. 2000, 48, 149–161. [Google Scholar]
- Monchatre-Leroy, E.; Murri, S.; Castel, G.; Calavas, D.; Boué, F.; Hénaux, V.; Marianneau, P. First insights into Puumala orthohantavirus circulation in a rodent population in Alsace, France. Zoonoses Public Health 2018, 65, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Van Apeldoorn, R.C.; Oostenbrink, W.T.; van Winden, A.; van der Zee, F.F. Effects of habitat fragmentation on the bank vole, Clethrionomys glareolus, in an agricultural landscape. Oikos 1992, 65, 265–274. [Google Scholar] [CrossRef]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.-F.; Plyusnin, A.; Marianneau, P.; et al. Complete genome and phylogeny of puumala hantavirus isolates circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef] [Green Version]
- Hörling, J.; Lundkvist, Å.; Jaarola, M.; Plyusnin, A.; Tegelström, H.; Persson, K.; Lehväslaiho, H.; Hörnfeldt, B.; Vaheri, A.; Niklasson, B. Distribution and genetic heterogeneity of Puumala virus in Sweden. J. Genet. Virol. 1996, 77, 2555–2562. [Google Scholar] [CrossRef]
- Nemirov, K.; Leirs, H.; Lundkvist, Å.; Olsson, G.E. Puumala hantavirus and Myodes glareolus in northern Europe: No evidence of co-divergence between genetic lineages of virus and host. J. Genet. Virol. 2010, 91, 262–1274. [Google Scholar] [CrossRef]
- Razzauti, M.; Plysunina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between wild-type strains. J. Genet. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A Russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef]
- Reynes, J.M.; Carli, D.; Thomas, D.; Castel, G. Puumala hantavirus genotypes in humans, France, 2012-2016. Emerg. Infect. Dis. 2019, 25, 140–143. [Google Scholar] [CrossRef]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kühnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Genet. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [Green Version]
- Razzauti-Sanfeliu, M. Microevolution of Puumala Hantavirus in Its Host, the Bank Vole (Myodes glareolus). Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 24 February 2012. Available online: https://helda.helsinki.fi/handle/10138/29616 (accessed on 12 July 2021).
- Dearing, M.D.; Dizney, L. Ecology of hantavirus in a changing world. Ann. N. Y. Acad. Sci. 2010, 1195, 99–112. [Google Scholar] [CrossRef]
- Voutilainen, L.; Savola, S.; Kallio, E.R.; Laakkonen, J.; Vaheri, A.; Vapalahti, O.; Henttonen, H. Environmental change and disease dynamics: Effects of intensive forest management on Puumala hantavirus infection in boreal bank vole populations. PLoS ONE 2012, 7, e39452. [Google Scholar] [CrossRef] [Green Version]
- Escutenaire, S.; Chalon, P.; de Jaegere, F.; Karelle-Bui, L.; Mees, G.; Brochier, B.; Rozenfeld, F.; Pastoret, P.P. Behavioral, physiologic, and habitat influences on the dynamics of Puumala virus infection in bank voles (Clethrionomys glareolus). Emerg. Infect. Dis. 2002, 8, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Deter, J.; Chaval, Y.; Galan, M.; Gauffre, B.; Morand, S.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Charbonnel, N.; Cosson, J.-F. Kinship, dispersal and hantavirus transmission in bank and common voles. Arch. Virol. 2008, 153, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, J.M.; Kawałko, A.; Marková, S.; Searle, J.B.; Kotlík, P. Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: A case study of bank voles using museum specimens. J. Zool. 2010, 281, 249–262. [Google Scholar] [CrossRef]
- Guivier, E.; Galan, M.; Salvador, A.R.; Xuéreb, A.; Chaval, Y.; Olsson, G.E.; Essbauer, S.; Henttonen, H.; Voutilainen, L.; Cosson, J.-F.; et al. Tnf-α expression and promoter sequences reflect the balance of tolerance/resistance to Puumala hantavirus infection in European bank vole populations. Infect. Genet. Evol. 2010, 10, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Drewes, S.; Ali, H.S.; Saxenhofer, M.; Rosenfeld, U.M.; Binder, F.; Cuypers, F.; Schlegel, M.; Röhrs, S.; Heckel, G.; Ulrich, R.G. Host-associated absence of human Puumala virus infections in Northern and Eastern Germany. Emerg. Infect. Dis. 2017, 23, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Kallio, E.R.; Klingström, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, Å. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef]
- Sauvage, F.; Langlais, M.; Yoccoz, N.G.; Pontier, D. Modelling hantavirus in fluctuating populations of bank voles: The role of indirect transmission on virus persistence. J. Anim. Ecol. 2003, 72, 1–13. [Google Scholar] [CrossRef]
- Glass, G.E.; Cheek, J.E.; Patz, J.A.; Shields, T.M.; Doyle, T.J.; Thoroughman, D.A.; Hunt, D.K.; Enscore, R.E.; Gare, K.L.; Irland, C.; et al. Using remote sensing data to identify areas at risk for hantavirus pulmonary syndrome. Emerg. Infect. Dis. 2000, 6, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Goodin, D.G.; Koch, D.E.; Owen, R.D.; Chu, Y.K.; Hutchinson, J.M.S.; Jonsson, C.B. Land cover associated with hantavirus presence in Paraguay. Glob. Ecol. Biogeogr. 2006, 15, 519–527. [Google Scholar] [CrossRef]
- Curran, P.J.; Atkinson, P.M.; Foody, G.M.; Milton, E.J. Linking remote sensing, land cover and disease. Adv. Parasitol. 2000, 47, 37–80. [Google Scholar]
- Olsson, G.E.; White, N.; Hjalten, J.; Ahlm, C. Habitat factors associated with bank voles (Clethrionomys glareolus) and concomitant hantavirus in Northern Sweden. Vector Borne Zoonotic Dis. 2005, 5, 315–323. [Google Scholar] [CrossRef]
- Kitron, U. Landscape ecology and epidemiology of vector-borne diseases: Tools for spatial analysis. J. Med. Entomol. 1998, 35, 435–445. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Vanmechelen, B.; Tersago, K.; Baele, G.; Lemey, P.; Leirs, H.; Dellicour, S.; Maes, P. Identifying the patterns and drivers of Puumala hantavirus enzootic dynamics using reservoir sampling. Virus Evol. 2019, 5, vez009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, J.P.; Fahrig, L.; Merriam, G.; Artsob, H. Landscape structure influences continental distribution of hantavirus in deer mice. Landsc. Ecol. 2001, 16, 255–266. [Google Scholar] [CrossRef]
- Ostfeld, R.S.; Glass, G.E.; Keesing, F. Spatial epidemiology: An emerging (or re-emerging) discipline. Trends Ecol. Evol. 2005, 20, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Bellocq, J.G.; Wasimuddin; Ribas, A.; Bryja, J.; Piálek, J.; Baird, S.J.E. Holobiont suture zones: Parasite evidence across the European house mouse hybrid zone. Mol. Ecol. 2018, 27, 5214–5227. [Google Scholar] [CrossRef] [PubMed]
- Saxenhofer, M.; Schmidt, S.; Ulrich, R.G.; Heckel, G. Secondary contact between diverged host lineages entails ecological speciation in a European hantavirus. PLoS Biol. 2019, 17, e3000142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guivier, E.; Galan, M.; Chaval, Y.; Xuéreb, A.; Ribas Salvador, A.; Poulle, M.L.; Voutilainen, L.; Henttonen, H.; Charbonnel, N.; Cosson, J.-F. Landscape genetics highlights the role of bank vole metapopulation dynamics in the epidemiology of Puumala hantavirus. Mol. Ecol. 2011, 20, 3569–3583. [Google Scholar] [CrossRef] [PubMed]
- McGarigal, K.; Cushman, S.A.; Ene, E. FRAGSTATS Volume 4: Spatial Pattern Analysis Program for Categorical and Continuous Maps. Available online: http://www.umass.edu/landeco/research/fragstats/fragstats.html (accessed on 12 July 2021).
- Kozakiewicz, M.; Chołuj, A.; Kozakiewicz, A. Long-distance movements of individuals in a free-living bank vole population: An important element of male breeding strategy. Acta Theriol. 2007, 52, 339–348. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree (Version 1.4.0). Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 July 2021).
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RStudio Team (2020). RStudio: Integrated Development for R; RStudio, PBC, Boston, MA, USA. Available online: http://www.rstudio.com/ (accessed on 12 July 2021).
- Barton, K. Mu-MIn: Multi-Model Inference. R Package Version 0.12.2/r18. Available online: http://R-Forge.R-project.org/projects/mumin/ (accessed on 12 July 2021).
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. R Package Version 3.1-140. Available online: https://CRAN.R-project.org/package=nlme (accessed on 12 July 2021).
- Johnson, J.; Omland, K. Model selection in ecology and evolution. Trends Ecol. Evol. 2004, 19, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Burnham, K.P.; Anderson, D.R. Model Selection and Inference. A Practical Information-Theoretic Approach; Springer: Heidelberg, Germany; New-York, NY, USA, 1998. [Google Scholar]
- Graham, M.H. Confronting multicollinearity in ecological multiple regression. Ecology 2003, 84, 2809–2815. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Simko, V. R Package ‘Corrplot’: Visualization of a Correlation Matrix (Version 0.90). Package Corrplot for R Software. 2021. Available online: https://github.com/taiyun/corrplot (accessed on 12 July 2021).
- Barrett, L.G.; Thrall, P.H.; Burdon, J.J.; Linde, C.C. Life history determines genetic structure and evolutionary potential of host-parasite interactions. Trends Ecol. Evol. 2008, 23, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Abbate, J.L.; Galan, M.; Razzauti, M.; Sironen, T.; Voutilainen, L.; Henttonen, H.; Gasqui, P.; Cosson, J.-F.; Charbonnel, N. Pathogen community composition and co-infection patterns in a wild community of rodents. BioRxiv 2020. preprint. [Google Scholar] [CrossRef] [Green Version]
- Penczykowski, R.M.; Laine, A.L.; Koskella, B. Understanding the ecology and evolution of host-parasite interactions across scales. Evol. Appl. 2015, 9, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Blanchong, J.A.; Samuel, M.D.; Scribner, K.T.; Weckworth, B.V.; Langenberg, J.A.; Filcek, K.B. Landscape genetics and the spatial distribution of chronic wasting disease. Biol. Lett. 2008, 4, 103–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sites | Longitude | Latitude | Proportion of Forested Habitat (%) | Forest Contiguity Index | Shape Index | Edge Density (m/km2) | No. of Patches | Distance to Next Patch (m) |
|---|---|---|---|---|---|---|---|---|
| Hargnies | 4.7950 | 49.9961 | 0.838 | 0.93 | 37.3 | 883.7 | 14 | 200 |
| Woiries | 4.7664 | 49.9037 | 0.861 | 0.93 | 37.3 | 936.4 | 9 | 200 |
| Renwez | 4.6113 | 49.8590 | 0.632 | 0.93 | 37.3 | 14484 | 10 | 200 |
| Cliron | 4.6204 | 49.8032 | 0.022 | 0.85 | 19.4 | 169.7 | 2 | 956 |
| Elan | 4.7674 | 49.6536 | 0.585 | 0.71 | 4.1 | 1203.3 | 3 | 200 |
| Cassine | 4.7945 | 49.5752 | 0.421 | 0.84 | 3.5 | 1226.3 | 3 | 208 |
| Sauville | 4.8003 | 49.5454 | 0.252 | 0.82 | 3.0 | 901.9 | 4 | 411 |
| Croix-aux-Bois | 4.8385 | 49.4166 | 0.596 | 0.82 | 2.8 | 900.2 | 9 | 355 |
| Boult-aux-Bois | 4.9215 | 49.4136 | 0.187 | 0.76 | 2.5 | 1311.8 | 8 | 445 |
| Briquenay | 4.9109 | 49.3937 | 0.169 | 0.78 | 2.7 | 904.5 | 7 | 493 |
| Genome Segment | No. of Strains | S (43–1341 nt) | M (2180–2632 nt) | L (577–987 nt) | |
|---|---|---|---|---|---|
| Analyzed length | Nucleotides (nt) | 33 | 1299 | 453 | 411 |
| Amino acids (aa) | 33 | 433 | 151 | 137 | |
| No. of substitutions | Nt polymorphic sites (total no. of mutations) | 33 | 178 (185) | 62 (63) | 69 (71) |
| Nt singleton variable sites | 33 | 29 | 10 | 2 | |
| Nt parsimony informative sites | 33 | 149 | 52 | 67 | |
| Aa variable sites | 33 | 11 | 4 | 5 | |
| Genetic diversity (%) | Nucleotides (nt) | 33 | 14.2 | 13.9 | 17.3 |
| Amino acids (aa) | 33 | 2.5 | 2.7 | 2.7 | |
| No. of nt substitutions (%) | Hargnies | 10 | 29 (2.2) | 11 (2.4) | 13 (3.2) |
| Woiries | 3 | 60 (4.6) | 25 (5.5) | 28 (6.8) | |
| Renwez | 5 | 10 (0.8) | 0 | 0 | |
| Cliron | 2 | 0 | 0 | 0 | |
| Elan | 2 | 4 (0.3) | 1 (0.2) | 1 (0.2) | |
| Cassine | 6 | 10 (0.8) | 2 (0.4) | 0 | |
| Croix-aux-Bois | 3 | 8 (0.6) | 4 (0.9) | 3 (0.7) | |
| Boult-aux-Bois | 2 | 4 (0.3) | 1 (0.2) | 0 | |
| No. of aa substitutions (%) | Hargnies | 10 | 3 (0.7) | 0 | 0 |
| Woiries | 3 | 1 (0.2) | 0 | 2 (1.5) | |
| Renwez | 5 | 2 (0.5) | 0 | 0 | |
| Cliron | 2 | 0 | 0 | 0 | |
| Elan | 2 | 0 | 0 | 0 | |
| Cassine | 6 | 1 (0.2) | 0 | 0 | |
| Croix-aux-Bois | 3 | 0 | 1 (0.7) | 1 (0.7) | |
| Boult-aux-Bois | 2 | 0 | 0 | 0 | |
| Segment | Strains Group | n | π | h | Hd | Tajima’s D | Fu and Li’s D | Fu and Li’s F |
|---|---|---|---|---|---|---|---|---|
| Ardennes | 33 | 0.036 | 30 | 0.994 | 0.10 | 0.64 | 0.50 | |
| S | CE | 99 | 0.098 | 89 | 0.997 | 0.53 | 0.62 | 0.70 |
| All PUUV | 189 | 0.138 | 178 | 0.999 | 0.46 | −0.04 | 0.25 | |
| Ardennes | 33 | 0.043 | 14 | 0.922 | 0.99 | 0.59 | 0.86 | |
| M | CE | 63 | 0.096 | 29 | 0.960 | 0.75 | 1.12 | 1.17 |
| All PUUV | 117 | 0.147 | 76 | 0.987 | 0.62 | 0.88 | 0.91 | |
| Ardennes | 33 | 0.051 | 12 | 0.898 | 0.75 | 1.70 *(p < 0.02) | 1.63 *( p < 0.05) | |
| L | CE | 52 | 0.106 | 26 | 0.953 | 0.49 | 0.68 | 0.73 |
| All PUUV | 88 | 0.160 | 61 | 0.984 | 0.31 | 0.67 | 0.62 |
| Source | df | SS | MS | Est. var. | % | Stat. | p (Rand ≥ Data) | |
|---|---|---|---|---|---|---|---|---|
| PUUV | Among forest massifs | 2 | 896.9 | 448 | 41.3 | 65 | PhiRT = 0.647 | 0.001 |
| Among localities | 5 | 313.1 | 62.6 | 14.8 | 23 | PhiPR = 0.661 | 0.001 | |
| Within localities | 25 | 190.9 | 7.6 | 7.6 | 12 | PhiPR = 0.661 | 0.001 | |
| Total | 32 | 1400 | 63.8 | 100 | PhiPT = 0.880 | 0.001 | ||
| Bank voles | Among forest massifs | 2 | 49.8 | 24.9 | 0.04 | 1 | FRT = 0.006 | 0.001 |
| Among localities | 7 | 111.9 | 16 | 0.1 | 2 | FSR = 0.020 | 0.001 | |
| Within localities | 300 | 2199.2 | 7.3 | 0.3 | 4 | FSR = 0.020 | 0.001 | |
| Total | 619 | 4435.8 | 7.2 | 100 | FST = 0.026 | 0.001 |
| Source | Landscape | Vole | |||
|---|---|---|---|---|---|
| % Forest | Contiguity | Shape | Genetic Diversity | ||
| Voles | Genetic diversity | + ** | NS | NS | |
| PUUV prevalence | + ** | + ** | NS | + *** | |
| PUUV | Nucleotide diversity (NtDiv) | + ** | + * | NS | NS |
| Amino-acid diversity (AaDiv) | NS | NS | NS | NS | |
| Genome Segment | PUUV Lineages | No. of Strains | No. of nt Substitutions (Variable Sites) | Genetic Diversity (%) | aa Substitutions (%) |
|---|---|---|---|---|---|
| S (43–1341 nt; 1299 nt, 433 aa) | All known PUUV | 189 | 913 (584) | 70.3 | 73 (16.9) |
| Danish (DAN) | 3 | 87 (85) | 6.7 | 17 (3.9) | |
| Latvian (LAT) | 5 | 178 (174) | 13.7 | 1 (0.2) | |
| North-Scandinavian (N-SCA) | 23 | 335 (294) | 25.8 | 20 (4.6) | |
| South-Scandinavian (S-SCA) | 8 | 261 (251) | 20.1 | 17 (3.9) | |
| Russian (RUS) | 16 | 364 (319) | 28.0 | 29 (6.7) | |
| Finnish (FIN) | 27 | 429 (361) | 33.0 | 52 (12.0) | |
| Alpe-Adrian (ALAD) | 8 | 138 (133) | 10.6 | 13 (3.0) | |
| Central European (CE) | 99 | 567 (435) | 43.6 | 31 (7.2) | |
| M (2180–2632 nt; 453 nt, 151 aa) | All knwon PUUV | 117 | 258 (167) | 57.0 | 13 (8.6) |
| Danish (DAN) | 3 | 21 (21) | 4.6 | 3 (2.0) | |
| Latvian (LAT) | 1 | - | - | - | |
| North-Scandinavian (N-SCA) | 8 | 128 (113) | 31.8 | 9 (6.0) | |
| South-Scandinavian (S-SCA) | 4 | 81 (77) | 17.9 | 3 (2.0) | |
| Russian (RUS) | 11 | 145 (121) | 32.0 | 8 (5.3) | |
| Finnish (FIN) | 15 | 144 (124) | 31.8 | 8 (5.3) | |
| Alpe-Adrian (ALAD) | 12 | 80 (71) | 17.7 | 6 (4.0) | |
| Central European (CE) | 63 | 146 (127) | 32.2 | 12 (7.9) | |
| L (577–987 nt; 411 nt, 137 aa) | All known PUUV | 88 | 304 (202) | 74.0 | 21 (15.3) |
| Danish (DAN) | 3 | 32 (32) | 7.8 | 4 (2.9) | |
| Latvian (LAT) | 4 | 91 (83) | 22.1 | 8 (5.8) | |
| North-Scandinavian (N-SCA) | 2 | 61 (61) | 14.8 | 10 (7.3) | |
| South-Scandinavian (S-SCA) | 0 | - | - | - | |
| Russian (RUS) | 7 | 122 (108) | 29.7 | 12 (8.8) | |
| Finnish (FIN) | 17 | 105 (95) | 25.5 | 8 (5.8) | |
| Alpe-Adrian (ALAD) | 3 | 21 (21) | 7.8 | 0 | |
| Central European (CE) | 52 | 173 (140) | 42.1 | 15 (10.9) | |
| North-Scandinavian (N-SCA) | 23 | 335 (294) | 25.8 | 20 (4.6) | |
| South-Scandinavian (S-SCA) | 8 | 261 (251) | 20.1 | 17 (3.9) | |
| Alpe-Adrian (ALAD) | 8 | 138 (133) | 10.6 | 13 (3.0) | |
| Central European (CE) | 99 | 567 (435) | 43.6 | 31 (7.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razzauti, M.; Castel, G.; Cosson, J.-F. Impact of Landscape on Host–Parasite Genetic Diversity and Distribution Using the Puumala orthohantavirus–Bank Vole System. Microorganisms 2021, 9, 1516. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071516
Razzauti M, Castel G, Cosson J-F. Impact of Landscape on Host–Parasite Genetic Diversity and Distribution Using the Puumala orthohantavirus–Bank Vole System. Microorganisms. 2021; 9(7):1516. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071516
Chicago/Turabian StyleRazzauti, Maria, Guillaume Castel, and Jean-François Cosson. 2021. "Impact of Landscape on Host–Parasite Genetic Diversity and Distribution Using the Puumala orthohantavirus–Bank Vole System" Microorganisms 9, no. 7: 1516. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9071516