1. Introduction
Pseudomonas aeruginosa (P. aeruginosa) is a gram-negative bacterium, considered to be an opportunistic pathogen [
1,
2]. It is ubiquitous in the environment and is frequently responsible for causing nosocomial infections in humans [
1,
2].
P. aeruginosa represents one of the most common bacterial pathogens in an upper and a lower airway infection and is of particular significance in cystic fibrosis (CF) patients [
1]. CF patients lack a functioning CF transmembrane conductance regulator gene, resulting in a change to the airway mucosa that is favourable to
P. aeruginosa infection [
2]. It is one of the first pathogens to infect and dominate the airways during a chronic infection and has been associated with worsening lung function in CF patients [
2,
3]. CF patients are also frequently affected by chronic rhinosinusitis (CRS), in which colonisation of the paranasal sinuses with
P. aeruginosa has been found to be a reservoir for a recurrent lung infection [
1].
P. aeruginosa has the capacity to produce diverse and difficult to treat infections due to the release of virulence factors and the formation of a biofilm [
2,
4]. Phenotypic changes in the infecting strain over time result in the conversion to antibiotic-resistant phenotypes, which have been correlated with increased morbidity and mortality [
2]. The primary treatment modality for CF patients involves systemic and inhaled broad-spectrum antibiotics from early in their life, which can lead to the development of multidrug-resistant (MDR) strains of
P. aeruginosa [
2,
3,
5]. Due to the emerging threat of infections with antibiotic-resistant strains and the lack of research and development of new antibiotics, there is an urgent need for new treatment options that are effective against such infections [
6,
7].
One potential treatment option that is being investigated as an alternative or complimentary treatment to antibiotics is bacteriophage (phage) therapy [
8,
9]. Phages were discovered over a century ago and have recently regained interest for their potential to treat MDR infections [
10,
11]. Lytic phages are viruses that infect, replicate within and eventually lyse bacteria, causing cell death [
1].
P. aeruginosa phages have shown promise in several in vitro and in vivo preclinical studies [
1,
12,
13,
14]. Some advantages of phage therapy compared to conventional antibiotics include the ability to target specific bacterial species, activity against antibiotic-resistant strains, replication at the site of infection and biofilm eradication [
1,
2]. Phages are species specific, and therefore target the pathogenic bacteria without disturbing non-harmful commensal bacteria, resulting in less systemic side effects than antibiotics [
1,
15]. Due to this specificity, treating bacterial infections requires access to phages that are biologically active against the patient’s bacterial strains [
4]. CF patients are often infected with several
P. aeruginosa strains that coexist within a patient, therefore, the development of a phage cocktail will increase the antibiofilm activity by expanding the target host range and decreasing the potential for phage resistance [
1,
2,
12].
P. aeruginosa phages have shown to be effective against planktonic and biofilm
P. aeruginosa, however, the use of phages in Australia is currently not available [
1,
4,
14]. Therapeutic phage candidates will be required to meet quality and safety requirements for human application [
16]. Hence, the isolation and characterisation of phages will follow the recommended minimal requirements for sustainable phage therapy products as proposed by Pirnay et al. as a guide towards the clinical application [
16]. There is an urgent need for the development of new treatment options that are effective at reducing infections in CF patients, and phage therapy presents a promising option in targeting
P. aeruginosa infections [
1,
4].
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
The Central Adelaide Local Health Network Human Research Ethics Committee approved the use of clinical isolates for this study (HREC/15/TQEH/132). P. aeruginosa reference strain ATCC15692 (PA01) was obtained from the American Type Culture Collection (Manassas, VA, USA). The P. aeruginosa clinical isolates of patients with CF (n = 9) were kindly donated by the Department of Otorhinolaryngology, Academic Medical Centre (Amsterdam, The Netherlands) and the P. aeruginosa clinical isolates from non-CF (n = 11) isolated from the sinonasal cavities of CRS patients with and without nasal polyps were kindly donated by the Department of Otolaryngology-Head and Neck Surgery, University of Adelaide at The Queen Elizabeth Hospital (TQEH; Woodville, SA, Australia). The presence of P. aeruginosa had been confirmed and the bacteria were isolated by Adelaide Pathology Partners (Mile End, SA, Australia). P. aeruginosa clinical isolates had been confirmed again using Cetrimide selective agar plates (Oxoid, Thebarton, SA, Australia) and an oxidase reagent test (BioMérieux, Marcy l’Etoile, France). The clinical isolates used to characterise phages were further confirmed using MALDI-TOF (Bruker, VIC, Australia).
The P. aeruginosa clinical isolates (n = 20) were stored in 25% glycerol in tryptone soya broth (TSB) (Oxoid, Thebarton, SA, Australia) at −80 °C. The clinical isolates were plated from frozen glycerol stocks onto 1.5% tryptone soya agar (TSA) plates and broth cultures were grown in TSB (Oxoid, Thebarton, SA, Australia). Unless otherwise stated, the agar plates and broth cultures for non-CF clinical isolates were incubated at 37 °C (Orbital Mixer Incubator, Ratek, Boronia, VIC, Australia) and the CF clinical isolates were incubated at 37 °C supplied with 5% extra CO2 (Panasonic Healthcare Co., Tokyo, Japan) with 180 rpm shaking for the broth cultures.
2.2. Bacteriophage Isolation
The
P. aeruginosa phages (n = 15) were obtained from TQEH wastewater. Wastewater samples were collected at 5 different time points every 30–60 min from the palliative care, respiratory, dermatology and infectious disease ward and intensive care unit departments. Samples of 50 mL from each time point were centrifuged at 4000 rpm for 10–30 min to remove cellular debris and faecal matter (Allegra X-30R Centrifuge, Beckman Coulter, NSW, Australia). The supernatant was filtered using a 0.2 μm filter (PALL Acrodisc, NY, USA) to remove the bacteria and debris. An amount of 0.5 McFarland Units (McF) of PA01 diluted 1:100 in TSB was grown overnight at 37 °C with 180 rpm shaking. An amount of 100 μL of filtered solution was combined with 100 μL of the PA01 overnight culture and 4 mL of molten (0.4%) TSA and plated on 1.5% TSA plates via the double agar overlay method as described [
17]. Following overnight incubation, the formation of plaques confirmed the presence of phage. Individual plaques with different morphology were picked using pipette tips and placed into a 1.5 mL screw neck vial (Thermo Fisher Scientific, Waltham, MA, USA) containing sodium magnesium buffer (SM; 100 mM sodium chloride, (Oxoid, Thebarton, SA, Australia); 8 mM magnesium sulfate heptahydrate, (Sigma-Aldrich, Castle Hill, NSW, Australia); 50 mM 1 M Tris HCL, pH 7.5), respectively, and vortexed vigorously before storage for 2–6 days at 4 °C for propagation.
2.3. Bacteriophage Propagation
A two-step propagation was applied to amplify and purify the isolated phage. For small-scale amplification of phage, 50 μL of PA01 overnight culture was added into 5 mL TSB and incubated for 1 h. An amount of 100 μL of phage in SM buffer was added into the coculture and incubated overnight. The supernatant containing phage was harvested after centrifugation (4000 rpm, 30 min) and filtration (0.2 μm syringe filter). Phage titration was performed before large-scale amplification.
For large-scale amplification of phage, 500 μL of the PA01 overnight culture was incubated with 50 mL of TSB for 1 h. Phage was then added into the coculture at a multiplicity of infection (MOI) = 1. The phage lysis was harvested as described above.
2.4. Bacteriophage Titration and Concentration
The phage titer was determined via a double agar spot assay as a modified protocol [
17]. Briefly, 100 μL of the PA01 overnight broth culture was added to 4 mL of molten (0.7%) TSA and overlaid onto a 1.5% TSA plate. Phages were serially diluted in SM Buffer and spot assays were performed in triplicate with a drop size of 3 μL. The plates were incubated at 37 °C overnight. The SM buffer solution alone was assessed as a negative control. The phage titer was counted and calculated. The samples requiring a higher titer were concentrated using a 100k MWCO Pierce
TM Protein Concentrator PES (Thermo Fisher Scientific, Waltham, MA, USA) according to the manual.
2.5. Bacteriophage Host Range Analysis
The ability of phage isolates to lyse 20
P. aeruginosa clinical isolates (9 CF and 11 non-CF) was tested using the double agar spot assay as described [
17]. Briefly, 100 μL of overnight culture for each clinical isolate was added to 4 mL of molten (0.7%) TSA and overlaid onto 1.5% TSA plates. Phage isolates at 1 × 10
9 plaque forming units per mL (PFU mL
−1) were serially diluted in SM buffer and 3 μL spots applied in triplicate on top of the plates. The phage sensitivity was determined as described previously [
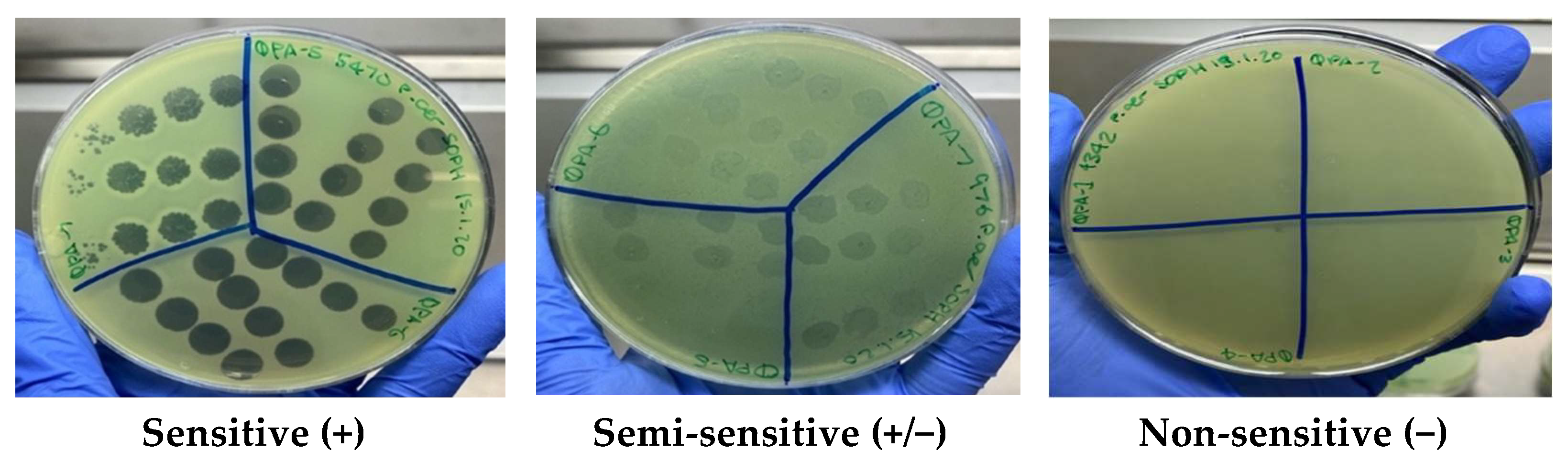
17,
18] using the clarity of plaques and classified as sensitive (+) for a clear lysis, semi-sensitive (+/−) for partial lysis and non-sensitive (−) for no lysis.
2.6. Bacteriophage Stability Testing
The stability of isolated phages was tested against a wide pH (3–12) and temperature (−80 °C to +80 °C) range using a working stock in TSB with an initial phage titer of around 1 × 10
9 PFU mL
−1. The pH stability testing was studied according to the methods described by Bae et al. [
19] Briefly, 10 μL working stock of each phage was suspended in 90 μL SM buffer previously adjusted with 1 M NaOH or HCL (Thermo Fisher Scientific, Winsford, United Kingdom) to yield a pH range of 3–12. The samples were incubated at room temperature (RT) for 1 h. For thermal stability, 80 μL of working stock phage was added to an Eppendorf tube and incubated for 1 h in an Eppendorf Thermomixer Compact (Sigma-Aldrich, Castle Hill, NSW, Australia) at 4 °C, 30 °C, 37 °C, 40 °C, 50 °C, 60 °C, 70 °C and 80 °C. The long-term storage of phage was examined over a 6-month period. An amount of 100 μL of phage at 1 × 10
9 PFU mL
−1 previously diluted in TSB were stored at RT, 4 °C, −20 °C and −80 °C. The phage stability for each experiment was determined by measuring the phage titration.
2.7. Minimum Inhibitory Concentration (MIC) Assays
The resistance to commonly used antibiotics was determined on aerobic, planktonic cultures using broth microdilution minimum inhibitory concentration (MIC) assays, as described [
20]. The antibiotics tested were ciprofloxacin, gentamicin, imipenem, tobramycin, obtained from Sigma-Aldrich (Castle Hill, NSW, Australia) and netilmicin (Chem-Supply, Gillman, SA, Australia). The clinical isolates were designated as being resistant as per the Clinical and Laboratory Standards Institute (CLSI) recommendations (Adelaide Pathology Partners, Mile End, VIC, Australia) [
21].
2.8. Bacteriophage Adsorption Assays
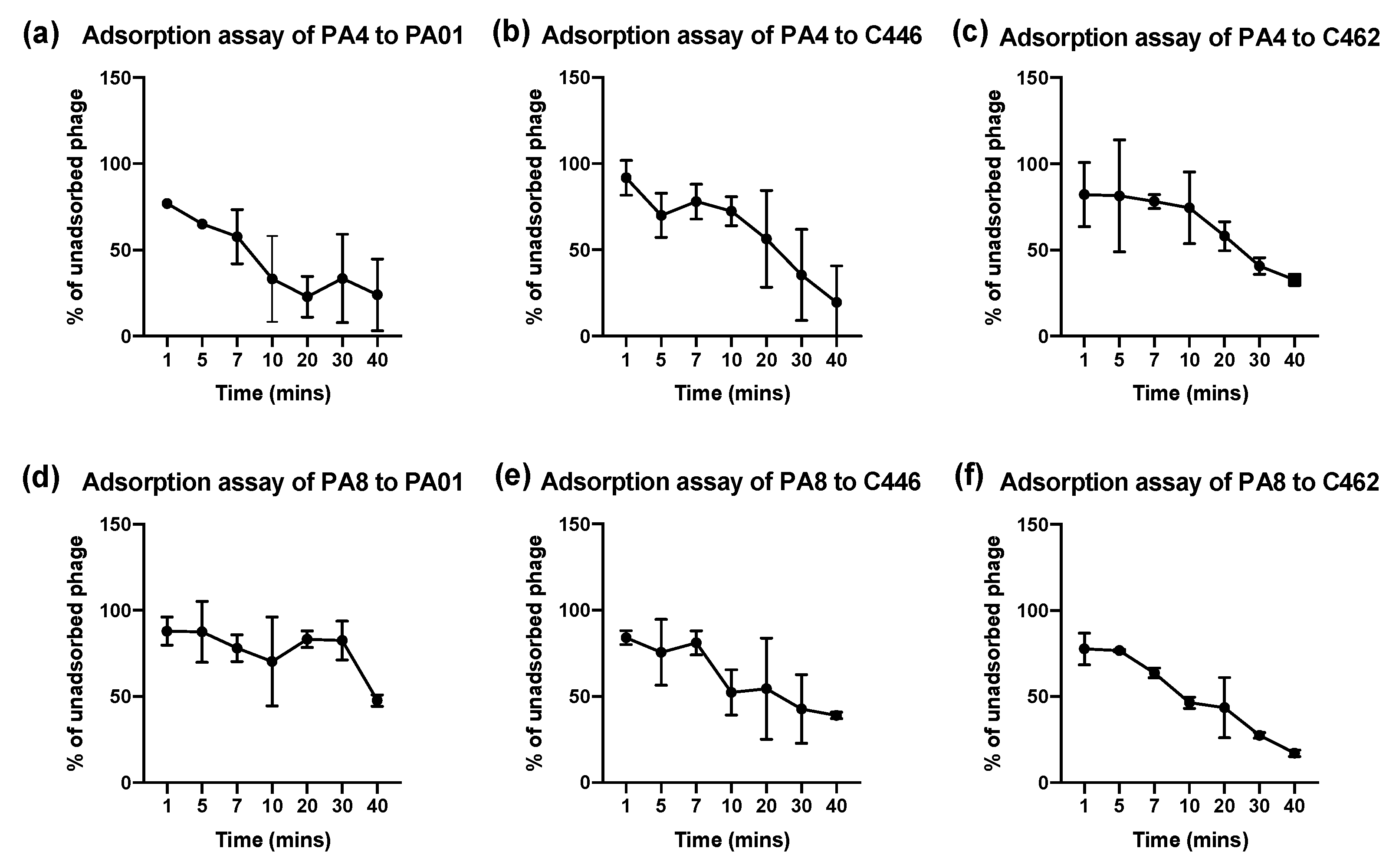
Phages PA4 and PA8 were selected for planktonic testing against 1 CF (C462) and 1 non-CF (C446) clinical isolate along with the PA01 reference strain. The optical density (OD) was measured at 600 nm in a spectrophotometer (SmartSpec 3000, Bio-Rad, CA, USA) for each overnight culture of CF and non-CF P. aeruginosa CIs and 400 μL of each overnight culture was diluted 1:100 in fresh TSB in a total volume of 40 mL. Phages were added at MOI = 0.1. The bacterial suspension was incubated at 37 °C with 180 rpm shaking. After taking an initial 3 mL sample at 0 min, 3 mL samples were taken at 1 min, 5 min, 7 min, 10 min and every 10 min thereafter for 40 min. The samples were centrifuged at 4000 rpm for 10 min. The supernatant containing the unadsorbed phages was filtered through a 0.2 μm filter and plated using the double agar spot assay method. The percent of unadsorbed phages of initial phage inoculum at each time interval was calculated. The adsorption experiments were repeated 3 times.
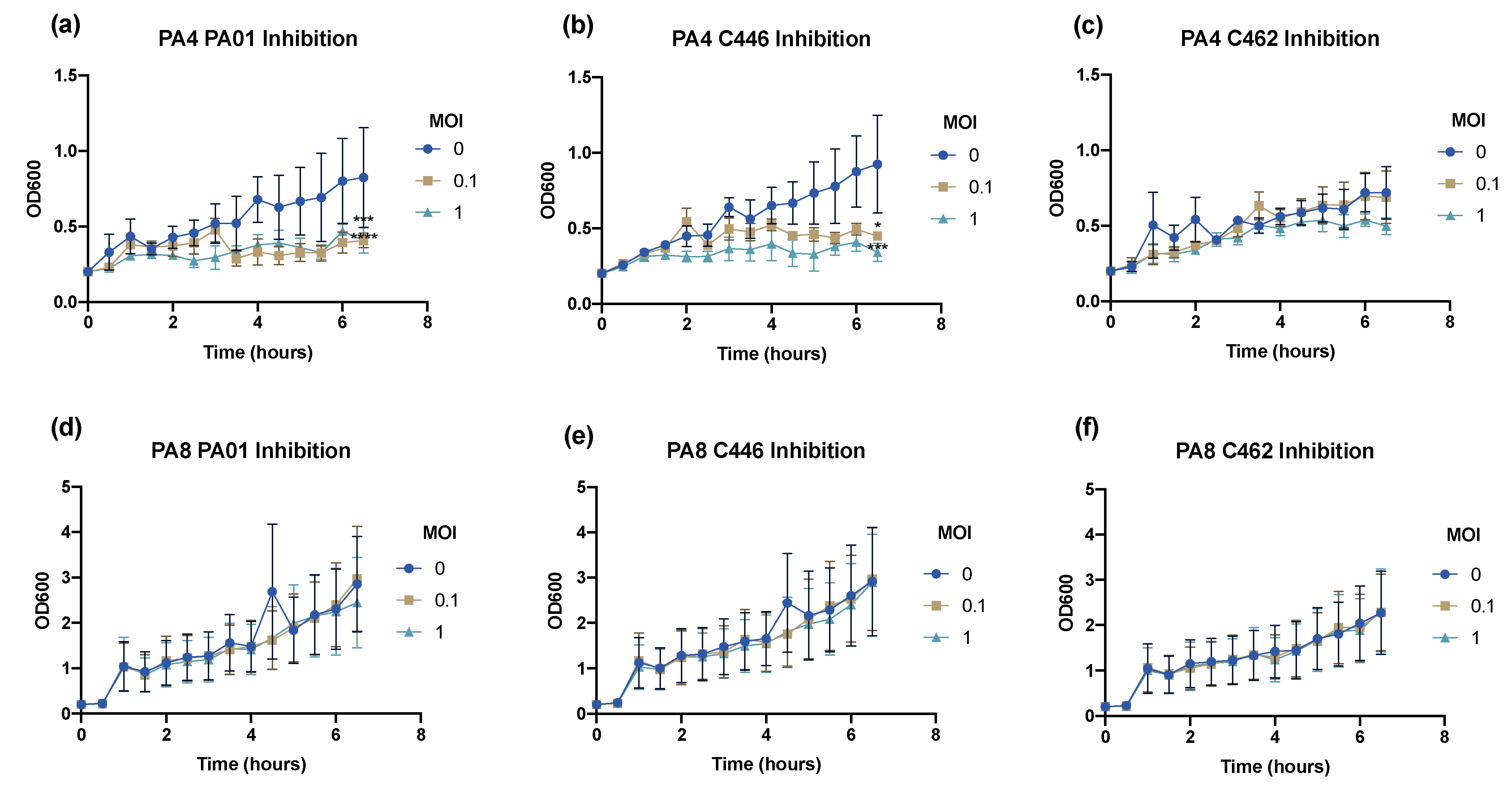
2.9. Bacteriophage Inhibition Assays
The infectivity profile of the 2 selected phages was assessed at MOI = 0, 0.1 and 1 using inhibition assays. In brief, the overnight cultures of each clinical isolate were prepared and the required volume of phage stock solution (~108 PFU mL−1) was added to the corresponding suspension to achieve an MOI = 0, 0.1 and 1. The samples were incubated at 37 °C with 180 rpm shaking. Every 30 min for 6.5 h, the OD600 value was measured.
2.10. Crystal Violet Biofilm Assay
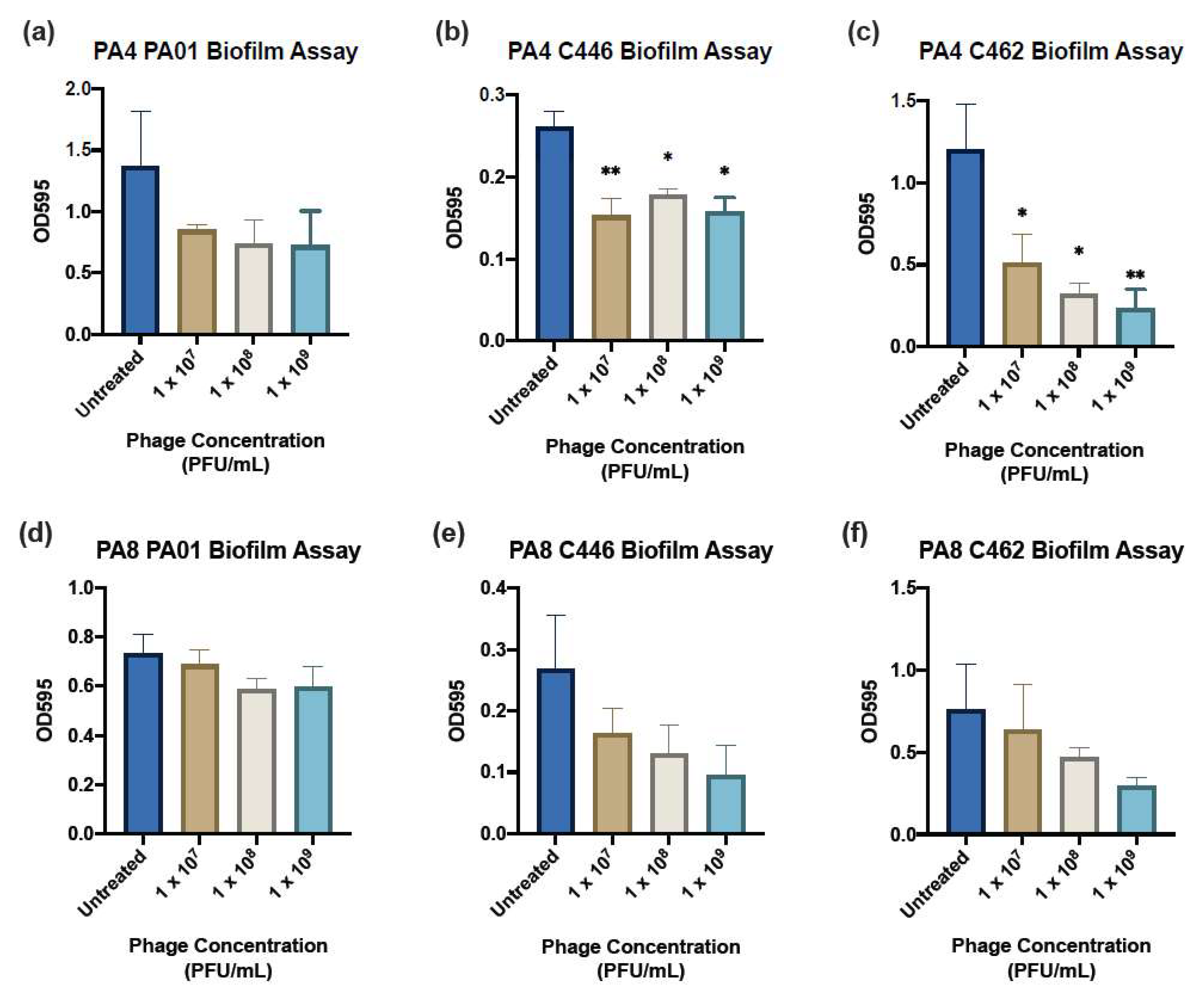
A 1.0 McF suspension in 0.9% saline of the clinical isolate was diluted 1:15 in TSB and gently mixed by inversion. An amount of 150 μL/well of the resulting suspension was plated into a FluoroNunc, U-Shaped 96-well Microplate (Thermo Fisher Scientific, Roskilde, Denmark). Wells adjacent to the top and bottom edge of the plate were filled with 200 μL of sterile phosphate buffer solution (PBS) to prevent dehydration during incubation. Positive and negative controls were added to each plate containing no phage treatment and only TSB solution, respectively. The plates were incubated for 24 h on a gyratory mixer (Ratek, VIC, Australia). Following 24 h, the wells were gently aspirated and washed twice with sterile PBS to remove any remaining planktonic cells. An amount of 180 μL/well of each treatment (phages PA4 and PA8) in TSB each at concentrations of 1 × 107, 108 and 109 PFU mL−1 were plated in six replicates and the biofilms were assessed after 24 h incubation. At 24 h post-treatment, the liquid contents of the wells were aspirated and washed gently twice with sterile PBS and stained with 180 μL/well 0.1% crystal violet (Sigma-Aldrich, Castle Hill, NSW, Australia) for 15 min. The stained plates were rinsed with distilled water and left to dry overnight. The crystal violet stain was eluted by the application of 180 μL/well 30% acetic acid (Chem-Supply, Adelaide, SA, Australia) and the plate was incubated at RT for 1 h. The acetic acid was pipetted into a new flat-bottom 96-well plate (BMG Labtech, Ortenberg, Germany) and the absorbance measured at 595 nm using the FLUOstar Optima microplate reader (BMG Labtech, Ortenberg, Germany).
2.11. LIVE/DEAD Staining
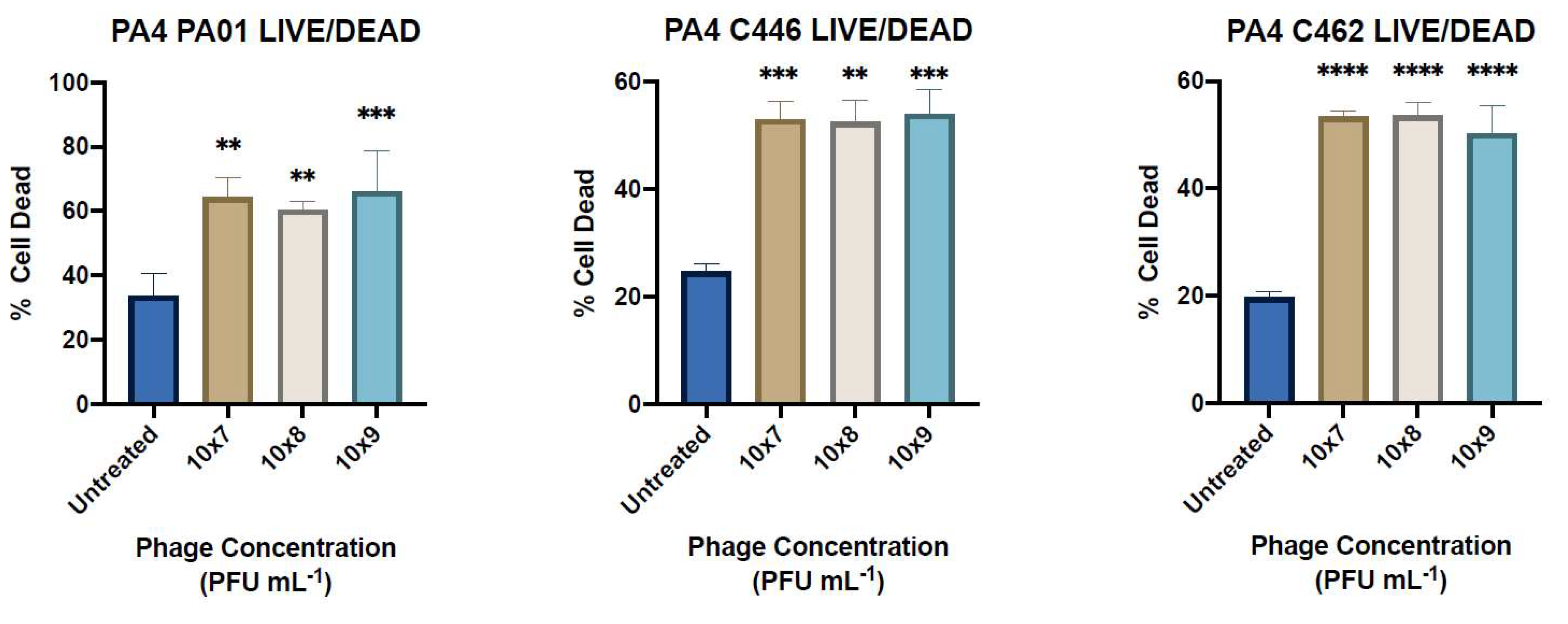
The biofilm viability was determined using the Invitrogen LIVE/DEAD BacLight Bacterial Viability Kit (Thermo Fisher Scientific, United Kingdom). Briefly, biofilms were grown over 24 h in 8-well cell imaging slides (Eppendorf, Hamburg, Germany) containing no bacteriophage as a positive control. The wells were aspirated and washed gently twice with 0.9% saline before application of 400 μL/well of PA4 treatment in TSB at concentrations of 1 × 107, 108 and 109 PFU mL−1. Following 24 h incubation, the wells were aspirated and washed again prior to the application of 180 μL/well 5% glutaraldehyde fixative (Sigma-Aldrich, Castle Hill, NSW, Australia) for 45 min. Prior to staining, the wells were aspirated and washed again. Two stock solutions of stain SYTO9 and propidium iodide (PI) were each diluted in MilliQ water to give a 1 mL:1.5 µL:1.5 µL ratio and 400 µL was applied to each well for 15 min before aspirating and washing the wells. Live SYTO9-stained cells and dead PI-stained cells were visualised with a confocal laser microscope at 20× magnification and the percentage of cell death was determined (Zeiss LSM700, Carl Zeiss AG, Oberkochen, Germany).
2.12. Bacteriophage Morphology
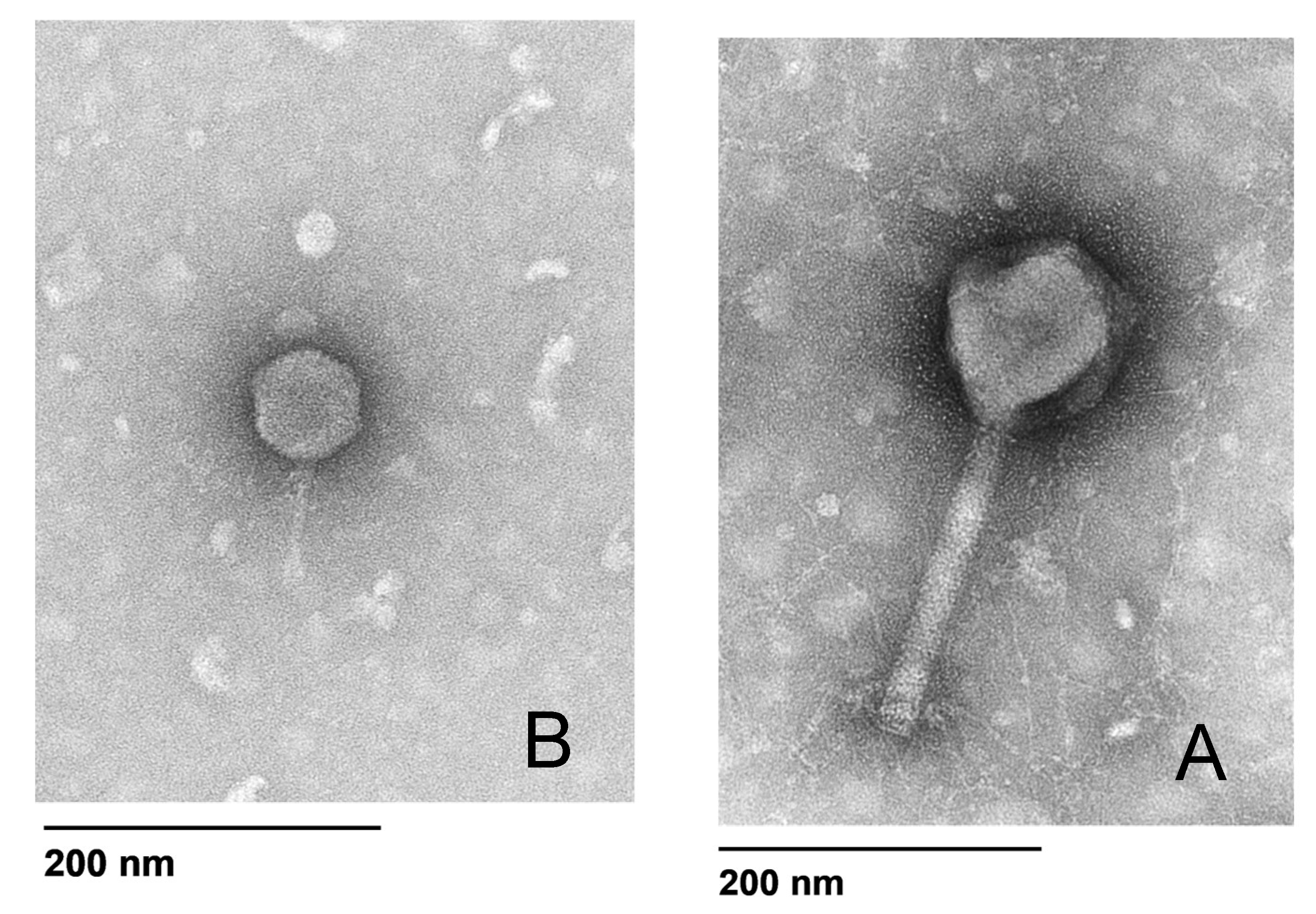
The phage morphology for PA4 and PA8 was determined by transmission electron microscopy (TEM) using a modified protocol [
22]. Briefly, 5 μL of phage diluted 1:10 in SM buffer was placed on the coated side of a carbon/formvar grid (ProSciTech Pty Ltd., Kirwan, QLD, Australia) for 3 min before being wicked dry with filter paper. An amount of 5 μL of electron microscopy (EM) fixative (1.25% glutaraldehyde, 4% paraformaldehyde in PBS to which 4% sucrose had been added) was then placed on the grid for 2 min before being wicked dry, followed by 5 μL of 2% uranyl acetate for 2 min. The samples were examined in a FEI Tecnai G2 Spirit 120 kV TEM (FEI Technologies Inc., Hillsboro, OR, USA).
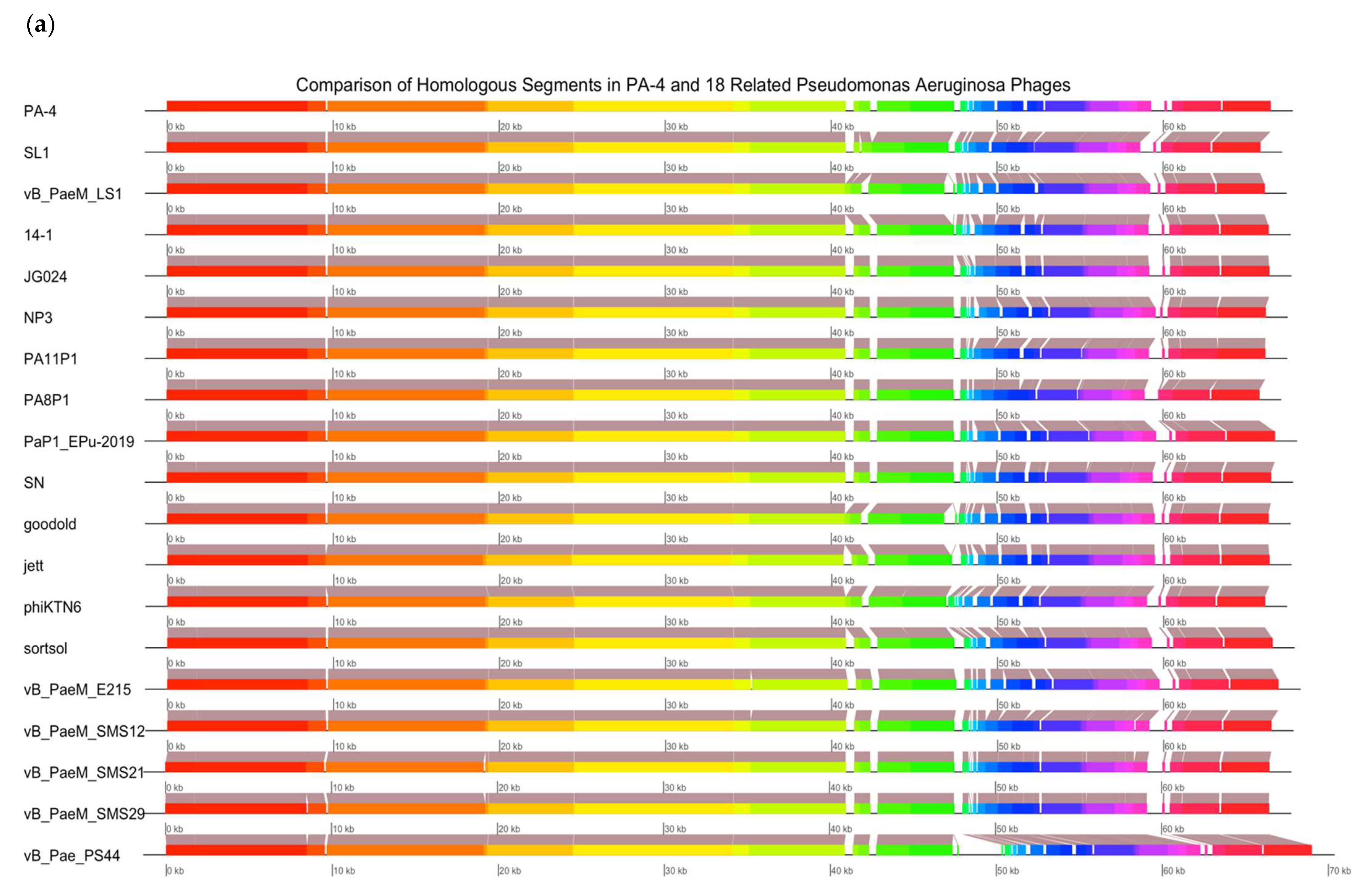
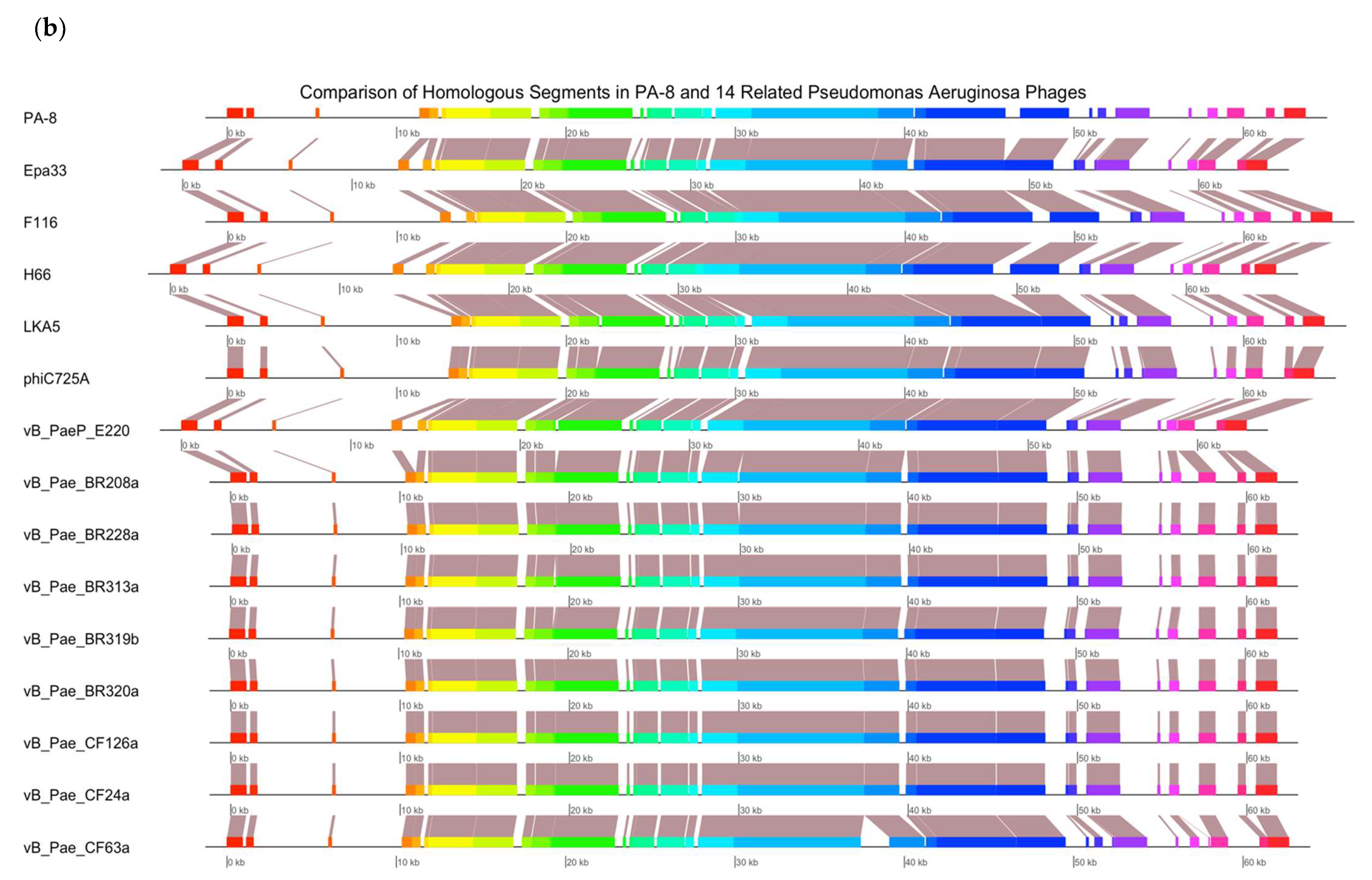
2.13. Bacteriophage Genomic Sequencing
Genomic sequencing was carried out for PA4 and PA8. A DNA extraction from a 1 mL aliquot of phage (>1 × 10
10 PFU mL
−1) was performed using the Phage DNA Isolation Kit (Norgen Biotek Corp, Thorold, ON, Canada), following the protocol provided by the manufacturer. A genomic sequencing of the isolated DNA was conducted by an external laboratory (SA Pathology, Adelaide, SA, Australia). Paired-end sequence reads were obtained on an Illumina sequencer. Poor reads were trimmed using Trimmomatic [
23]. The genome assembly was performed using the De Bruijn graph-based short read de novo assembler ABYSS 2.0 [
24]. The sequences were verified using QUAST [
25] and annotated with PROKKA [
26] and RASTtk [
27]. A genomic analysis was conducted with the non-redundant (nr) NCBI database using MEGABLAST. Genome alignments were obtained using Mauve and plotted in R using genoplotR. An individual analysis of the phages included the phage in question and 14–18 closely related
P. aeruginosa phage genomes available in the nr NCBI database.
2.14. Statistical Analysis
A Statistical analysis was performed using GraphPad Prism v.9 (GraphPad Prism, version 9.0.0 (86); Macintosh Version by Software MacKiev © 2021-2020 GraphPad Software, LLC.; San Diego, CA, USA). To determine the effect of pH, high temperature and long-term storage on phage viability, as well as planktonic and biofilm efficacy, data was analysed using a one-way analysis of variance (ANOVA) followed by Tukey’s and Dunnett’s post hoc multiple comparison test. Significant differences were determined at p < 0.05. All of the experiments were repeated three times and performed in triplicate or six replicates. The mean values of the three or six replicates obtained with standard error of the means (SEM).
4. Discussion
A
P. aeruginosa airway infection is associated with worsening lung function in CF patients [
2]. Treatment with systemic and inhaled broad-spectrum antibiotics from early in the life of those patients can lead to the development of antibiotic-resistant strains of
P. aeruginosa [
2,
3,
4]. Due to the emerging threat of MDR infections and lack of research and development of new antibiotics, there is an urgent need for new therapeutic treatment options [
6].
P. aeruginosa phages have shown to be effective against planktonic and biofilm
P. aeruginosa; however, clinical-grade phages are currently not available in Australia [
1,
4,
14,
30].
In this study,
P. aeruginosa phages isolated from wastewater displayed a broad host range to the 20 clinical isolates tested (9 CF and 11 non-CF), with a total of 18/20 (90%) isolates susceptible (sensitive or semi-sensitive) to at least 1/15 phages. The infective properties of two selected phages (PA4 and PA8) demonstrated variability in bacterial killing, and therefore the potential to increase the target host range when combined in a cocktail and were selected for further analysis. One particular concern with the use of phages is indeed the emergence of phage-resistant bacteria [
18,
31]. The combination of phages in cocktails that are regularly changed with new or different phages could potentially address this issue by maintaining selective pressure on the bacterial host [
31].
Phage applications in humans require them to be stable in acidic, alkaline and high- or low-temperature conditions. In a therapeutic setting, pH levels for the administration of phages need to be considered [
4]. For example, oral administration of phages in a tablet form would pass through a very acidic environment in the gastrointestinal tract [
4]. A nasal spray would expose the phages to a different environment with pH levels around 5.5–6.5 dependent on the nasal mucosa [
4]. Results from this study show phages were stable at a wide pH range (3–11).
Temperature is an important factor in phage survivability and plays a significant role in phage infective properties, such as attachment and replication [
32]. Phage activity at different temperatures varies amongst strains, and studies have shown that high temperatures can lead to nucleic acid and protein denaturation and an inactivate phage [
31]. In the current study, the phages were viable between 4–50 °C with seven of the fifteen phages maintaining viability at 60 °C and 70 °C; however, exposure to 80 °C for 1 h inactivated all of the phages. This variation may be due to different strains being more sensitive to higher temperatures, or phages developing heat resistance through mutations [
31].
The long-term storage and stability of selected phages is desirable, and a good candidate for phage therapy should be one that maintains an infective ability upon storage [
31]. The phage long-term storage viability was determined at RT, 4 °C, −20 °C and −80 °C to ensure their integrity during storage and transfer [
31]. The phages survived well at −80 °C, with minimal activity loss over 6 months of storage. The phage activity at 4 °C and −20 °C demonstrated similar results with a greater reduction in the viability compared to −80 °C; however, the viability of seven phages at RT began to decline after 1month of storage. This is in line with previous studies which suggested the storage of phages at RT is recommended for no longer than 40 days and long-term storage of phages is recommended at −80 °C [
32].
The efficacy of phages to infect planktonic bacterial cells was measured using phage adsorption and inhibition assays. The phage binding properties varied amongst clinical isolates; however, PA4 demonstrated a strong attachment to the target pathogen, suggesting it had efficient binding properties towards the host surface. This phage, belonging to the Myoviridae family, varies substantially in its structure compared to PA8, belonging to the Podoviridae family, as confirmed by TEM. The activity of PA4 showed an inhibitory effect towards clinical isolates over 6.5 h, unlike PA8, which showed negligible effects. Sequencing of these phages revealed the lytic ability of PA4, which supports these findings, whilst PA8 is characteristic of a temperate phage and instead has the capacity to integrate into the bacterial host genome. This phage was therefore excluded for inclusion in future clinical-grade phage cocktails.
One of the main advantages of phage treatments is their activity against MDR pathogens and their supposed efficacy against biofilms [
2]. In this study, some of the clinical isolates (e.g., C458) were resistant to at least two classes of commonly used antibiotics in the treatment of
P. aeruginosa infection and 14/20 of the isolates (including C458) were sensitive or semi-sensitive to PA4. The lytic effect of PA4 on planktonic bacteria translated well to a reduction in the biofilm biomass, given the strictly lytic nature of the phage. This is consistent with previous studies showing the efficacy of phages to reduce biofilm in vitro and
in vivo [
17]. The results show a slightly better killing of the C462 strain at 1 × 10
9 PFU mL
−1 after 24 h of treatment. However, the ability of PA4 to reduce the biomass of the C446 strain tended to be greater at a lower concentration. This is supported by other studies of
P. aeruginosa phages, which have shown that a higher concentration does not necessarily translate to a greater antibiofilm activity [
33,
34]. This non-linear relationship may be expected for some phages due to their self-replicating nature [
33]. The advantage of screening phages at various concentrations is the possibility of identifying the lowest acceptable concentration range since the application of phages at large volumes in a therapeutic setting may be problematic and costly [
33]. Therefore, the ability of PA4 to effectively reduce the biomass at a low concentration makes it a suitable candidate for phage therapy. This is demonstrated in whole by its broad infectivity profile, good stability in various pH and temperature conditions, lytic ability and the absence of antibiotic-resistant, toxic or lysogenic genes.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}