
Identification of the Key Genes Associated with Different Hair Types in the Inner Mongolia Cashmere Goat

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Construction of Weighted Gene Coexpression Network
2.3. Screening of Candidate Modules
2.4. Enrichment Analysis of Candidate Modules
2.5. Candidate Gene Screening
2.6. Sample Collection
2.7. Quantitative Real-Time PCR (qRT-PCR)
2.8. Data Analysis
3. Results
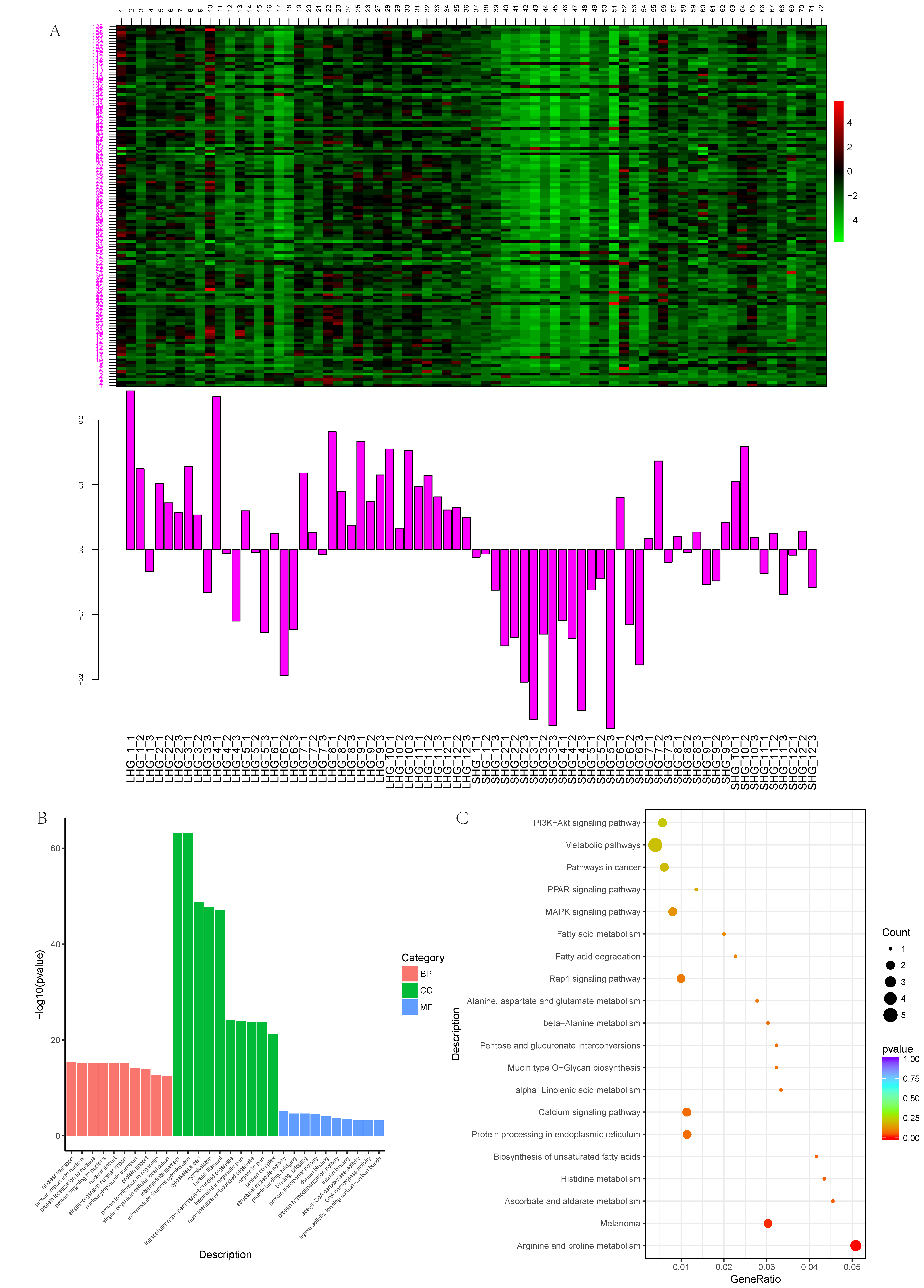
3.1. Construction of Weighted Gene Coexpression Network
3.2. Key Modules Related to Hair Follicle Cycle Development
3.3. Magenta Module Function Analysis
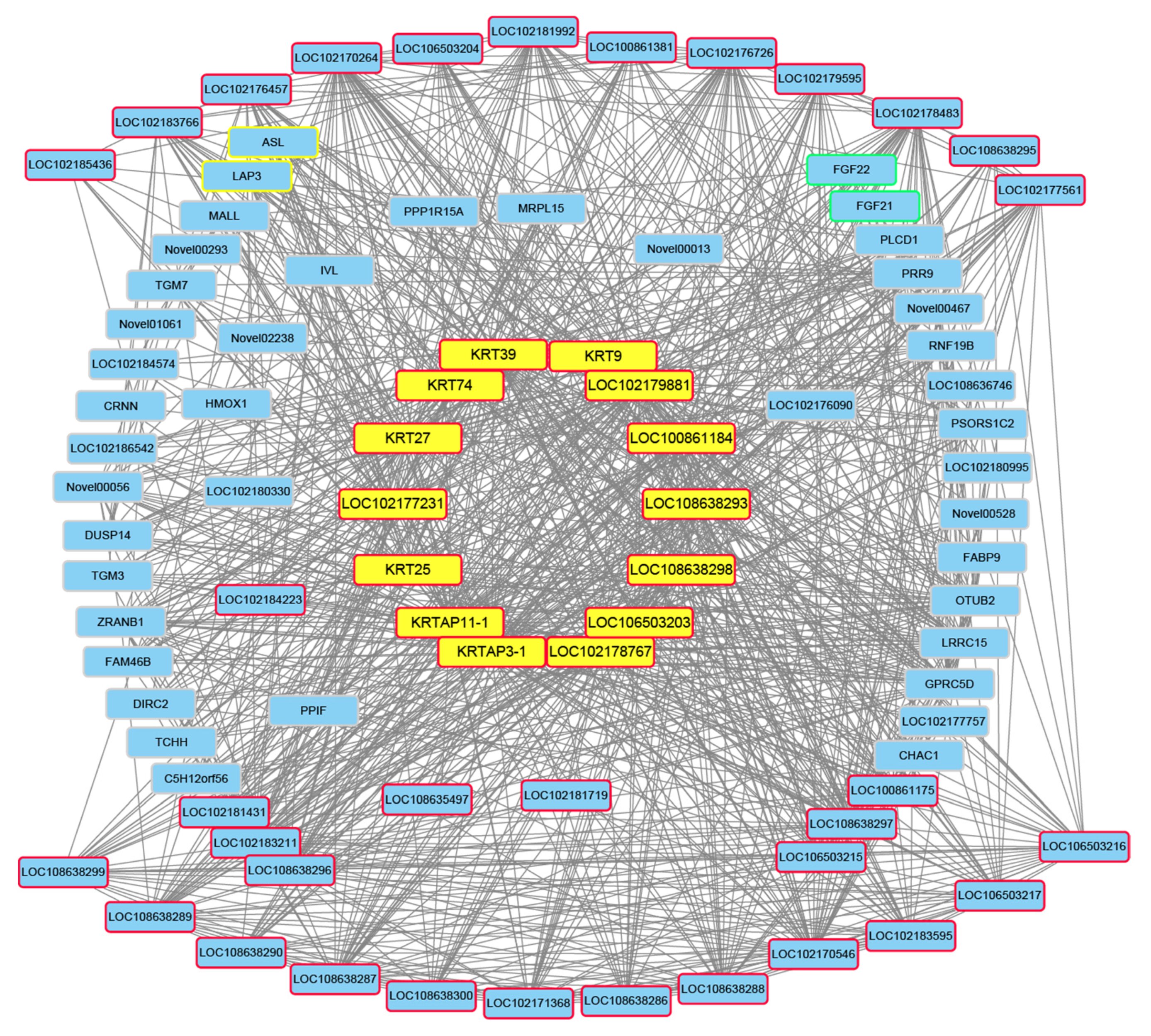
3.4. Screening of Candidate Genes for Different Hair Types
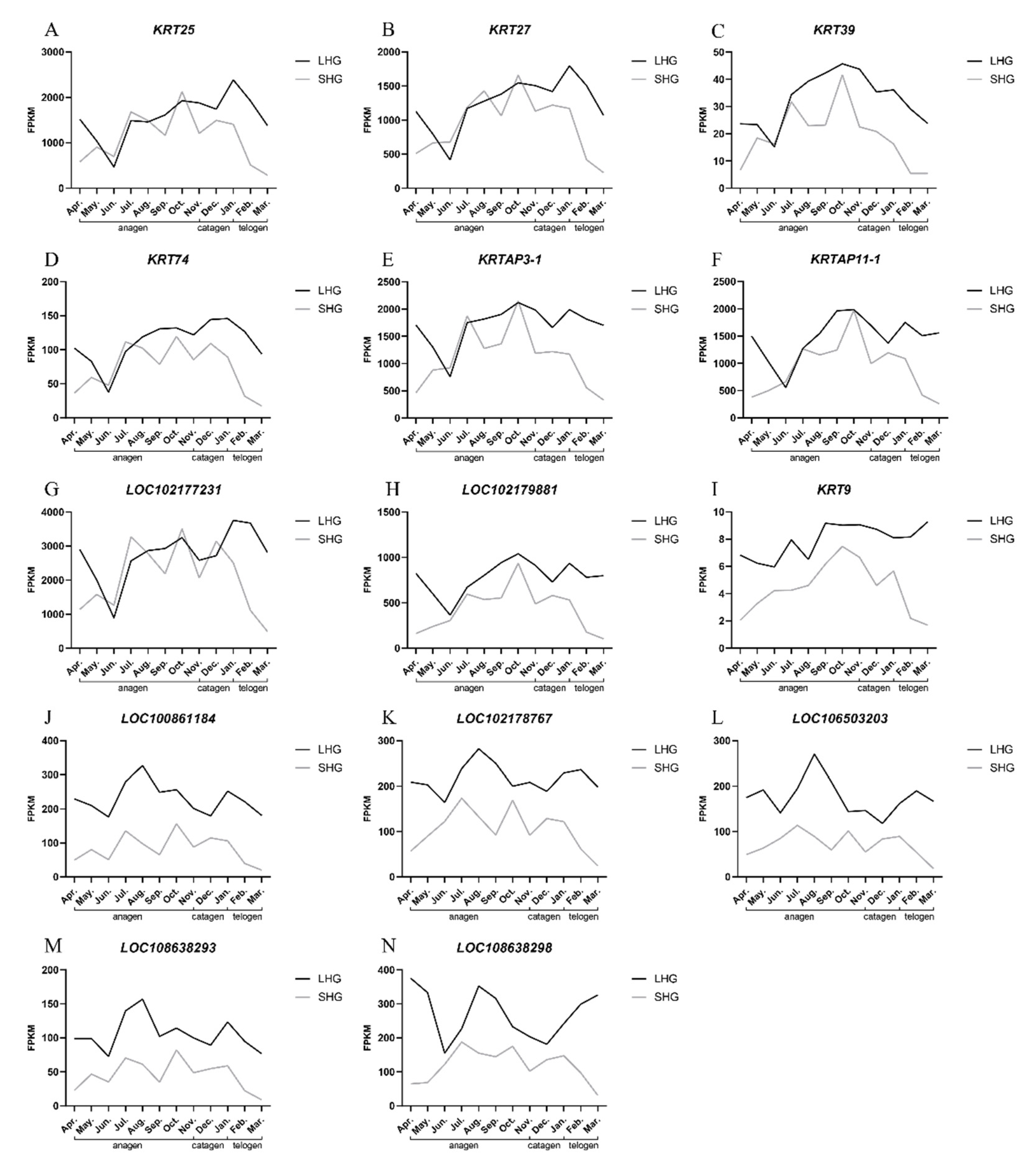
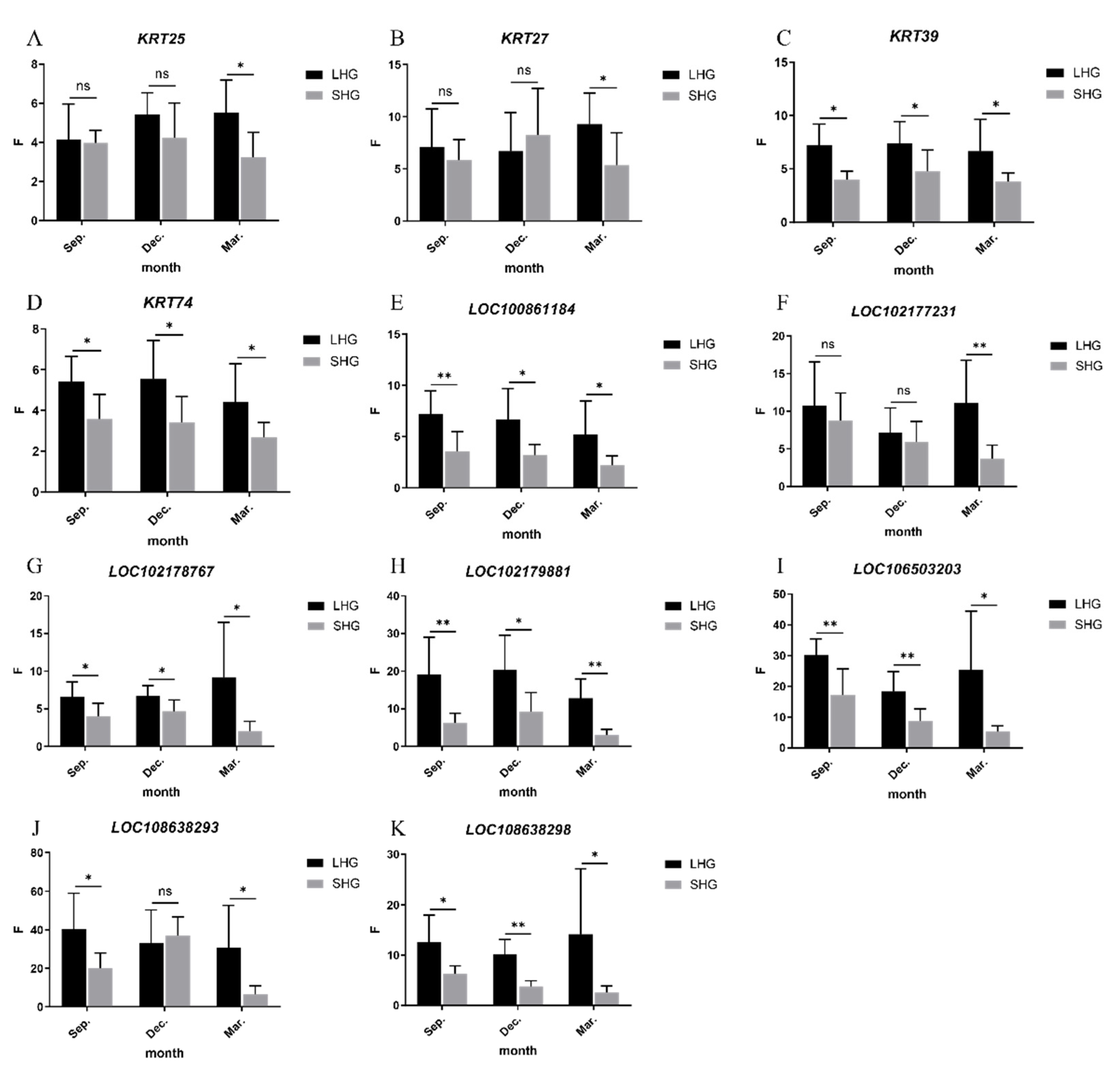
3.5. Detection of Relative Expression of Candidate Genes by qRT-PCR
3.6. Correlation Analysis between Gene Expression and Hair Length
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berger, J.; Buuveibaatar, B.; Mishra, C. Globalization of the cashmere market and the decline of large mammals in central Asia. Conserv. Biol. 2013, 27, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Dubeuf, J. Situation, changes and future of goat industry around the world. Small Rumin. Res. 2003, 51, 165–173. [Google Scholar] [CrossRef]
- Ge, W.; Zhang, W.; Zhang, Y.; Zheng, Y.; Li, F.; Wang, S.; Liu, J.; Tan, S.; Yan, Z.; Wang, L.; et al. A Single-cell Transcriptome Atlas of Cashmere Goat Hair Follicle Morphogenesis. Genom. Proteom. Bioinform. 2021, 19, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Cotsarelis, G. The biology of hair follicles. N. Engl. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Su, R.; Gong, G.; Zhang, L.; Yan, X.; Wang, F.; Zhang, L.; Qiao, X.; Li, X.; Li, J. Screening the key genes of hair follicle growth cycle in Inner Mongolian Cashmere goat based on RNA sequencing. Arch. Anim. Breed. 2020, 63, 155–164. [Google Scholar] [CrossRef]
- Yuan, C.; Wang, X.; Geng, R.; He, X.; Qu, L.; Chen, Y. Discovery of cashmere goat (Capra hircus) microRNAs in skin and hair follicles by Solexa sequencing. BMC Genom. 2013, 14, 511. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, R.L.; Zhu, X.P.; Kleernann, D.O.; Yue, C.W.; Jia, Z.H. Effects of dietary copper on ruminal fermentation, nutrient digestibility and fibre characteristics in Cashmere goats. Asian-Australas. J. Anim. Sci. 2007, 20, 1843–1848. [Google Scholar] [CrossRef]
- Song, S.; Yao, N.; Yang, M.; Liu, X.X.; Dong, K.Z.; Zhao, Q.J.; Pu, Y.B.; He, X.H.; Guan, W.J.; Yang, N.; et al. Exome sequencing reveals genetic differentiation due to high-altitude adaptation in the Tibetan cashmere goat (Capra hircus). BMC Genom. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Todini, L. Thyroid hormones in small ruminants: Effects of endogenous environmental and nutritional factors. Animal 2007, 1, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Shi, J.; Zhang, Y.; Lian, A.; Han, X.; Zuo, K.; Liu, M.; Zheng, T.; Zou, F.; Liu, X.; et al. NANOG Attenuates Hair Follicle-Derived Mesenchymal Stem Cell Senescence by Upregulating PBX1 and Activating AKT Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 4286213. [Google Scholar] [CrossRef] [Green Version]
- Bhat, B.; Yaseen, M.; Singh, A.; Ahmad, S.M.; Ganai, N.A. Identification of potential key genes and pathways associated with the Pashmina fiber initiation using RNA-Seq and integrated bioinformatics analysis. Sci. Rep. 2021, 11, 1766. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Zhou, H.; Wang, J.; Li, S.; Luo, Y.; Hickford, J.G.H. Characterisation of an Ovine Keratin Associated Protein (KAP) Gene, Which Would Produce a Protein Rich in Glycine and Tyrosine, but Lacking in Cysteine. Genes 2019, 10, 848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, M.; Komi-Kuramochi, A.; Asada, M.; Suzuki, M.; Oki, J.; Jiang, J.; Imamura, T. Comprehensive analysis of FGF and FGFR expression in skin: FGF18 is highly expressed in hair follicles and capable of inducing anagen from telogen stage hair follicles. J. Investig. Dermatol. 2005, 124, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibraheem, M.; Galbraith, H.; Scaife, J.; Ewen, S. Growth of secondary hair follicles of the Cashmere goat in vitro and their response to prolactin and melatonin. J. Anat. 1994, 185 Pt 1, 135–142. [Google Scholar]
- Dicks, P.; Russel, A.J.; Lincoln, G.A. The role of prolactin in the reactivation of hair follicles in relation to moulting in cashmere goats. J. Endocrinol. 1994, 143, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q. Study on Breeding Methods in Inner Mongolia Cashmere Goats. Ph.D. Thesis, China Agricultural University, Beijing, China, 2005. [Google Scholar]
- Na, Q. Genetic Pattern of Different Types of Hair Coat in Inner Mongolia Cashmere Goat and Their Effect on Important Economic Traits. Master’s Thesis, Inner Mongolia Agricultural University, Hohhot, China, 2016. [Google Scholar]
- Li, X.W.; Wang, R.J.; Wang, Z.Y.; Li, H.W.; Wang, Z.Y.; Su, R.; Zhang, Y.J.; Liu, Z.H.; Li, J.Q. Genetic parameter estimation of cashmere yield and body weight at different staple types of Inner Mongolian cashmere goats. Sci. Agric. Sin. 2018, 23, 53–59. [Google Scholar]
- Li, X.W. Study on Genetic Law and Indirect Selection for Important Economic Traits of Inner Mongolian Cashmere Goats at Different Staple Types. Ph.D. Thesis, Inner Mongolia Agricultural University, Hohhot, China, 2019. [Google Scholar]
- Hao, D.; Wang, X.; Yang, Y.; Thomsen, B.; Holm, L.E.; Qu, K.; Huang, B.; Chen, H. Integrated Analysis of mRNA and MicroRNA Co-expressed Network for the Differentiation of Bovine Skeletal Muscle Cells After Polyphenol Resveratrol Treatment. Front. Vet. Sci. 2021, 8, 777477. [Google Scholar] [CrossRef]
- Bo, D.; Jiang, X.; Liu, G.; Hu, R.; Chong, Y. RNA-Seq Implies Divergent Regulation Patterns of LincRNA on Spermatogenesis and Testis Growth in Goats. Animals 2021, 11, 625. [Google Scholar] [CrossRef]
- Zhang, X.; Bao, P.; Ye, N.; Zhou, X.; Zhang, Y.; Liang, C.; Guo, X.; Chu, M.; Pei, J.; Yan, P. Identification of the Key Genes Associated with the Yak Hair Follicle Cycle. Genes 2021, 13, 32. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.M.; Mao, X.Z.; Cai, T.; Luo, J.C.; Wei, L.P. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.C.; Gillespie, J.M. Comparison of samples of human hair by two-dimensional electrophoresis. J. Forensic Sci. Soc. 1982, 22, 377–385. [Google Scholar] [CrossRef]
- Zhou, H.; Gong, H.; Wang, J.; Luo, Y.; Li, S.; Tao, J.; Hickford, J.G.H. The Complexity of the Ovine and Caprine Keratin-Associated Protein Genes. Int. J. Mol. Sci. 2021, 22, 2838. [Google Scholar] [CrossRef] [PubMed]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Wildermoth, J.E.; Wallace, O.A.; Gordon, S.W.; Maqbool, N.J.; Maclean, P.H.; Nixon, A.J.; Pearson, A.J. Annotation of sheep keratin intermediate filament genes and their patterns of expression. Exp. Dermatol. 2011, 20, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Langbein, L.; Rogers, M.A.; Praetzel-Wunder, S.; Helmke, B.; Schirmacher, P.; Schweizer, J. K25 (K25irs1), K26 (K25irs2), K27 (K25irs3), and K28 (K25irs4) represent the type I inner root sheath keratins of the human hair follicle. J. Investig. Dermatol. 2006, 126, 2377–2386. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, Y.; Hayashi, R.; Tsuruta, D.; Shimomura, Y.; Sugawara, K. Novel splice site mutation in the LIPH gene in a patient with autosomal recessive woolly hair/hypotrichosis: Case report and published work review. J. Dermatol. 2018, 45, 613–617. [Google Scholar] [CrossRef]
- Ansar, M.; Raza, S.I.; Lee, K.; Shahi, S.; Acharya, A.; Dai, H.; Smith, J.D.; University of Washington Center for Mendelian Genomics; Shendure, J.; Bamshad, M.J.; et al. A homozygous missense variant in type I keratin KRT25 causes autosomal recessive woolly hair. J. Med. Genet. 2015, 52, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Liu, G.; Liu, Y.; Xu, Q.; Zhang, M.; Fang, M. Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep. PLoS ONE 2013, 8, e71763. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Almohanna, H.; Griggs, J.; Tosti, A. Genetic Hair Disorders: A Review. Dermatol. Ther. 2019, 9, 421–448. [Google Scholar] [CrossRef] [Green Version]
- Salmela, E.; Niskanen, J.; Arumilli, M.; Donner, J.; Lohi, H.; Hytonen, M.K. A novel KRT71 variant in curly-coated dogs. Anim. Genet. 2019, 50, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, B.; Alhaddad, H.; Joslin, S.E.; Khan, R.; Filler, S.; Brem, G.; Lyons, L.A. A splice variant in KRT71 is associated with curly coat phenotype of Selkirk Rex cats. Sci. Rep. 2013, 3, 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langbein, L.; Rogers, M.A.; Praetzel, S.; Winter, H.; Schweizer, J. K6irs1, K6irs2, K6irs3, and K6irs4 represent the inner-root-sheath-specific type II epithelial keratins of the human hair follicle. J. Investig. Dermatol. 2003, 120, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, Y.; Wajid, M.; Petukhova, L.; Kurban, M.; Christiano, A.M. Autosomal-dominant woolly hair resulting from disruption of keratin 74 (KRT74), a potential determinant of human hair texture. Am. J. Hum. Genet. 2010, 86, 632–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Gordon, S.W.; Nixon, A.J.; Bawden, C.S.; Rogers, M.A.; Wildermoth, J.E.; Maqbool, N.J.; Pearson, A.J. Expression patterns of keratin intermediate filament and keratin associated protein genes in wool follicles. Differentiation 2009, 77, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Langbein, L.; Rogers, M.A.; Winter, H.; Praetzel, S.; Beckhaus, U.; Rackwitz, H.R.; Schweizer, J. The catalog of human hair keratins. I. Expression of the nine type I members in the hair follicle. J. Biol. Chem. 1999, 274, 19874–19884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, Y.; Yokohama, M.; Wada, K.; Fujita, M.; Kotani, M.; Nagura, Y.; Kanno, M.; Nomura, K.; Amano, T.; Kikkawa, Y. Expression analysis of the type I keratin protein keratin 33A in goat coat hair. Anim. Sci. J. 2011, 82, 773–781. [Google Scholar] [CrossRef]
- Bao, Q.; Zhang, X.; Bao, P.; Liang, C.; Guo, X.; Yin, M.; Chu, M.; Yan, P. Genome-wide identification, characterization, and expression analysis of keratin genes (KRTs) family in yak (Bos grunniens). Gene 2022, 818, 146247. [Google Scholar] [CrossRef]
- Fu, X.; Zhao, B.; Tian, K.; Wu, Y.; Suo, L.; Ba, G.; Ciren, D.; De, J.; Awang, C.; Gun, S.; et al. Integrated analysis of lncRNA and mRNA reveals novel insights into cashmere fineness in Tibetan cashmere goats. PeerJ 2020, 8, e10217. [Google Scholar] [CrossRef]
- Choi, H.I.; Choi, G.I.; Kim, E.K.; Choi, Y.J.; Sohn, K.C.; Lee, Y.; Kim, C.D.; Yoon, T.J.; Sohn, H.J.; Han, S.H.; et al. Hair greying is associated with active hair growth. Br. J. Dermatol. 2011, 165, 1183–1189. [Google Scholar] [CrossRef]
- Plowman, J.; Thomas, A.; Perloiro, T.; Clerens, S.; de Almeida, A.M. Characterisation of white and black merino wools: A proteomics study. Animal 2019, 13, 659–665. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Chen, L.; Luo, F.; Zhou, H.; Wang, J.; Zhang, Q.; Lu, T.; Wu, S.; Hickford, J.G.H.; Tao, J. Differentially phosphorylated proteins in the crimped and straight wool of Chinese Tan sheep. J. Proteom. 2021, 235, 104115. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequences | Product Length (bp) | TM (°C) | |
|---|---|---|---|---|
| KRT25 | F: | AAGTCAGGACCGAGACCGAG | 129 | 61 |
| R: | CAGACTTGCAAGCCCCATCAT | |||
| KRT27 | F: | ACTTCACGGTAATTGACGACCT | 283 | 60 |
| R: | GCCTTCATCTCCTCCTCATGG | |||
| KRT39 | F: | TGAGATTGCCACATACCGCA | 169 | 59 |
| R: | CTCATGTATCCCACAGGGGC | |||
| KRT74 | F: | GATCAATCAGCGCACAGCAG | 152 | 60 |
| R: | CCACCTCCGCATCGTACAAA | |||
| LOC102179881 | F: | GTGCAGAGCCTGATCGTCAA | 252 | 60 |
| R: | TCCACACCGAGTACGTGAGA | |||
| LOC108638298 | F: | GCGCTCACCATCCCTAAGTA | 117 | 60 |
| R: | GGGTGCCTCCTAGTTTGAAGA | |||
| LOC102177231 | F: | TGTGGATGCTGCTTATGCCA | 171 | 60 |
| R: | AGGTTTAGGTCGCGGTTGTT | |||
| LOC100861184 | F: | CCCTGTGTACTGCCACAGAA | 198 | 60 |
| R: | TGACAGGAGGATCAGCAGGA | |||
| LOC108638293 | F: | GCCAAGCCCAGGAATGT | 219 | 60 |
| R: | CCAGTTGTCAGCAAAGTCTC | |||
| LOC106503203 | F: | AAACTCACCCAGAACCTCCA | 112 | 58 |
| R: | GGACGGTAGCAGGTCTCTT | |||
| LOC102178767 | F: | GTCTCCTGCCACACCACTTG | 164 | 60 |
| R: | CCTAAGGGTCAGCGCGAAA | |||
| GAPDH | F: | GCAAGTTCCACGGCACAG | 118 | 60 |
| R: | TCAGCACCAGCATCACCC | |||
| β-actin | F: | GGCAGGTCATCACCATCGG | 158 | 60 |
| R: | CGTGTTGGCGTAGAGGTCTTT | |||
| Module Color | Gene Numbers | Module Color | Gene Numbers |
|---|---|---|---|
| Black | 181 | Magenta | 128 |
| Blue | 1839 | Midnight blue | 90 |
| Brown | 620 | Pink | 168 |
| Cyan | 92 | Purple | 110 |
| Green | 343 | Red | 324 |
| Green-yellow | 102 | Salmon | 96 |
| Grey | 254 | Tan | 102 |
| Grey60 | 68 | Turquoise | 2207 |
| Light cyan | 79 | Yellow | 469 |
| Light green | 48 |
| Gene ID | Gene Name | Gene Description |
|---|---|---|
| 102178766 | KRT9 | Keratin 9 |
| 100861172 | KRT25 | Keratin 25 |
| 100861382 | KRT27 | Keratin 27 |
| 102178207 | KRT39 | Keratin 39 |
| 102176789 | KRT74 | Keratin 74 |
| 100861180 | KRTAP3-1 | Keratin-associated protein 3-1 |
| 100861173 | KRTAP11-1 | Keratin-associated protein 11-1 |
| 100861184 | LOC100861184 | Keratin-associated protein 9.2 |
| 102177231 | LOC102177231 | Keratin, type II cytoskeletal 71 |
| 102178767 | LOC102178767 | Keratin-associated protein 4-8-like |
| 102179881 | LOC102179881 | Keratin, type I microfibrillar, 47.6 kDa, KRT33A |
| 106503203 | LOC106503203 | Keratin-associated protein 4-9-like |
| 108638293 | LOC108638293 | Keratin-associated protein 9-9-like |
| 108638298 | LOC108638298 | Keratin, high-sulfur matrix protein, IIIA3-like |
| Hair Type | Gene Name | Sep. | Dec. | Mar. | Gene Name | Sep. | Dec. | Mar. |
|---|---|---|---|---|---|---|---|---|
| LHG | KRT25 | 4.16 ± 1.68 | 5.42 ± 1.04 | 5.54 ± 1.52 | LOC102178767 | 6.63 ± 1.79 | 6.72 ± 1.25 | 9.14 ± 6.79 |
| SHG | 3.98 ± 0.59 | 4.24 ± 1.65 | 3.23 ± 1.18 | 4.31 ± 1.59 | 4.86 ± 1.39 | 2.15 ± 1.18 | ||
| p-value | 0.0124 | 0.8059 | 0.1633 | 0.0208 | 0.0199 | 0.0269 | ||
| LHG | KRT27 | 7.08 ± 3.40 | 6.69 ± 3.43 | 9.28 ± 2.77 | LOC102179881 | 19.11 ± 9.17 | 20.30 ± 8.60 | 12.81 ± 4.78 |
| SHG | 6.20 ± 1.79 | 7.50 ± 4.12 | 6.01 ± 2.88 | 5.91 ± 2.30 | 8.41 ± 4.74 | 3.23 ± 1.41 | ||
| p-value | 0.4539 | 0.4878 | 0.0335 | 0.0062 | 0.017 | 0.0004 | ||
| LHG | KRT39 | 7.20 ± 1.85 | 7.40 ± 1.88 | 6.65 ± 2.75 | LOC106503203 | 30.11 ± 4.94 | 18.31 ± 6.01 | 25.26 ± 17.80 |
| SHG | 4.00 ± 0.73 | 4.79 ± 1.82 | 3.79 ± 0.78 | 19.21 ± 7.92 | 8.70 ± 3.70 | 4.65 ± 1.72 | ||
| p-value | 0.03 | 0.0019 | 0.0357 | 0.0052 | 0.0054 | 0.0387 | ||
| LHG | KRT74 | 5.41 ± 1.14 | 5.54 ± 1.75 | 4.41 ± 1.73 | LOC108638293 | 40.52 ± 17.15 | 32.94 ± 16.17 | 30.71 ± 20.37 |
| SHG | 3.58 ± 1.12 | 3.42 ± 1.16 | 2.69 ± 0.66 | 21.17 ± 7.44 | 33.81 ± 9.12 | 5.58 ± 4.20 | ||
| p-value | 0.0416 | 0.0159 | 0.0291 | 0.0194 | 0.6067 | 0.0144 | ||
| LHG | LOC100861184 | 7.19 ± 2.10 | 6.69 ± 2.79 | 5.22 ± 3.02 | LOC108638298 | 12.57 ± 4.99 | 10.12 ± 2.78 | 14.17 ± 12.02 |
| SHG | 3.80 ± 1.82 | 3.22 ± 0.99 | 3.86 ± 2.85 | 6.51 ± 1.41 | 3.79 ± 1.05 | 2.75 ± 1.16 | ||
| p-value | 0.0075 | 0.0131 | 0.037 | 0.0124 | 0.0002 | 0.0371 | ||
| LHG | LOC102177231 | 10.72 ± 5.43 | 7.16 ± 3.04 | 11.08 ± 5.28 | ||||
| SHG | 9.63 ± 3.40 | 5.38 ± 2.55 | 3.54 ± 1.65 | |||||
| p-value | 0.4687 | 0.448 | 0.0068 | |||||
| Gene Name | Correlation Coefficient between Hair Length and mRNA Expression | p-Value |
|---|---|---|
| KRT25 | 0.5066 | 0.0645 |
| KRT27 | 0.4060 | 0.1498 |
| KRT39 | 0.81085 | 0.0004 |
| KRT74 | 0.76605 | 0.0014 |
| LOC100861184 | 0.79705 | 0.0006 |
| LOC102177231 | 0.59344 | 0.0253 |
| LOC102178767 | 0.81826 | 0.0003 |
| LOC102179881 | 0.88643 | <0.0001 |
| LOC106503203 | 0.8966 | <0.0001 |
| LOC108638293 | 0.63469 | 0.0148 |
| LOC108638298 | 0.73014 | 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, G.; Fan, Y.; Li, W.; Yan, X.; Yan, X.; Zhang, L.; Wang, N.; Chen, O.; Zhang, Y.; Wang, R.; et al. Identification of the Key Genes Associated with Different Hair Types in the Inner Mongolia Cashmere Goat. Animals 2022, 12, 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12111456
Gong G, Fan Y, Li W, Yan X, Yan X, Zhang L, Wang N, Chen O, Zhang Y, Wang R, et al. Identification of the Key Genes Associated with Different Hair Types in the Inner Mongolia Cashmere Goat. Animals. 2022; 12(11):1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12111456
Chicago/Turabian StyleGong, Gao, Yixing Fan, Wenze Li, Xiaochun Yan, Xiaomin Yan, Ludan Zhang, Na Wang, Oljibilig Chen, Yanjun Zhang, Ruijun Wang, and et al. 2022. "Identification of the Key Genes Associated with Different Hair Types in the Inner Mongolia Cashmere Goat" Animals 12, no. 11: 1456. https://0-doi-org.brum.beds.ac.uk/10.3390/ani12111456