
Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cellular Processes Leading from MI to Fibrosis
2.1. Inflammatory Phase
2.2. Reparative Phase
2.3. Maturative Phase
3. Molecular Signaling Pathways
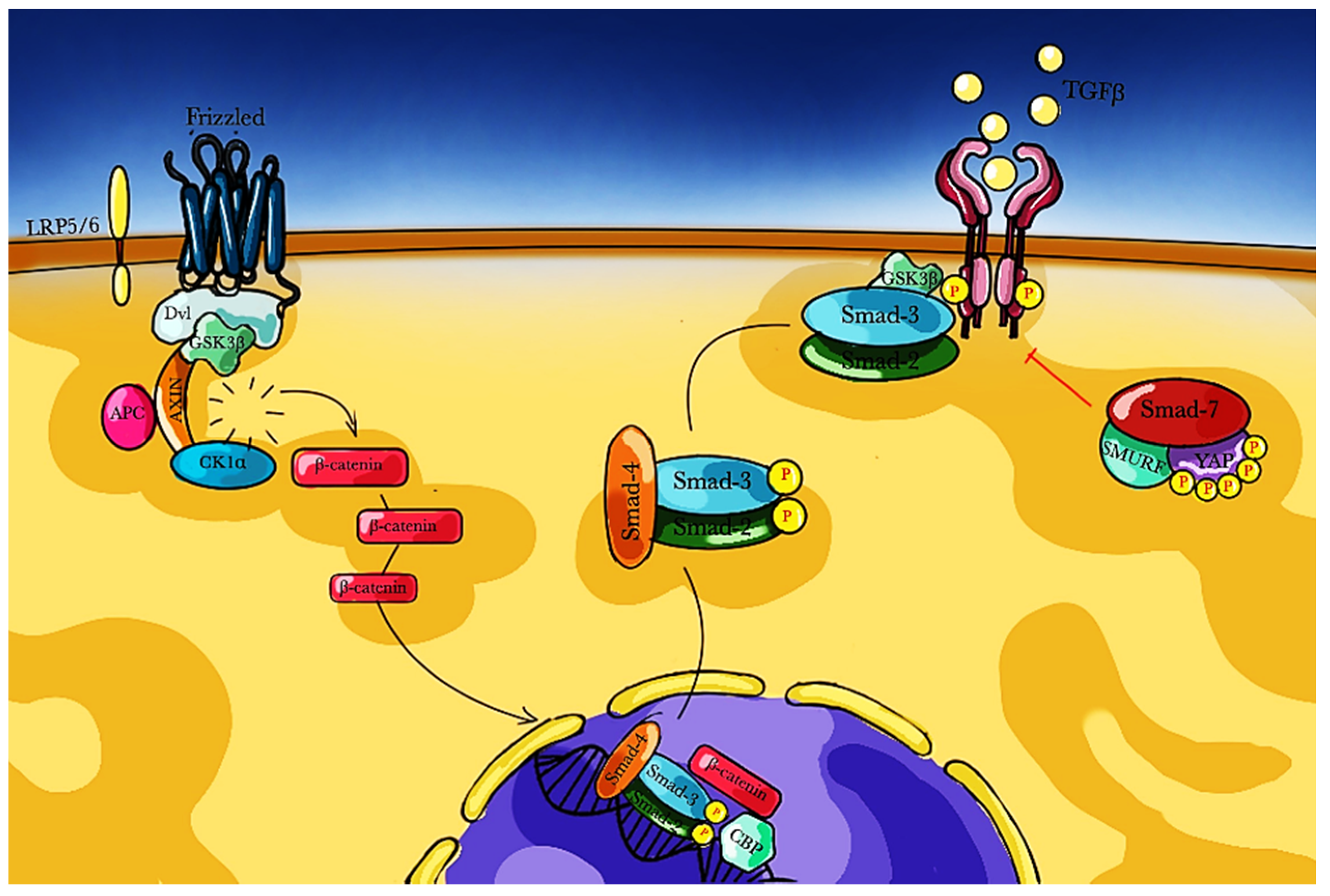
3.1. TGF-β Pathway
3.2. Angiotensin Pathway
3.3. Wnt/β-Catenin Pathway
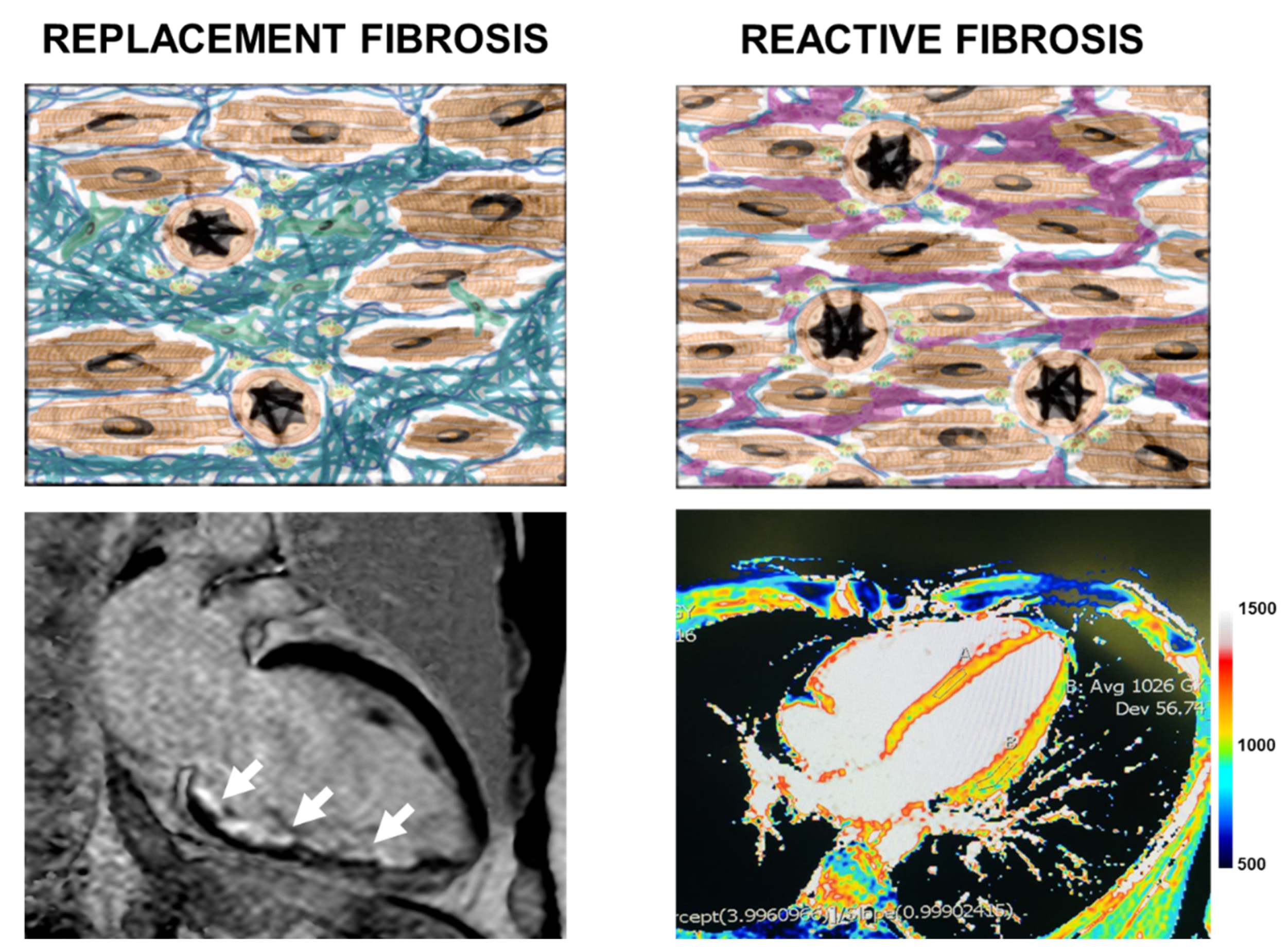
4. Cardiovascular Imaging for the Evaluation of Myocardial Fibrosis
4.1. Introduction to Cardiac Magnetic Resonance
4.2. Cardiac Magnetic Resonance Image Features
5. Prognostic Impact of Fibrosis Identified by CMR
6. Future Perspectives
6.1. Future Therapeutic Perspectives
6.2. Future Diagnostic Perspectives
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Roger, V.L. Epidemiology of Heart Failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lopez, E.F.; Lindsey, M.L. Macrophage roles following myocardial infarction. Int. J. Cardiol. 2008, 130, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage Depletion Impairs Wound Healing and Increases Left Ventricular Remodeling after Myocardial Injury in Mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.D. Post-infarction ventricular remodeling: An array of molecular events. J. Mol. Cell. Cardiol. 2005, 38, 547–550. [Google Scholar] [CrossRef]
- Sutton, M.G.S.J.; Sharpe, N. Left Ventricular Remodeling After Myocardial Infarction. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Stuart, S.D.F.; De Jesus, N.M.; Lindsey, M.L.; Ripplinger, C.M. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 114–122. [Google Scholar] [CrossRef] [Green Version]
- De Haan, J.J.; Smeets, M.B.; Pasterkamp, G.; Arslan, F. Danger Signals in the Initiation of the Inflammatory Response after Myocardial Infarction. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Van Der Borght, K.; Lambrecht, B.N. Heart macrophages and dendritic cells in sickness and in health: A tale of a complicated marriage. Cell. Immunol. 2018, 330, 105–113. [Google Scholar] [CrossRef]
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased Myocardial Dendritic Cells is Associated With Impaired Reparative Fibrosis and Development of Cardiac Rupture After Myocardial Infarction in Humans. J. Am. Heart Assoc. 2014, 3, e000839. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Jeong, S.-J.; Kim, S.; Chalifour, L.; Yun, T.J.; Alam Miah, M.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory Role of Dendritic Cells in Postinfarction Healing and Left Ventricular Remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Pennell, K.Y.; Tian, Y.; Zhang, B.; Cates, C.A.; Iyer, R.P.; Cannon, P.; Shah, P.; Aiyetan, P.; Halade, G.V.; Ma, Y.; et al. CD36 Is a Matrix Metalloproteinase-9 Substrate That Stimulates Neutrophil Apoptosis and Removal During Cardiac Remodeling. Circ. Cardiovasc. Genet. 2016, 9, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil Function: From Mechanisms to Disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.-U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Gonzalez, O.J.R.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Miura, T.; Whittaker, P.; Miki, T.; Sakamoto, J.; Nakamura, Y.; Ichikawa, Y.; Ikeda, Y.; Kobayashi, H.; Ohori, K.; et al. Macrophage Colony-Stimulating Factor Treatment After Myocardial Infarction Attenuates Left Ventricular Dysfunction by Accelerating Infarct Repair. J. Am. Coll. Cardiol. 2006, 47, 626–634. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef]
- Ulloa, L.; Doody, J.F.; Massagué, J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nat. Cell Biol. 1999, 397, 710–713. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 Inhibits Inflammation and Attenuates Left Ventricular Remodeling After Myocardial Infarction via Activation of STAT3 and Suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Boscá, L.; Zandbergen, H.R.; Kovacic, J.C.; Narula, N.; González-Ramos, S.; Fernandez-Velasco, M.; Agrawal, S.; Paz-García, M.; Gupta, S.; et al. Transition of Macrophages to Fibroblast-Like Cells in Healing Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 74, 3124–3135. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hong, T.; Ding, S.; Deng, L.; Abudupataer, M.; Zhang, W.; Tong, M.; Jia, J.; Gong, H.; Zou, Y.; et al. Aggravated myocardial infarction-induced cardiac remodeling and heart failure in histamine-deficient mice. Sci. Rep. 2017, 7, 44007. [Google Scholar] [CrossRef] [Green Version]
- Tatler, A.L.; Porte, J.; Knox, A.; Jenkins, G.; Pang, L. Tryptase activates TGFβ in human airway smooth muscle cells via direct proteolysis. Biochem. Biophys. Res. Commun. 2008, 370, 239–242. [Google Scholar] [CrossRef]
- Zhao, X.-Y.; Zhao, L.-Y.; Zheng, Q.-S.; Su, J.-L.; Guan, H.; Shang, F.-J.; Niu, X.-L.; He, Y.-P.; Lu, X.-L. Chymase induces profibrotic response via transforming growth factor-β1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2007, 310, 159–166. [Google Scholar] [CrossRef]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Zhou, G.; Rokosh, G.; Hamid, T.; Prabhu, S.D. Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy. Circulation 2019, 139, 206–221. [Google Scholar] [CrossRef]
- Shao, D.D.; Suresh, R.; Vakil, V.; Gomer, R.H.; Pilling, D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J. Leukoc. Biol. 2008, 83, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurujeyalakshmi, G.; Giri, S.N. Molecular Mechanisms of Antifibrotic Effect of Interferon Gamma in Bleomycin-Mouse Model of Lung Fibrosis: Downregulation of TGF- β and Procollagen I and III Gene Expression. Exp. Lung Res. 1995, 21, 791–808. [Google Scholar] [CrossRef]
- Hall, A.O.; Beiting, D.P.; Tato, C.M.; John, B.; Oldenhove, G.; Lombana, C.G.; Pritchard, G.H.; Silver, J.S.; Bouladoux, N.; Stumhofer, J.S.; et al. The Cytokines Interleukin 27 and Interferon-γ Promote Distinct Treg Cell Populations Required to Limit Infection-Induced Pathology. Immunity 2012, 37, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.G.; Mendoza, A.; Hemmers, S.; Moltedo, B.; Niec, R.E.; Schizas, M.; Hoyos, B.E.; Putintseva, E.V.; Chaudhry, A.; Dikiy, S.; et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nat. Cell Biol. 2017, 546, 421–425. [Google Scholar] [CrossRef]
- Shintani, Y.; Ito, T.; Fields, L.; Shiraishi, M.; Ichihara, Y.; Satoshi, K.; Podaru, M.; Kainuma, S.; Tanaka, H.; Suzuki, K. IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Hofmann, U.; Knorr, S.; Vogel, B.; Weirather, J.; Frey, A.; Ertl, G.; Frantz, S. Interleukin-13 Deficiency Aggravates Healing and Remodeling in Male Mice After Experimental Myocardial Infarction. Circ. Heart Fail. 2014, 7, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Miteva, K.; Tschöpe, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Crome, S.Q.; Clive, B.; Wang, A.Y.; Kang, C.Y.; Chow, V.; Yu, J.; Lai, A.; Ghahary, A.; Broady, R.; Levings, M.K. Inflammatory Effects of Ex Vivo Human Th17 Cells Are Suppressed by Regulatory T Cells. J. Immunol. 2010, 185, 3199–3208. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3 + CD4 + T Cells Improve Healing After Myocardial Infarction by Modulating Monocyte/Macrophage Differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; DeLeon-Pennell, K.Y. CD8+T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Circ. Physiol. 2019, 317, H581–H596. [Google Scholar] [CrossRef] [PubMed]
- Trial, J.; Baughn, R.E.; Wygant, J.N.; McIntyre, B.W.; Birdsall, H.H.; Youker, K.A.; Evans, A.; Entman, M.L.; Rossen, R.D. Fibronectin fragments modulate monocyte VLA-5 expression and monocyte migration. J. Clin. Investig. 1999, 104, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobaczewski, M.; Bujak, M.; Zymek, P.; Ren, G.; Entman, M.L.; Frangogiannis, N.G. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006, 324, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Trueblood, N.A.; Xie, Z.; Communal, C.; Sam, F.; Ngoy, S.; Liaw, L.; Jenkins, A.W.; Wang, J.; Sawyer, D.B.; Bing, O.H.L.; et al. Exaggerated Left Ventricular Dilation and Reduced Collagen Deposition After Myocardial Infarction in Mice Lacking Osteopontin. Circ. Res. 2001, 88, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aelst, L.N.; Voss, S.; Carai, P.; Van Leeuwen, R.; Vanhoutte, D.; Wijk, S.S.-V.; Eurlings, L.; Swinnen, M.; Verheyen, F.K.; Verbeken, E.; et al. Osteoglycin Prevents Cardiac Dilatation and Dysfunction After Myocardial Infarction Through Infarct Collagen Strengthening. Circ. Res. 2015, 116, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.; Michael, L.H.; Entman, M.L. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 2000, 48, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.-J.; Dangerfield, A.L.; Rattan, S.G.; Bathe, K.L.; Cunnington, R.H.; Raizman, J.E.; Bedosky, K.M.; Freed, D.H.; Kardami, E.; Dixon, I.M.C. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: Expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev. Dyn. 2010, 239, 1573–1584. [Google Scholar] [CrossRef]
- Voorhees, A.P.; DeLeon-Pennell, K.Y.; Ma, Y.; Halade, G.V.; Yabluchanskiy, A.; Iyer, R.P.; Flynn, E.; Cates, C.A.; Lindsey, M.L.; Han, H.-C. Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J. Mol. Cell. Cardiol. 2015, 85, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Koganti, T.; Yao, Z.; Cannon, P.; Shah, P.; Pietrovito, L.; Modesti, A.; Aiyetan, P.; DeLeon-Pennell, K.; Ma, Y.; et al. Cardiac extracellular proteome profiling and membrane topology analysis using glycoproteomics. Proteom. Clin. Appl. 2014, 8, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 Receptor Type I Signaling Critically Regulates Infarct Healing and Cardiac Remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2012, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Pennell, K.Y.; Brás, L.E.D.C.; Iyer, R.P.; Bratton, D.R.; Jin, Y.-F.; Ripplinger, C.M.; Lindsey, M.L.P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J. Mol. Cell. Cardiol. 2014, 76, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming Growth Factor-β Function Blocking Prevents Myocardial Fibrosis and Diastolic Dysfunction in Pressure-Overloaded Rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, S.M.; Allen, J.B.; Weeks, B.S.; Wong, H.L.; Klotman, P.E. Transforming growth factor beta enhances integrin expression and type IV collagenase secretion in human monocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 4577–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, N.; Piek, E.; Sundström, M.; Dijke, P.T.; Nilsson, G. Transforming growth factor-β-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell. Signal. 2001, 13, 483–490. [Google Scholar] [CrossRef]
- Wahl, S.M.; Hunt, D.A.; Wakefield, L.M.; McCartney-Francis, N.; Roberts, A.B.; Sporn, M.B. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. USA 1987, 84, 5788–5792. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.; Chantry, D.; Feldmann, M. Transforming growth factor β induces the production of interleukin 6 by human peripheral blood mononuclear cells. Cytokine 1990, 2, 211–216. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. TGF-β: A Master of All T Cell Trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. TRANSFORMING GROWTH FACTOR-β REGULATION OF IMMUNE RESPONSES. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.L.; Flavell, R.A. Mechanism of Transforming Growth Factor β–induced Inhibition of T Helper Type 1 Differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-G.; Dai, J.; Yuan, Y.-P.; Bian, Z.-Y.; Xu, S.-C.; Jin, Y.-G.; Zhang, X.; Tang, Q.-Z. T-bet deficiency attenuates cardiac remodelling in rats. Basic Res. Cardiol. 2018, 113, 19. [Google Scholar] [CrossRef]
- Ashcroft, G.S. Bidirectional regulation of macrophage function by TGF-β. Microbes Infect. 1999, 1, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Calderone, A.; Izzo, N.J.; Mäki, T.M.; Marsh, J.D.; Colucci, W.S. Hypertrophic stimuli induce transforming growth factor-beta 1 expression in rat ventricular myocytes. J. Clin. Investig. 1994, 94, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-Y.; Park, S.J.; Hwang, H.-Y.; Park, E.J.; Nam, J.H.; Kim, J. TGF-β1 induces cardiac hypertrophic responses via PKC-dependent ATF-2 activation. J. Mol. Cell. Cardiol. 2005, 39, 627–636. [Google Scholar] [CrossRef]
- Schiller, M.; Javelaud, D.; Mauviel, A. TGF-β-induced SMAD signaling and gene regulation: Consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004, 35, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Brooks, W.W.; Conrad, C.H. Myocardial Fibrosis in Transforming Growth Factor β1Heterozygous Mice. J. Mol. Cell. Cardiol. 2000, 32, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Flesch, M.; Amann, K.; Haeuseler, C.; Kilter, H.; Seeland, U.; Schlüter, K.-D.; Böhm, M. Alterations of β-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-β1. Am. J. Physiol. Circ. Physiol. 2002, 283, H1253–H1262. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Watabe, T. Roles of TGF-βSignals in Endothelial-Mesenchymal Transition during Cardiac Fibrosis. Int. J. Inflamm. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, G.; Hu, M.C.-T.; Yao, Z.; Tan, T.-H. Activation of the Hematopoietic Progenitor Kinase-1 (HPK1)-dependent, Stress-activated c-Jun N-terminal Kinase (JNK) Pathway by Transforming Growth Factor β (TGF-β)-activated Kinase (TAK1), a Kinase Mediator of TGF β Signal Transduction. J. Biol. Chem. 1997, 272, 22771–22775. [Google Scholar] [CrossRef] [Green Version]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 Receptors: Regulation, Signalling and Function. Blood Press. 2003, 12, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Gallinat, S.; Bohle, R.-M.; Reinecke, A.; Seebeck, J.; Franke, F.E.; Fink, L.; Zhu, M.; Sumners, C.; Unger, T. Expression of Angiotensin AT1 and AT2 Receptors in Adult Rat Cardiomyocytes after Myocardial Infarction. Am. J. Pathol. 2000, 157, 605–611. [Google Scholar] [CrossRef]
- Rompe, F.; Artuc, M.; Hallberg, A.; Alterman, M.; Ströder, K.; Thöne-Reineke, C.; Reichenbach, A.; Schacherl, J.; Dahlöf, B.; Bader, M.; et al. Direct Angiotensin II Type 2 Receptor Stimulation Acts Anti-Inflammatory Through Epoxyeicosatrienoic Acid and Inhibition of Nuclear Factor κB. Hypertension 2010, 55, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Meffert, S.; Stoll, M.; Steckelings, U.M.; Bottari, S.P.; Unger, T. The angiotensin II AT2 receptor inhibits proliferation and promotes differentiation in PC12W cells. Mol. Cell. Endocrinol. 1996, 122, 59–67. [Google Scholar] [CrossRef]
- Kawano, H.; Do, Y.S.; Kawano, Y.; Starnes, V.; Barr, M.; Law, R.E.; Hsueh, W.A. Angiotensin II Has Multiple Profibrotic Effects in Human Cardiac Fibroblasts. Circulation 2000, 101, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J.-I.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984. [Google Scholar] [CrossRef]
- Kamo, T.; Akazawa, H.; Komuro, I. Cardiac Nonmyocytes in the Hub of Cardiac Hypertrophy. Circ. Res. 2015, 117, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Naito, A.T.; Shiojima, I.; Komuro, I. Wnt Signaling and Aging-Related Heart Disorders. Circ. Res. 2010, 107, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, T.P.; Kühl, M. An Updated Overview on Wnt Signaling Pathways. Circ. Res. 2010, 106, 1798–1806. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.R.; Gao, E.; et al. Cardiac Fibroblast Glycogen Synthase Kinase-3β Regulates Ventricular Remodeling and Dysfunction in Ischemic Heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef]
- Xiang, F.-L.; Fang, M.; Yutzey, K.E. Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A Second Canon. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2011, 31, 429–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklepkiewicz, P.; Shiomi, T.; Kaur, R.; Sun, J.; Kwon, S.; Mercer, B.; Bodine, P.; Schermuly, R.T.; George, I.; Schulze, P.C.; et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ. Heart Fail. 2015, 8, 362–372. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, L.; Ni, A.; Zhang, Z.; Mirotsou, M.; Mao, L.; Pratt, R.E.; Dzau, V.J. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc. Natl. Acad. Sci. USA 2010, 107, 21110–21115. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yue, W.; Zhu, M.J.; Sreejayan, N.; Du, M. AMP-activated protein kinase (AMPK) cross-talks with canonical Wnt signaling via phosphorylation of β-catenin at Ser 552. Biochem. Biophys. Res. Commun. 2010, 395, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Hu, X.; Xu, X.; Fassett, J.; Zhu, G.; Viollet, B.; Xu, W.; Wiczer, B.M.; Bernlohr, D.A.; Bache, R.J.; et al. AMP Activated Protein Kinase-α2 Deficiency Exacerbates Pressure-Overload–Induced Left Ventricular Hypertrophy and Dysfunction in Mice. Hypertension 2008, 52, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-G.; Dai, J.; Wei, W.-Y.; Zhang, W.-B.; Xu, S.-C.; Liao, H.-H.; Yang, Z.; Tang, Q.-Z. Asiatic Acid Protects against Cardiac Hypertrophy through Activating AMPKα Signalling Pathway. Int. J. Biol. Sci. 2016, 12, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, F.; Zhou, J.; Liu, J.; Zhu, C.; Cui, B.; Lin, H.; Liu, Y.; Jin, W.; Yang, H.; Hu, Z. Glycogen synthase kinase-3β negatively regulates TGF-β1 and Angiotensin II-mediated cellular activity through interaction with Smad3. Eur. J. Pharmacol. 2010, 644, 17–23. [Google Scholar] [CrossRef]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.-R.; Veenstra, T.D.; Zhang, Y.E. A Negative Feedback Control of Transforming Growth Factor-β Signaling by Glycogen Synthase Kinase 3-mediated Smad3 Linker Phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.-G.; Dai, J.; Zhang, W.-B.; Yuan, Y.; Liao, H.-H.; Zhang, N.; Bian, Z.-Y.; Tang, Q.-Z. Protection against cardiac hypertrophy by geniposide involves the GLP-1 receptor / AMPKα signalling pathway. Br. J. Pharmacol. 2016, 173, 1502–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Cui, W.; Zhou, W.; Li, D.; Li, L.; Zhao, P.; Mo, X.; Zhang, Z.; Gao, J. Activation of Wnt/β-catenin signalling is required for TGF-β/Smad2/3 signalling during myofibroblast proliferation. J. Cell. Mol. Med. 2017, 21, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Noppe, G.; Dufeys, C.; Buchlin, P.; Marquet, N.; Castanares-Zapatero, D.; Balteau, M.; Hermida, N.; Bouzin, C.; Esfahani, H.; Viollet, B.; et al. Reduced scar maturation and contractility lead to exaggerated left ventricular dilation after myocardial infarction in mice lacking AMPKα1. J. Mol. Cell. Cardiol. 2014, 74, 32–43. [Google Scholar] [CrossRef]
- Itoh, S.; Dijke, P.T. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007, 19, 176–184. [Google Scholar] [CrossRef]
- Patel, A.R.; Kramer, C.M. Role of Cardiac Magnetic Resonance in the Diagnosis and Prognosis of Nonischemic Cardiomyopathy. JACC Cardiovasc. Imaging 2017, 10, 1180–1193. [Google Scholar] [CrossRef]
- Di Bella, G. Walking with Gianluca Di Bella during the development of clinical cardiac imaging. World J. Cardiol. 2010, 2, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Judd, R.M.; Lugo-Olivieri, C.H.; Arai, M.; Kondo, T.; Croisille, P.; Lima, J.A.; Mohan, V.; Becker, L.C.; Zerhouni, E.A. Physiological Basis of Myocardial Contrast Enhancement in Fast Magnetic Resonance Images of 2-Day-Old Reperfused Canine Infarcts. Circulation 1995, 92, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.J.; Chen, E.-L.; Lima, J.A.; Judd, R.M. Myocardial Gd-DTPA Kinetics Determine MRI Contrast Enhancement and Reflect the Extent and Severity of Myocardial Injury after Acute Reperfused Infarction. Circulation 1996, 94, 3318–3326. [Google Scholar] [CrossRef]
- Karamitsos, T.D.; Francis, J.M.; Myerson, S.; Selvanayagam, J.B.; Neubauer, S. The Role of Cardiovascular Magnetic Resonance Imaging in Heart Failure. J. Am. Coll. Cardiol. 2009, 54, 1407–1424. [Google Scholar] [CrossRef] [Green Version]
- Mahrholdt, H.; Wagner, A.; Judd, R.M.; Sechtem, U.; Kim, R.J. Delayed enhancement cardiovascular magnetic resonance assessment of non-ischaemic cardiomyopathies. Eur. Heart J. 2005, 26, 1461–1474. [Google Scholar] [CrossRef]
- Mahrholdt, H.; Wagner, A.; Holly, T.A.; Elliott, M.D.; Bonow, R.O.; Kim, R.J.; Judd, R.M. Reproducibility of Chronic Infarct Size Measurement by Contrast-Enhanced Magnetic Resonance Imaging. Circulation 2002, 106, 2322–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bella, G.; Aquaro, G.D.; Strata, E.; Deiana, M.; De Marchi, D.; Lombardi, M.; Pingitore, A. Simultaneous visualization of myocardial scar, no-reflow phenomenon, ventricular and atrial thrombi by cardiac magnetic resonance. Int. J. Cardiol. 2007, 115, E10–E11. [Google Scholar] [CrossRef]
- Spiewak, M.; Malek, L.A.; Misko, J.; Chojnowska, L.; Milosz, B.; Klopotowski, M.; Petryka, J.; Dąbrowski, M.; Kepka, C.; Ruzyllo, W. Comparison of different quantification methods of late gadolinium enhancement in patients with hypertrophic cardiomyopathy. Eur. J. Radiol. 2010, 74, e149–e153. [Google Scholar] [CrossRef]
- Mewton, N.; Liu, C.Y.; Croisille, P.; Bluemke, D.; Lima, J.A. Assessment of Myocardial Fibrosis With Cardiovascular Magnetic Resonance. J. Am. Coll. Cardiol. 2011, 57, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Kellman, P.; Wilson, J.R.; Xue, H.; Ugander, M.; Arai, A.E. Extracellular volume fraction mapping in the myocardium, part 1: Evaluation of an automated method. J. Cardiovasc. Magn. Reson. 2012, 14, 63. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Han, J.; Nacif, M.S.; Jones, J.; Kawel, N.; Kellman, P.; Sibley, C.T.; Bluemke, D.A. Diffuse myocardial fibrosis evaluation using cardiac magnetic resonance T1 mapping: Sample size considerations for clinical trials. J. Cardiovasc. Magn. Reson. 2012, 14, 90. [Google Scholar] [CrossRef] [Green Version]
- Robbers, L.F.; Baars, E.N.; Brouwer, W.P.; Beek, A.M.; Hofman, M.B.; Niessen, H.W.; Van Rossum, A.C.; Marcu, C.B. T1 Mapping Shows Increased Extracellular Matrix Size in the Myocardium Due to Amyloid Depositions. Circ. Cardiovasc. Imaging 2012, 5, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Bauner, K.U.; Biffar, A.; Theisen, D.; Greiser, A.; Zech, C.J.; Nguyen, E.T.; Reiser, M.F.; Wintersperger, B.J. Extracellular Volume Fractions in Chronic Myocardial Infarction. Investig. Radiol. 2012, 47, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.M.; Piechnik, S.K.; Dall’Armellina, E.; Karamitsos, T.D.; Francis, J.M.; Choudhury, R.P.; Friedrich, M.G.; Robson, M.D.; Neubauer, S. Non-contrast T1-mapping detects acute myocardial edema with high diagnostic accuracy: A comparison to T2-weighted cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2012, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Di Bella, G.; Siciliano, V.; Aquaro, G.D.; Molinaro, S.; Lombardi, M.; Carerj, S.; Landi, P.; Rovai, D.; Pingitore, A. Scar extent, left ventricular end-diastolic volume, and wall motion abnormalities identify high-risk patients with previous myocardial infarction: A multiparametric approach for prognostic stratification. Eur. Heart J. 2013, 34, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Symons, R.; Masci, P.G.; Goetschalckx, K.; Doulaptsis, K.; Janssens, S.; Bogaert, J. Effect of Infarct Severity on Regional and Global Left Ventricular Remodeling in Patients with Successfully Reperfused ST Segment Elevation Myocardial Infarction. Radiology 2015, 274, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masci, P.G.; Pavon, A.G.; Pontone, G.; Symons, R.; Lorenzoni, V.; Francone, M.; Zalewski, J.; Barison, A.; Guglielmo, M.; Aquaro, G.D.; et al. Early or deferred cardiovascular magnetic resonance after ST-segment-elevation myocardial infarction for effective risk stratification. Eur. Heart J. Cardiovasc. Imaging 2019, 21, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, Y.; Yang, Z.; Yang, M.; Diao, K.; Zhou, X. Cardiac magnetic resonance T1 mapping allows for predicting adverse left ventricular remodeling post-reperfused st-elevation myocardial infarction. Eur. Heart J. 2020, 41, 41. [Google Scholar] [CrossRef]
- Baritussio, A.; Biglino, G.; Scatteia, A.; De Garate, E.; Dastidar, A.G.; Palazzuoli, A.; Harries, I.; Strange, J.W.; Diab, I.; Bucciarelli-Ducci, C. Long-term outcome of myocardial scarring and deformation with cardiovascular magnetic resonance in out of hospital cardiac arrest survivors. Eur. Heart J. Cardiovasc. Imaging 2020, jeaa293. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Pellicori, P.; Khan, M.J.I.; Graham, F.J.; Cleland, J.G.F. New perspectives and future directions in the treatment of heart failure. Heart Fail. Rev. 2020, 25, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-Mediated Regression of Myocardial Fibrosis in Patients With Hypertensive Heart Disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef] [Green Version]
- López, B.; Querejeta, R.; Varo, N.; González, A.; Larman, M.; Ubago, J.L.M.; Díez, J. Usefulness of Serum Carboxy-Terminal Propeptide of Procollagen Type I in Assessment of the Cardioreparative Ability of Antihypertensive Treatment in Hypertensive Patients. Circulation 2001, 104, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Díez, J.; Querejeta, R.; López, B.; González, A.; Larman, M.; Ubago, J.L.M. Losartan-Dependent Regression of Myocardial Fibrosis Is Associated With Reduction of Left Ventricular Chamber Stiffness in Hypertensive Patients. Circulation 2002, 105, 2512–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, Y.J.; Passeri, J.J.; Baggish, A.L.; O’Callaghan, C.; Lowry, P.A.; Yannekis, G.; Abbara, S.; Ghoshhajra, B.B.; Rothman, R.D.; Ho, C.Y.; et al. Effects of Losartan on Left Ventricular Hypertrophy and Fibrosis in Patients With Nonobstructive Hypertrophic Cardiomyopathy. JACC Heart Fail. 2013, 1, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; Ito, H.; Onuki, T.; Miyoshi, F.; Watanabe, N.; Asano, T.; Tanno, K.; Kobayashi, Y. Candesartan Decreases Type III Procollagen-N-Peptide Levels and Inflammatory Marker Levels and Maintains Sinus Rhythm in Patients With Atrial Fibrillation. J. Cardiovasc. Pharmacol. 2010, 55, 511–517. [Google Scholar] [CrossRef]
- Kosmala, W.; Przewlocka-Kosmala, M.; Szczepanik-Osadnik, H.; Mysiak, A.; Marwick, T.H. Fibrosis and cardiac function in obesity: A randomised controlled trial of aldosterone blockade. Heart 2013, 99, 320–326. [Google Scholar] [CrossRef]
- Kosmala, W.; Przewlocka-Kosmala, M.; Szczepanik-Osadnik, H.; Mysiak, A.; O’Moore-Sullivan, T.; Marwick, T.H. A Randomized Study of the Beneficial Effects of Aldosterone Antagonism on LV Function, Structure, and Fibrosis Markers in Metabolic Syndrome. JACC Cardiovasc. Imaging 2011, 4, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, G.J.; Ledwidge, M.T.; Watson, C.J.; Phelan, D.M.; Dawkins, I.R.; Murphy, N.F.; Patle, A.K.; Baugh, J.A.; McDonald, K.M. Natural History of Markers of Collagen Turnover in Patients With Early Diastolic Dysfunction and Impact of Eplerenone. J. Am. Coll. Cardiol. 2009, 54, 1674–1682. [Google Scholar] [CrossRef]
- Kobayashi, M.; Machida, N.; Mitsuishi, M.; Yamane, Y. β-blocker improves survival, left ventricular function, and myocardial remodeling in hypertensive rats with diastolic heart failure. Am. J. Hypertens. 2004, 17, 1112–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busseuil, D.; Shi, Y.; Mecteau, M.; Brand, G.; Gillis, M.-A.; Thorin, E.; Asselin, C.; Romeo, P.; Leung, T.K.; Latour, J.-G.; et al. Heart Rate Reduction by Ivabradine Reduces Diastolic Dysfunction and Cardiac Fibrosis. Cardiology 2010, 117, 234–242. [Google Scholar] [CrossRef]
- Becher, P.M.; Lindner, D.; Miteva, K.; Savvatis, K.; Zietsch, C.; Schmack, B.; Van Linthout, S.; Westermann, D.; Schultheiss, H.-P.; Tschöpe, C. Role of Heart Rate Reduction in the Prevention of Experimental Heart Failure. Hypertension 2012, 59, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.-A.; Kim, Y.-J.; Lee, H.-W.; Kim, D.-H.; Kim, H.-K.; Chang, H.-J.; Sohn, D.-W.; Oh, B.-H.; Park, Y.-B. Effect of Rosuvastatin on Cardiac Remodeling, Function, and Progression to Heart Failure in Hypertensive Heart With Established Left Ventricular Hypertrophy. Hypertension 2009, 54, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Krum, H.; Ashton, E.; Reid, C.; Kalff, V.; Rogers, J.; Amarena, J.; Singh, B.; Tonkin, A. Double-Blind, Randomized, Placebo-Controlled Study of High-Dose HMG CoA Reductase Inhibitor Therapy on Ventricular Remodeling, Pro-Inflammatory Cytokines and Neurohormonal Parameters in Patients With Chronic Systolic Heart Failure. J. Card. Fail. 2007, 13, 1–7. [Google Scholar] [CrossRef]
- Ashton, E.; Windebank, E.; Skiba, M.; Reid, C.; Schneider, H.; Rosenfeldt, F.; Tonkin, A.; Krum, H. Why did high-dose rosuvastatin not improve cardiac remodeling in chronic heart failure? Mechanistic insights from the UNIVERSE study. Int. J. Cardiol. 2011, 146, 404–407. [Google Scholar] [CrossRef]
- Ogata, T.; Miyauchi, T.; Sakai, S.; Takanashi, M.; Irukayama-Tomobe, Y.; Yamaguchi, I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-b pathway. J. Am. Coll. Cardiol. 2004, 43, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Edgley, A.J.; Krum, H.; Kelly, D.J. Targeting Fibrosis for the Treatment of Heart Failure: A Role for Transforming Growth Factor-β. Cardiovasc. Ther. 2010, 30, e30–e40. [Google Scholar] [CrossRef]
- Holmesjr, D.R.; Savage, M.; Lablanche, J.-M.; Grip, L.; Serruys, P.; Fitzgerald, P.; Fischman, D.; Goldberg, S.; Brinker, J.A.; Zeiher, A.; et al. Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) Trial. Circulation 2002, 106, 1243–1250. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Lupu, F.; Terclavers, S.; Vanwetswinkel, B.; Herbert, J.-M.; Baker, A.; Collen, D.; Carmeliet, P.; Moons, L. Loss or Inhibition of uPA or MMP-9 Attenuates LV Remodeling and Dysfunction after Acute Pressure Overload in Mice. Am. J. Pathol. 2005, 166, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, H.; Ide, T.; Matsushima, S.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Kinugawa, S.; Tsutsui, H. Targeted Deletion of Matrix Metalloproteinase 2 Ameliorates Myocardial Remodeling in Mice With Chronic Pressure Overload. Hypertension 2006, 47, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Hudson, M.P.; Armstrong, P.W.; Ruzyllo, W.; Brum, J.; Cusmano, L.; Krzeski, P.; Lyon, R.; Quinones, M.; Theroux, P.; Sydlowski, D.; et al. Effects of Selective Matrix Metalloproteinase Inhibitor (PG-116800) to Prevent Ventricular Remodeling After Myocardial Infarction. J. Am. Coll. Cardiol. 2006, 48, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, C.S.; Bodaragama, H.; Chew, J.Y.; Widdop, R.E.; Royce, S.G.; Hewitson, T.D. Serelaxin Is a More Efficacious Antifibrotic Than Enalapril in an Experimental Model of Heart Disease. Hypertension 2014, 64, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.S.; Cendrawan, S.; Gao, X.-M.; Ming, Z.; Zhao, C.; Kiriazis, H.; Xu, Q.; Tregear, G.W.; Bathgate, R.A.D.; Du, X.-J. Relaxin remodels fibrotic healing following myocardial infarction. Lab. Investig. 2011, 91, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.S.; Hewitson, T.D.; Zhang, Y.; Kelly, D.J. Relaxin Ameliorates Fibrosis in Experimental Diabetic Cardiomyopathy. Endocrinology 2008, 149, 3286–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lekgabe, E.D.; Kiriazis, H.; Zhao, C.; Xu, Q.; Moore, X.L.; Su, Y.; Bathgate, R.A.; Du, X.-J.; Samuel, C.S. Relaxin Reverses Cardiac and Renal Fibrosis in Spontaneously Hypertensive Rats. Hypertension 2005, 46, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.L.; Gabris, B.; Li, Q.; Martin, B.; Giannini, M.; Parikh, A.; Patel, D.; Haney, J.; Schwartzman, D.S.; Shroff, S.G.; et al. Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na+ channels. Heart Rhythm. 2016, 13, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Voors, A.A.; et al. Effects of Serelaxin in Patients with Acute Heart Failure. N. Engl. J. Med. 2019, 381, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Riegler, J.; Tiburcy, M.; Ebert, A.D.; Tzatzalos, E.; Raaz, U.; Abilez, O.J.; Shen, Q.; Kooreman, N.G.; Neofytou, E.; Chen, V.C.; et al. Human Engineered Heart Muscles Engraft and Survive Long Term in a Rodent Myocardial Infarction Model. Circ. Res. 2015, 117, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberger, F.; Breckwoldt, K.; Pecha, S.; Kelly, A.; Geertz, B.; Starbatty, J.; Yorgan, T.; Cheng, K.-H.; Lessmann, K.; Stolen, T.; et al. Cardiac repair in guinea pigs with human engineered heart tissue from induced pluripotent stem cells. Sci. Transl. Med. 2016, 8, 363ra148. [Google Scholar] [CrossRef]
- Menasche, P.; Vanneaux, V.; Hagege, A.; Bel, A.; Cholley, B.; Cacciapuoti, I.; Parouchev, A.; Benhamouda, N.; Tachdjian, G.; Tosca, L.; et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical casereport. Eur. Heart J. 2015, 36, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Coats, A.J.; Cristian, G.; Dragomir, D.; Pusineri, E.; Piredda, M.; Bettari, L.; Dowling, R.; Volterrani, M.; Kirwan, B.-A.; et al. A prospective comparison of alginate-hydrogel with standard medical therapy to determine impact on functional capacity and clinical outcomes in patients with advanced heart failure (AUGMENT-HF trial). Eur. Heart J. 2015, 36, 2297–2309. [Google Scholar] [CrossRef] [Green Version]
- Januzzi, J.L.; Chandrashekhar, Y. Strain Echocardiography. J. Am. Coll. Cardiol. 2017, 70, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Kalam, K.; Otahal, P.; Marwick, T.H. Prognostic implications of global LV dysfunction: A systematic review and meta-analysis of global longitudinal strain and ejection fraction. Heart 2014, 100, 1673–1680. [Google Scholar] [CrossRef]
- Shah, A.M.; Claggett, B.; Sweitzer, N.K.; Shah, S.J.; Anand, I.S.; Liu, L.; Pitt, B.; Pfeffer, M.A.; Solomon, S.D. Prognostic Im-portance of Impaired Systolic Function in Heart Failure with Preserved Ejection Fraction and the Impact of Spironolactone. Circulation 2015, 132, 402–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalique, Z.; Ferreira, P.F.; Scott, A.D.; Nielles-Vallespin, S.; Wage, R.; Firmin, D.N.; Pennell, D.J. Diffusion Tensor Cardiovascular Magnetic Resonance of Microstructural Recovery in Dilated Cardiomyopathy. JACC Cardiovasc. Imaging 2018, 11, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Nielles-Vallespin, S.; Khalique, Z.; Ferreira, P.F.; de Silva, R.; Scott, A.D.; Kilner, P.; McGill, L.-A.; Giannakidis, A.; Gatehouse, P.D.; Ennis, D.; et al. Assessment of Myocardial Microstructural Dynamics by In Vivo Diffusion Tensor Cardiac Magnetic Resonance. J. Am. Coll. Cardiol. 2017, 69, 661–676. [Google Scholar] [CrossRef]
- Eriksson, J.; Bolger, A.F.; Ebbers, T.; Carlhäll, C.-J. Four-dimensional blood flow-specific markers of LV dysfunction in dilated cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2012, 14, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalise, R.F.M.; De Sarro, R.; Caracciolo, A.; Lauro, R.; Squadrito, F.; Carerj, S.; Bitto, A.; Micari, A.; Bella, G.D.; Costa, F.; et al. Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value. Med. Sci. 2021, 9, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9010016
Scalise RFM, De Sarro R, Caracciolo A, Lauro R, Squadrito F, Carerj S, Bitto A, Micari A, Bella GD, Costa F, et al. Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value. Medical Sciences. 2021; 9(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9010016
Chicago/Turabian StyleScalise, Renato Francesco Maria, Rosalba De Sarro, Alessandro Caracciolo, Rita Lauro, Francesco Squadrito, Scipione Carerj, Alessandra Bitto, Antonio Micari, Gianluca Di Bella, Francesco Costa, and et al. 2021. "Fibrosis after Myocardial Infarction: An Overview on Cellular Processes, Molecular Pathways, Clinical Evaluation and Prognostic Value" Medical Sciences 9, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9010016