
Common Pitfalls in Ewing Sarcoma and Desmoplastic Small Round Cell Tumor Diagnosis Seen in a Study of 115 Cases


Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lettieri, C.K.; Garcia-Filion, P.; Hingorani, P. Incidence and outcomes of desmoplastic small round cell tumor: Results from the surveillance, epidemiology, and end results database. J. Cancer Epidemiol. 2014, 2014, 680126. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, I.M.; Cote, G.M.; Hornick, J.L. Contemporary Sarcoma Diagnosis, Genetics, and Genomics. J. Clin. Oncol. 2018, 36, 101–110. [Google Scholar] [CrossRef]
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th ed.; IARC: Lyon, France, 2020. [Google Scholar]
- Ahmed, S.K.; Robinson, S.I.; Okuno, S.H.; Rose, P.S.; Laack, N.N.I. Adult ewing sarcoma: Survival and local control outcomes in 102 patients with localized disease. Sarcoma 2013, 2013, 681425. [Google Scholar] [CrossRef]
- Ahmed, S.K.; Robinson, S.I.; Okuno, S.H.; Rose, P.S.; Issa Laack, N.N. Adult Ewing sarcoma: Survival and local control outcomes in 36 patients with metastatic disease. Am. J. Clin. Oncol. 2014, 37, 423–429. [Google Scholar] [CrossRef]
- Hayes-Jordan, A.; LaQuaglia, M.P.; Modak, S. Management of desmoplastic small round cell tumor. Semin. Pediatr. Surg. 2016, 25, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Lal, D.R.; Su, W.T.; Wolden, S.L.; Loh, K.C.; Modak, S.; La Quaglia, M.P. Results of multimodal treatment for desmoplastic small round cell tumors. J. Pediatr. Surg. 2005, 40, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Puls, F.; Niblett, A.; Marland, G.; Gaston, C.L.; Douis, H.; Mangham, D.C.; Sumathi, V.P.; Kindblom, L.G. BCOR-CCNB3 (Ewing-like) sarcoma: A clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am. J. Surg. Pathol. 2014, 38, 1307–1318. [Google Scholar] [CrossRef]
- Kao, Y.C.; Owosho, A.A.; Sung, Y.S.; Zhang, L.; Fujisawa, Y.; Lee, J.C.; Wexler, L.; Argani, P.; Swanson, D.; Dickson, B.C.; et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am. J. Surg. Pathol. 2018, 42, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Di Mauro, I.; Rapp, J.; Pierron, G.; Auger, N.; Alberti, L.; Chibon, F.; Escande, F.; Voegeli, A.C.; Ghnassia, J.P.; et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): A prospective, multicentre, observational study. Lancet Oncol. 2016, 17, 532–538. [Google Scholar] [CrossRef]
- Terrier-Lacombe, M.J.; Guillou, L.; Chibon, F.; Gallagher, G.; Benhattar, J.; Terrier, P.; Ranchère, D.; Coindre, J.M. Superficial primitive Ewing’s sarcoma: A clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod. Pathol. 2009, 22, 87–94. [Google Scholar] [CrossRef]
- Ptaszyński, K.; Szumera-Ciećkiewicz, A.; Pekul, M.; Nowecki, Z. Differential diagnosis of small round cell tumours (SRCT), fluorescence in situ hybridization (FISH) and immunohistochemical (IHC) study. Pol. J. Pathol. 2009, 60, 151–162. [Google Scholar]
- Olsen, S.H.; Thomas, D.G.; Lucas, D.R. Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod. Pathol. 2006, 19, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Rekhi, B.; Vogel, U.; Basak, R.; Desai, S.B.; Jambhekar, N.A. Clinicopathological and molecular spectrum of ewing sarcomas/PNETs, including validation of EWSR1 rearrangement by conventional and array FISH technique in certain cases. Pathol. Oncol. Res. 2014, 20, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.; Navarro, S.; López-Guerrero, J.A.; Verdini, L.; Picci, P.; Giner, F.; Llombart-Bosch, A. Neuroendocrine differentiation in a large series of genetically-confirmed Ewing’s sarcoma family tumor: Does it provide any diagnostic or prognostic information? Pathol. Res. Pract. 2021, 219, 153362. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.N.; Guo, Z.; Baus, R.M.; Werner, H.; Rehrauer, W.M.; Lloyd, R.V. INSM1: A Novel Immunohistochemical and Molecular Marker for Neuroendocrine and Neuroepithelial Neoplasms. Am. J. Clin. Pathol. 2015, 144, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Krishnan, C.; Charville, G.W. INSM1 Expression in Peripheral Neuroblastic Tumors and Other Embryonal Neoplasms. Pediatr. Dev. Pathol. 2019, 22, 440–448. [Google Scholar] [CrossRef]
- Yoshida, A.; Makise, N.; Wakai, S.; Kawai, A.; Hiraoka, N. INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Mod. Pathol. 2018, 31, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.K.; Hornick, J.L.; Vivero, M. INSM1 expression in a subset of thoracic malignancies and small round cell tumors: Rare potential pitfalls for small cell carcinoma. Mod. Pathol. 2020, 33, 1571–1580. [Google Scholar] [CrossRef]
- Collini, P.; Sampietro, G.; Bertulli, R.; Casali, P.G.; Luksch, R.; Mezzelani, A.; Sozzi, G.; Pilotti, S. Cytokeratin immunoreactivity in 41 cases of ES/PNET confirmed by molecular diagnostic studies. Am. J. Surg. Pathol. 2001, 25, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Antonescu, C.R.; Guiter, G.; Huvos, A.G.; Ladanyi, M.; Zakowski, M.F. Cytokeratin immunoreactivity in Ewing’s sarcoma: Prevalence in 50 cases confirmed by molecular diagnostic studies. Am. J. Surg. Pathol. 2000, 24, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Pfeifer, J.D.; Marley, E.F.; Dehner, L.P.; Humphrey, P.A.; Zhu, X.; Swanson, P.E. WT1 staining reliably differentiates desmoplastic small round cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am. J. Clin. Pathol. 2000, 114, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez, N.G. Desmoplastic small round cell tumor: II: An ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am. J. Surg. Pathol. 1998, 22, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Gerald, W.L.; Ladanyi, M.; de Alava, E.; Cuatrecasas, M.; Kushner, B.H.; LaQuaglia, M.P.; Rosai, J. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its variants. J. Clin. Oncol. 1998, 16, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef] [Green Version]


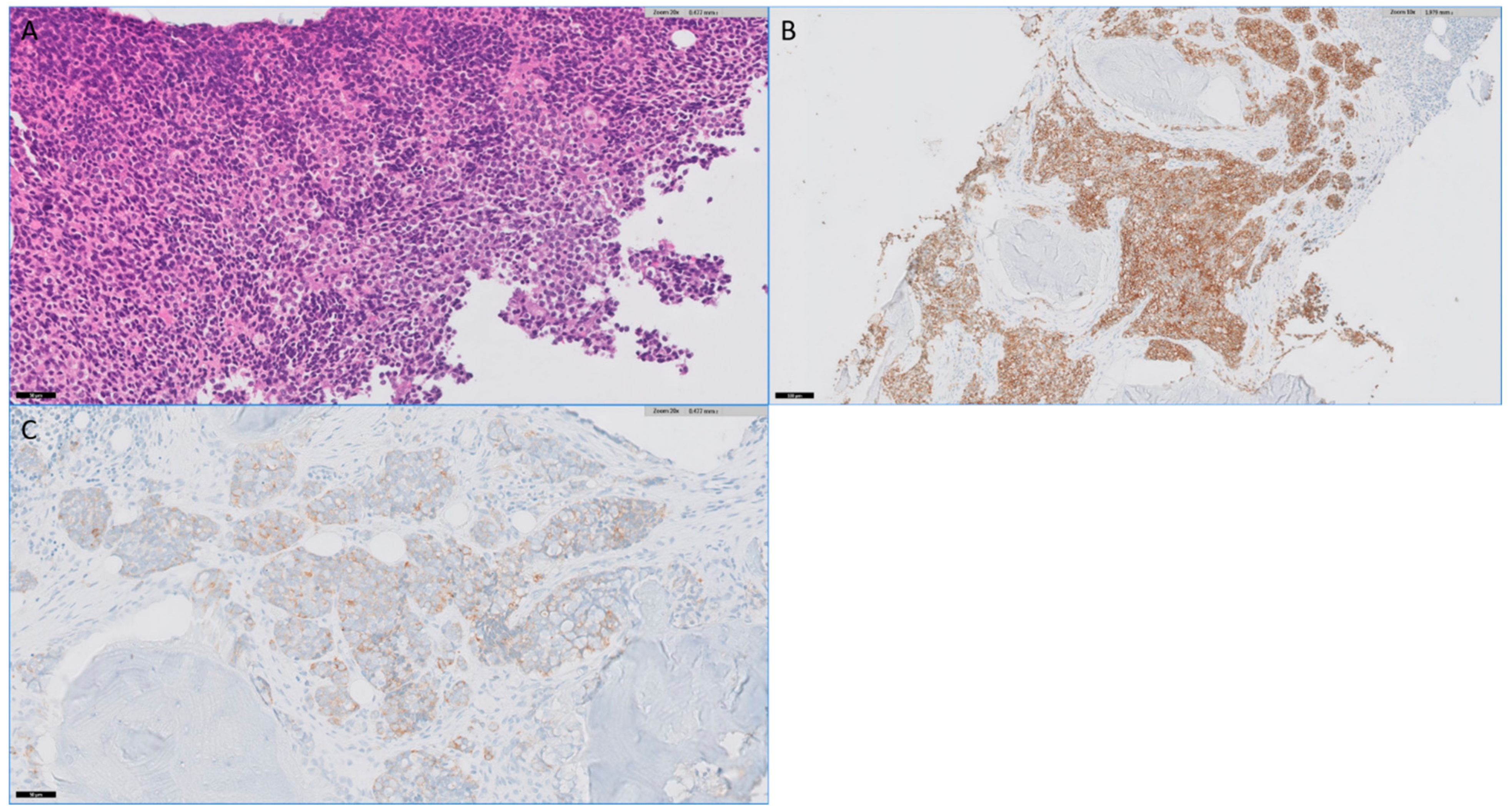{kind=link}
| ID | Sex (M/F) | Year of Dx | Age at Diagnosis | Initial Biopsy Site | Primary Tumor Site | Initial Diagnosis | Final Diagnosis | Initial Treatment | Status as of Last Contact (Alive/Dead) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 2013 | 63 | Neck | Mediastinum | Undifferentiated cancer, then PNET/lung NEC (small cell) | ELS | VAC and radiation | Alive |
| 2 | F | 2013 | 54 | Lung | Thigh | NHL (T-cell) | ELS | VAC and radiation | Alive |
| 3 | M | 2013 | 36 | Rib | Chest wall | Poorly differentiated NEC (small cell) | ES | VAC | Alive |
| 4 | M | 2016 | 11 | Bone marrow | Femur | Poorly differentiated NEC (small cell) | ES | N/A | N/A |
| 5 | M | 2017 | 25 | Mesentery | Mesentery | Poorly differentiated NEC (small cell) | ES | Etoposide and cisplatin | Dead |
| 6 | M | 2018 | 57 | Chest wall | Chest wall | Poorly differentiated NEC (small cell) | ES | Etoposide and cisplatin, avelumab | Dead |
| 7 | M | 2006 | 29 | Peritoneum | Peritoneum | Small cell cancer | DSCRT | VAC | Dead |
| 8 | F | 2008 | 42 | Uterus | Involvement of uterus, adnexa and omentum | High-grade endometrial stromal sarcoma | DSCRT | Carboplatin and paclitaxel with megestrol acetate | Dead |
| 9 | M | 2011 | 41 | Peritoneum | Peritoneum | Poorly differentiated cancer | DSCRT | N/A | Dead |
| 10 | M | 2019 | 60 | Peritoneum | Peritoneum | Poorly differentiated NEC | DSCRT | Carboplatin and etoposide, followed by pembrolizumab | Alive |
| 11 | M | 2020 | 32 | Peritoneum | Peritoneum | Small cell cancer with possible neuroendocrine differentiation | DSCRT | Cisplatin and etoposide | Alive |
| ID | Molecular Diagnostics | Panel | Misdiagnosis at Sarcoma/Tertiary Care Center? | Diagnostic Challenge |
|---|---|---|---|---|
| 1 | FISH negative for EWSR1 rearrangement | Abbott/Washington University | No | Expression of CAM5.2 and synaptophysin |
| 2 | Not done | N/A | Yes | Numerous admixed lymphocytes |
| 3 | FISH positive for EWSR1 rearrangement | Washington University | No | Expression of CD56 and synaptophysin |
| 4 | Initial FISH negative, subsequent FISH positive for EWSR1 rearrangement | N/A | Yes | Molecular testing initially negative, and expression of synaptophysin and pankeratin (diffuse) |
| 5 | FISH positive for EWSR1 rearrangement, EWSR1-FLI1 fusion reportedly also detected | Integrated Oncology Laboratories | Yes | Mesenteric origin, expression of CD56, chromogranin and synaptophysin |
| 6 | Targeted NGS panel positive for EWSR1-FLI1 fusion | FoundationOne NGS | Yes | Expression of CD56 and synaptophysin |
| 7 | RT-PCR negative for EWSR1-WT1 fusion | N/A | No | Expression of CD56 and chromogranin |
| 8 |
nuc ish 22q12(EWSRIx3)(5′EWSRI sep 3′EWSRIx1)[14/110]/ 22q12(EWSRIx2)(5′EWSRI sep 3′EWSRIx1)[55/110]/ 22q12(EWSRIx3)[6/110]/22q12(EWSRIx2)[25/110] nuc ish 11p13(WTIx2),22q12(EWSx3),(WTI con EWSx1)[12/50]/ 11p13(WTIx2),22q12(EWSx2),(WTI con EWSx1)[11/50]/ 11p13(WTIx2),22q12(EWSx2)[21/50] | Vysis, Inc./Washington University | No | Origin in uterus |
| 9 | Negative WT1/nuc ish(EWSR1x3)(5′EWSR1 sep 3′EWSR1x2)[19/200]/(EWSR1x3)(5′EWSR1 sep 3′EWSR1x1)[10/200]/ (EWSR1x2)(5′EWSR1 sep 3′EWSR1x1)[123/200]/(EWSR1x2)[29/200] | Abbott/Washington University | Yes | Keratin expression |
| 10 | EWSR1 EWSR1(NM_005243)-WT1(NM_000378) fusion (E9;W7) | FoundationOne NGS and Washington University cytogenetics | Yes | Expression of CD56, chromogranin and synaptophysin |
| 11 | nuc ish (5′EWSR1x2-3, 3′EWSR1x2-3) (5′EWSR1 con 3′EWSR1x1) [97/100] | EWSR1 probe/Mayo clinic laboratories | No | Expression of CD56, chromogranin and synaptophysin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trikalinos, N.A.; Chrisinger, J.S.A.; Van Tine, B.A. Common Pitfalls in Ewing Sarcoma and Desmoplastic Small Round Cell Tumor Diagnosis Seen in a Study of 115 Cases. Med. Sci. 2021, 9, 62. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9040062
Trikalinos NA, Chrisinger JSA, Van Tine BA. Common Pitfalls in Ewing Sarcoma and Desmoplastic Small Round Cell Tumor Diagnosis Seen in a Study of 115 Cases. Medical Sciences. 2021; 9(4):62. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9040062
Chicago/Turabian StyleTrikalinos, Nikolaos A., John S. A. Chrisinger, and Brian A. Van Tine. 2021. "Common Pitfalls in Ewing Sarcoma and Desmoplastic Small Round Cell Tumor Diagnosis Seen in a Study of 115 Cases" Medical Sciences 9, no. 4: 62. https://0-doi-org.brum.beds.ac.uk/10.3390/medsci9040062