
Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue

1
Institut du Savoir Montfort, 1E103, 713 Montreal Road, Ottawa, ON K1K 0T2, Canada
2
Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON K1N 6N5, Canada
3
Clinical Diabetes and Metabolism, Department of Medical Sciences, Uppsala University, 752 36 Uppsala, Sweden
4
School of Human Kinetics, Faculty of Health Sciences, University of Ottawa, Ottawa, ON K1N 6N5, Canada
5
Interdisciplinary School of Health Sciences, Faculty of Health Sciences, University of Ottawa, Ottawa, ON K1N 6N5, Canada
*
Author to whom correspondence should be addressed.
Environments 2021, 8(4), 35; https://doi.org/10.3390/environments8040035
Submission received: 20 January 2021
/
Revised: 12 March 2021
/
Accepted: 2 April 2021
/
Published: 19 April 2021
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Bisphenol A (BPA) and bisphenol S (BPS) are environmental contaminants that have been associated with the development of insulin resistance and type 2 diabetes (T2D). Two organs that are often implicated in the development of insulin resistance are the skeletal muscle and the adipose tissue, however, seldom studies have investigated the effects of bisphenols on their metabolism. In this review we discuss metabolic perturbations that occur in both the skeletal muscle and adipose tissue affected with insulin resistance, and how exposure to BPA or BPS has been linked to these changes. Furthermore, we highlight the possible effects of BPA on the cross-talk between the skeletal muscle and adipose tissue.
1. Introduction
Diabetes is a chronic disease that is marked by lack of insulin production by the pancreatic β-cells and/or the inability of tissues and cells to appropriately respond to insulin [1]. Type 2 diabetes (T2D) accounts for the majority of diabetes cases and has been shown to be the result of a variety of factors, including genetic predisposition, diet, obesity, lack of physical activity, and environmental conditions such as chemicals [1]. Whilst the T2D epidemic has been greatly focused on carbohydrate consumption and physical activity, recent studies emphasized the association between pollutant exposure and the development of T2D. Increased awareness can be attributed to the growing number of studies highlighting a positive association between the accumulated levels of persistent organic pollutants (POPs) in the body and metabolic dysfunction [2,3,4]. In addition, there is a synergistic association between the body load of POPs and the development of metabolic syndrome [5]. It is hypothesized that increased production and exposure to environmental contaminants can contribute and accelerate the development of T2D.
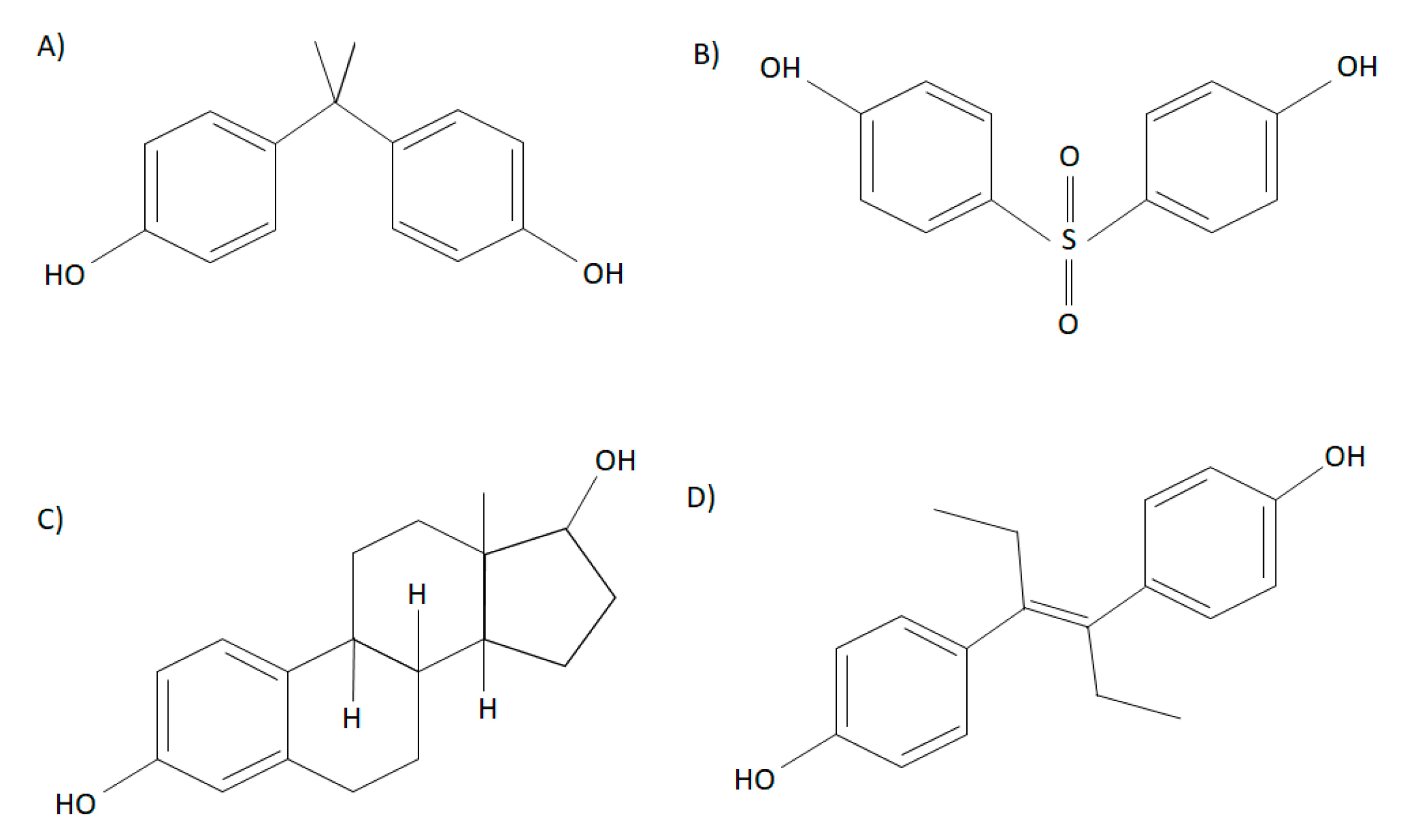
Bisphenols are high-production chemicals that are commonly used in the manufacturing of plastic products and are characterized by two hydroxyphenol groups. The most abundantly used bisphenol, bisphenol A (BPA) (2,2-bis(4-hydroxyphenyl)propane) contains a dimethyl derivative, and is soluble in organic solvents (Figure 1) [6]. Repeating BPA monomers are used in the production of polycarbonate plastics and epoxy resins [6]. Although the carbonate linking of BPA monomers is often stable, BPA can leach due to incomplete polymerization or degradation of polymers at high temperatures or altered acidity [7,8]. In the 1930’s, Edward Charles Dodd discovered that BPA had estrogenic effects similar to the synthetic estrogen diethylstilbestrol, a drug that is structurally similar to BPA, and used as a nonsteroidal estrogen to support pregnancies in women with recurrent miscarriages (Figure 1). Structurally, BPA resembles 17β-estradiol (E2) and has been shown to have hormone-mimicking properties [9,10]. Nonetheless, since the 1950’s, BPA has been produced in large quantities, due to many factors such as its cost-efficient method of producing products, including plastic bottles, food containers, thermal receipts, and dental sealants [6]. Early research demonstrated that BPA had low toxicity and was rapidly metabolized, ultimately leading to safety approval of use in plastic production [11]. It was later found in both in vivo and in vitro studies that BPA acts as an endocrine disruptor, leading to adverse endocrine effects such as increased prostate weight, growth of mammary glands, and altered postnatal development; therefore, challenging previous beliefs that BPA was safe at low doses [12]. Due to increasing concerns about the adverse effects of BPA on health over the last few decades, it has recently been replaced by an analog known as bisphenol S (BPS). Bisphenol S (4,4′-sulfonylbisphenol) is structurally similar to BPA and has two hydroxyphenol groups around a sulfonyl group. Polymers of BPS are known as polyether sulfones [6,13] (Figure 1). The research on the toxicological effects of BPS is sparse, and thus, there are fewer regulations on its use in consumer products. Interestingly, it has been shown that the effects BPS may be more potent than BPA [14]. For example, it has been shown that BPS has a more potent effect on lipid metabolism in 3T3-L1 cells than BPA, such as on adipogenesis and peroxisome proliferator-activated receptor (PPARγ) activation [15]. In addition to BPA and BPS, other analogs have been less frequently studied, including bisphenol AF (BPAF), bisphenol F (BPF), bisphenol B (BPB), dimethylbisphenol A (DMBPA), bisphenol C (BPC), and tetrabromobisphenol A (TBBPA), some of which have been shown to also have endocrine activity. Although BPS and BPF are the most commonly used BPA analogs, exposure to other analogs have been demonstrated in human populations [14]. For instance, studies have demonstrated that BPA analogs such as BPF, BPB, BPAF, bisphenol Z (BPZ), and bisphenol E (BPE), among others, have been found in food products [16,17]. In addition, BPS, BPF, BPAF, and BPC are reported to have estrogenic activity similar to or greater than BPA [18]. Some of these analogs, such as BPB, BPE, and BPF can also bind other hormonal receptors, such as the androgen receptor [18,19]. It was reported that BPF decreased basal testosterone secretion in human fetal testes, highlighting its anti-androgenic effects [18]. Further research is required to understand whether BPA and its analogs are safe for human health in order to improve manufacturing regulations.
2. Exposure, Absorption and Metabolism
It has been reported that BPA exposure is widespread, as BPA has been detected in over 90% of urinary samples in the populations of the United States, Europe, and Asia [20,21,22]. The Environmental Protection Agency (United States) states that the reference dose (maximal acceptable oral dose of a substance) of BPA is 50 μg/kg/day, which is 1000 times lower than the lowest observed adverse effect level (LOAEL) [23]. Interestingly, the European Food Safety Authority (EFSA) states that the tolerable daily intake for BPA is 4 μg/kg/day, however concentrations below this are scarcely used in experimental studies [24]. Interestingly, animal models have shown that there are effects at doses lower than the reference dose. In a study by Timms et al., 2005, showing that pregnant mice exposed to 0.25 μg/kg/day of BPA had disturbed mammary gland growth, postnatal growth, and rate of sexual maturation [25]. Similarly, in a different study, rats exposed to a low dose of BPA (2.5 μg/kg/day) in utero had changes in the mevalonate pathway and a reduction in the neurotrophic precursor brain derived neurotrophic factor (pro-BDNF), which is implicated in brain development and function [26]. The levels of BPA found in humans often depend on various factors such as sample type (serum, tissue, etc.). However, levels measured (i.e., environmentally-relevant) are often in the low nanomolar range (below 50 nM) [27,28,29]. Low concentrations of BPA used in animal studies are often similar to levels found regarding human exposure from plastic containers, cans, dental sealants, and even drinking water (low nanomolar range) [23]. In one study, the amount of BPA that leached from polycarbonate water bottles at room temperature was found to be 0.2 to 0.79 ng of BPA per hour, and boiling increased the rate by 55-fold [30]. Additionally, this study showed that levels of BPA corresponding to levels in the water were able to stimulate rapid non-genomic pathways in cerebellar neurons [30]. Hence, it is apparent that experimental studies using low concentrations of BPA may provide a proxy for understanding in vivo effects following human exposure to this compound.
The most common form of bisphenol exposure is oral ingestion, as food is the most important source of BPA exposure in the general population [31]. After oral ingestion, bisphenols are absorbed in the gastrointestinal tract and metabolized in the liver into BPA-glucuronide (BPA-G) by UDP-glucuronosyltransferases (UGT) for excretion in the urine [32]. BPA-glucuronide can be reactivated by cleavage, primarily by bacterial enzymes, in the intestinal tract and can then enter the bloodstream [33]. UDP-glucuronosyltransferase 2B1 (UGT2B1) is an enzyme that glucuronidates BPA for excretion, and interestingly is reduced in pregnant women, whereas it is absent in fetuses, and slowly appears after birth [23]. Fetuses and neonates have been reported to have higher potency of BPA due to the limited capacity of the liver to conjugate BPA to BPA-G [34]. In addition, evidence shows there is BPA bioaccumulation during pregnancy, and this does not occur in non-pregnant females, or in males [34]. Thus, it is apparent that pregnant women, fetuses, and newborns may have higher levels of metabolically active BPA in circulation due to reduced UGT2B1 thus reduced excretion [23].
3. Potential Mechanisms of Action
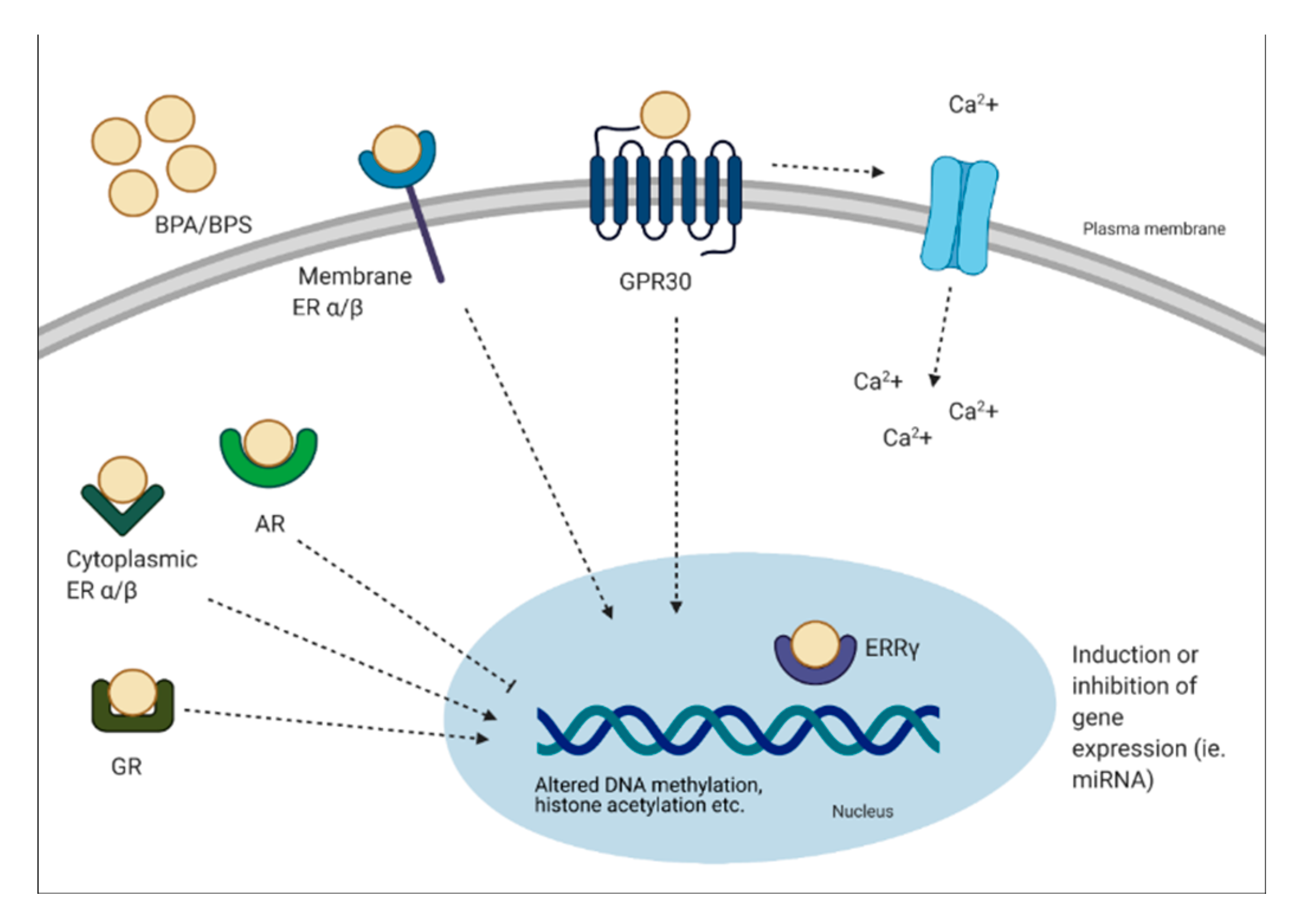
Bisphenols can have genomic and non-genomic effects. Multiple mechanisms have been identified, including binding to the estrogen receptors (ER-α, ER-β, G protein-coupled receptor 30 (GPR30)) and estrogen-related receptor (ERR-γ) [6]. BPA can bind to classical and non-classical ER and trigger genomic responses by activating gene expression. It has been demonstrated that BPA can bind ER-α and -β, but at an affinity at least 10,000-fold less than E2 [35]. This process involves the formation of a complex with the ER, translocation to the nucleus and binding to the ER element to alter transcriptional activity [36]. Although BPA can bind to ER, it has a weak affinity to these receptors. Interestingly, BPA has been shown to have a strong affinity for ERR-γ at low nanomolar ranges [37,38]. The expression pattern of ERR-γ is important to note, as it is expressed in a tissue-specific manner during development [37]. In other studies, BPA has been shown to act as an antagonist to the thyroid hormone receptor (TR) and suppress transcriptional activity in a dose-dependent manner [38,39,40]. Other receptors BPA can bind include the AR and GR [41,42]. Non-genomic effects of BPA include formation of multimolecular complexes with ER-α and ER-β at the plasma membrane. This results in rapid non-genomic responses, such as ER-α-mediated phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathways, or ER-β-mediated mitogen-activated protein kinase (MAPK) signaling [36]. In relation to rapid signaling, the binding of BPA to GPR30 is linked to altered oscillation of Ca2+, which can lead to events such as endoplasmic reticulum stress and insulin release from pancreatic islets [36]. It has been shown that 1 pM of BPA stimulates Ca2+ influx within 30 seconds in rat pituitary tumor cells [43]. Furthermore, prenatal BPA exposure has been linked to epigenetic changes, such as decreased DNA methylation of Grin2b, a gene important for neuronal function in the brain of adult mice [44]. BPA exposure has also been shown to alter microRNA (miRNA) expression, which are non-coding RNA molecules that function in RNA silencing. For instance, BPA treatment was associated with the induction of miR-146a in placental cells, which lead to a reduction in cell proliferation and higher sensitivity to DNA damage [45]. Other non-genomic pathways BPA has been shown to alter include histone acetylation, such as histone acetylation in cerebral cortex and hippocampus of mice offspring’s, a key regulator of transcription and memory formation [46].
It is important to briefly highlight the sex differences in response to bisphenol exposure and metabolism. Studies have reported that women show differential expressions of the ER in comparison to men. For instance, in one study, it was shown that estradiol signaling mediates sex differences in visceral adipose tissue, as males have lower levels of ER-α than females, which corresponds to higher visceral adiposity in males [47]. Prenatal exposure to low doses of BPA can alter the transcriptome of the amygdala of neonates [48]. Specifically, it is believed that the female amygdala is more sensitive to BPA than males, highlighting possible sex differences in response to BPA exposure [48]. Therefore, it is evident that since BPA and BPS can exert their effects through ER signaling pathways, there may be sex differences in exposure to these compounds.
In brief, it is apparent that BPA can exert its effects by binding to various hormonal receptors, however, the overall effects may be due to various factors such as the type of receptor expressed and the type of tissue exposed. A schematic diagram describing the mechanisms of action of BPA is presented in Figure 2.
4. Implications of Bisphenol Exposure on the Development of Type 2 Diabetes
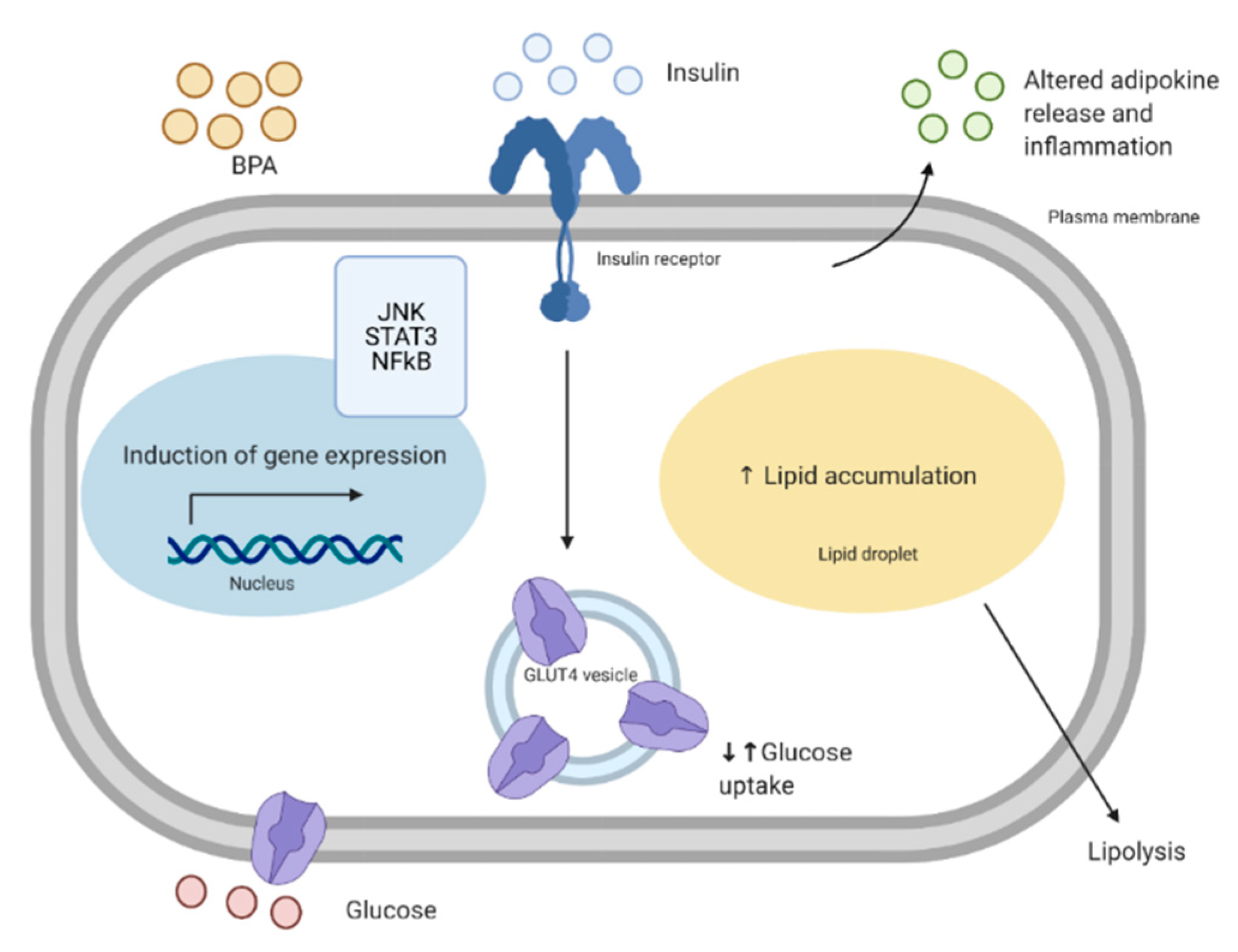
In the past few decades, there has been a growing list of studies showing the link between BPA exposure and the development of T2D. For example, analyzed urine samples from the National Health and Nutrition Examination Survey (NHANES), a US survey, has shown a positive association between high urinary BPA (>4.20 ng/mL) and T2D development, independent of common diabetes risk factors [49]. Other studies have demonstrated similar trends that show a positive association between urinary BPA and the development of T2D [50,51]. A sub-study, which included 3 NHANES cycles, showed that urinary BPA was associated with elevated glycated hemoglobin (HbA1C) and T2D incidence when data were pooled from all three cycles, but there was only a significant association in one of the cycles [52]. They concluded that it was likely that the other two cycles were not significant due to reduced statistical power [52]. They emphasized the importance of focusing on longitudinal studies to determine the association of urinary BPA and the development of T2D [52]. Interestingly, however, a 2012 study highlighted that NHANES datasets should not be solely used to draw conclusions about short-lived chemicals and their link to the development of diabetes, since they did not find such association. Thus, this highlights the importance of investigating the effects of chemicals such as BPA in vivo and in vitro [53]. Animal studies have investigated the in vivo effects of BPA exposure on glucose metabolism. Mice exposed to a single low dose (10 μg/kg) of BPA intraperitoneally show increased insulin release followed by a rapid reduction in glycemia [54]. Furthermore, long-term BPA exposure in mice (intraperitoneal twice daily for 4 days) resulted in an increase of β-cell insulin content, hyperinsulinemia, and insulin resistance [54]. This is supported by in vitro studies showing that exposure of a mouse β-cell line to BPA (100 ng/mL) for 1 h leads to enhanced glucose-stimulated insulin secretion, which is associated with mitochondrial stress and activation of B-cell lymphoma 2 (Bcl-2) family members as well as caspases which are responsible for apoptosis in pancreatic β-cells [55]. Contrastingly, long-term exposure (72 h) to high concentrations of BPA can lead to reduced insulin secretion of INS-1E cells [56]. Therefore, it is evident that BPA can alter β-cell function, which can then contribute to the development of T2D.
Two major sites of metabolic dysfunction in T2D are the skeletal muscle and adipose tissue. Skeletal muscle uptakes approximately 80% of postprandial glucose disposal, therefore, is vital for the regulation of glucose homeostasis [57]. Adipose tissue plays an important role in whole-body homeostasis, as it serves not only as a storage reservoir, but also acts as an endocrine organ that produces and secretes several hormones, peptides, and enzymes, that can act locally but also enter the circulation and mediate inter-organ cross-talk [58]. Although there is a clear association between bisphenol exposure, insulin resistance and T2D, there are few studies that investigate the effects of bisphenols on human skeletal muscle and adipose tissue metabolism. Both tissues play important roles in whole-body glucose homeostasis, understanding the effects of bisphenols on their metabolism would further elucidate the mechanisms bisphenols exert.
5. The Effects of Bisphenols on Skeletal Muscle Glucose Metabolism
5.1. Insulin Signaling Pathway
Insulin resistance of the skeletal muscle is an important feature in T2D. Under healthy conditions, the binding of insulin to the insulin receptor leads to phosphorylation of the insulin receptor substrate 1 and 2 (IRS1/IRS2), in which tyrosine phosphorylation of the IRS by the insulin receptor allows for the recruitment of PI3K [59]. This is followed by the recruitment and phosphorylation of Akt, inactivation of AS160 (Akt substrate of 160 kDa), GLUT4 translocation to the cell membrane, and glucose uptake [60]. Furthermore, Akt phosphorylates and inactivates glycogen synthase kinase 3 (GSK3). Inhibition of GSK3 leads to active glycogen synthase, increased glycogen synthesis, and reduced blood glucose. Patients with T2D show skeletal muscle insulin resistance and impaired muscle glucose uptake, glycogen synthesis, and glycogen synthase activation [61]. Interestingly, increased serine phosphorylation of IRS1 leads to reduced Akt phosphorylation and inhibition of the insulin signaling pathway, which is exhibited in muscle biopsy samples from insulin resistant offspring of patients with T2D [62]. Serine phosphorylation of IRS1 has also been shown to be involved in fat-induced insulin resistance [63]. Altered glucose homeostasis may be due to defects in the insulin signaling pathway [61]. It is proposed that early stages of T2D are linked to reduced IRS and PI3K signaling, whereas late stages of T2D are characterized by reduced GLUT4 translocation [64,65].
Although bisphenols have been linked to the development of insulin resistance in the skeletal muscle, it is still not completely clear how this occurs. In one study, rats orally treated with 10–400 mg/kg body weight (bw)/day of BPA for 30 days had reduced protein level of insulin receptor β and its tyrosine phosphorylated form in the gastrocnemius muscle [66]. Additionally, the total and phosphorylated forms of IRS1 and Akt were reduced [66]. These changes in the insulin signaling pathway of the muscle corresponded to hyperglycemia and insulin resistance in these rats [66]. Moreover, they also showed that BPA was able to accumulate in the gastrocnemius muscle of these rats, following a non-monotonic dose response. It is possible that at high concentrations, BPA becomes toxic to the skeletal muscle, leading to accelerated clearance. Similarly, in another study, mice given a lower dose of BPA (50 μg/kg bw/day) orally for 12 weeks had reduced phosphorylation of Akt and GSK3 in the skeletal muscle, which corresponded to glucose intolerance in these mice [67]. Furthermore, these mice had reduced levels of adiponectin, an adipokine known to improve insulin sensitivity in the skeletal muscle. In another study, mice treated with higher concentrations (20 and 200 mg/kg bw/day) had reduced glucose oxidation, which corresponded to reduced protein levels of Akt, IRS1, and GLUT4, with no effect at the mRNA level [68]. This resulted in hyperinsulinemia and insulin resistance in the mice [68]. This highlights the whole-body effect of bisphenols and the possible changes in communication between the adipose tissue and the skeletal muscle (see Section 6 for an in-depth discussion about of the role of BPA on altered cross-talk between the adipose tissue and the skeletal muscle) [67]. Contrastingly, Alonso-Magedelena et al. (2006) showed that a single low dose of BPA (10 μg/kg bw/day) administered subcutaneously rapidly increased plasma insulin and decreased blood glucose 30, 60, and 90 minutes after injection, corresponding to improved insulin sensitivity [54]. Similarly, in one of our studies, we showed that L6 myotubes treated with high levels of BPA (105 nM), have an increased glycolytic rate, glucose uptake, and levels of insulin-induced phosphorylation of Akt [69]. Increased glucose uptake of L6 myotubes exposed to BPA brings into question how BPA exposure is linked to the development of muscle insulin resistance. Some studies investigating acute BPA exposure appear to show improved insulin sensitivity and release, whereas studies investigating longer, or “chronic” BPA exposure appear to show a reduction in insulin sensitivity and insulin [54,55]. It is possible that acutely, there is a compensatory response against the effects of BPA, however, this response is lost with chronic exposure. This could be explained by the fact that BPA can alter the mitochondrial function in skeletal muscle, as discussed in the following section.
5.2. Mitochondrial Function
Following a meal, insulin release and subsequent glucose uptake in peripheral tissues provide fuel for mitochondrial respiration and ATP production by oxidative phosphorylation. Specifically, a rise in cytoplasmic ATP/ADP in pancreatic β-cells signals insulin release [70]. The skeletal muscle is rich in mitochondria and is strongly reliant on oxidative phosphorylation for energy production [71]. Mitochondrial dysfunction can be a result of different factors such as reduced mitochondrial activity, or changes in reactive oxygen species (ROS) production [72]. Several studies have shown that the skeletal muscle of individuals with T2D or those with a family history of T2D have reduced mitochondrial function [71,73,74]. In healthy individuals, hyperinsulinemia results in increased ATP production in skeletal muscle, whereas individuals with insulin resistance often have a reduced response to insulin [75]. Morphological abnormalities in the mitochondria have been shown in individuals with T2D. For instance, Kelley et al., 2002 showed that skeletal muscle mitochondria were smaller in obesity and T2D and, in some cases, the mitochondria were damaged in the T2D subjects [71]. Furthermore, overall oxidative capacity of the mitochondria, measured by NADH:O2 oxidoreductase activity, was found to be reduced in the skeletal muscle of individuals with T2D [71]. Mutations in mitochondrial genes such as those that encode tRNA lead to impaired insulin secretion [76]. Moreover, increased levels of free fatty acids in the plasma lead to intracellular lipid accumulation and reduced oxidative capacity, which is associated with insulin resistance in muscle [77]. We have previously shown that L6 myotubes acutely (24 h) exposed to 105 nM of BPA have reduced basal and maximal mitochondrial function determined by oxygen consumption rate measurement [69]. These findings aligned with an in vitro study, where 24 h BPA exposure induced depolarization of the mitochondrial membrane potential, inhibition of the mitochondrial respiratory chain activity, and reduction of ATP production in intestinal goblet cells (LS174T cells) [78]. Furthermore, BPA (105 nM) leads to increased mitochondrial proton leak in L6 myotubes [69]. This aligns with patients with T2D, whose muscles show increased proton leak, and reduced basal and maximal respiration [79,80]. Specifically, mitochondrial proton leak can reduce mitochondrial membrane potential, which reduces ROS [81]. Mitochondrial UCPs are anion carrier proteins and play an important role in reducing ROS production [81]. It is possible that UCP3, which is the skeletal muscle isoform, may have increased activity by 24 h BPA exposure, which could explain increased proton leak. Therefore, by altering mitochondrial activity, BPA exposure may lead to insulin resistance in the skeletal muscle. The growth and division of pre-existing mitochondria, known as mitochondrial biogenesis, is driven by peroxisome proliferator-activated receptor (PPAR) coactivator (PGC)-1α and levels of PGC-1α are higher under conditions of increased ATP demand (i.e., exercise) [82]. It has been shown that in individuals with insulin resistance and T2D, PGC-1α levels are reduced in the skeletal muscle, highlighting a link between mitochondrial number and insulin resistance [74]. Interestingly, there are few studies investigating the effects of BPA on human skeletal muscle biogenesis. However, there are studies that have investigated other cell types, such as rat insulinoma (INS-1) cells, where BPA exposure leads to a reduction in Tfam expression, which is an important regulator of mitochondrial biogenesis [83].
6. The Effects of Bisphenols on Adipose Tissue Metabolism
6.1. Adipogenesis
In recent years, obesity has been considered a serious health concern due to its association with numerous health conditions such as T2D and is thought to be one of the greatest risk factors for the development of insulin resistance [84,85,86]. Adipose tissue plays a significant role in whole-body homeostasis, functioning as a storage reservoir and endocrine organ [87]. Specifically, adipose tissue secretes adipokines, cytokines, and chemokines that can act locally to regulate adipose tissue function or enter the circulation and signal to organs such as the skeletal muscle to regulate metabolism, thus can mediate inter-organ cross-talk [87].
Both in vivo and in vitro studies often show conflicting results regarding the effects of BPA on adipocyte metabolism [88]. For instance, it has been shown that BPA increases adipogenesis in a concentration-dependent manner in human adipose-derived stromal cells (ASCs) [88]. Boucher et al. verified that one metabolite of BPA, BPA-G, which was formerly perceived to be an inactive metabolite, can modify adipocyte metabolism. They demonstrated that chronic exposure to 0.05 and 0.25 μM of BPA-G induces differentiation in 3T3-L1 adipocytes (mouse embryonic fibroblast cells), and in primary human adipocytes. Additionally, 10 μM of BPA-G increased mRNA expression of adipogenic factors such as sterol regulatory element-binding protein 1 (SREBF1) and lipoprotein lipase (LPL), both of which are required in mature adipocyte phenotype [89]. In a different study, BPA was shown to increase adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children [90]. This may be due to an increase in PPARγ expression [91]. Interestingly, other studies have found that BPA fails to induce adipogenesis at comparable concentrations used in previous studies. For example, De Filippis et al. (2018) used the same concentrations of BPA that have been shown to enhance adipogenesis in 3T3-L1 adipocytes and found no effect on adipogenesis in 3T3-L1 adipocytes [92,93]. This inconsistency was also captured in a different study, where BPA did not induce adipogenesis in mesenchymal stem cells (MSCs), but instead only in 3T3-L1 cells [93]. These dissimilarities may be due to various differences such as in mechanisms of action BPA and BPA-G, differences in cell types and species (3T3-L1, primary human adipocytes, etc.), or the use of more improved techniques. Moreover, the differentiation cocktail used by Boucher et al. (2015) comprised of either BPA-G or dexamethasone, in which dexamethasone is normally required for adipocyte differentiation [89]. Contrastingly, the studies by De Filippis et al. (2018) and Chamorro-García et al. (2012) use both BPA and dexamethasone in the differentiation cocktail [92,93]. The presence of dexamethasone results in fully differentiated adipocytes. Therefore, it is also possible that differentiation is further increased when using both BPA and dexamethasone.
6.2. Insulin Signaling
Adipose tissue derived from individuals with T2D have been shown to have reduced insulin receptor kinase activity, which corresponds with reduced insulin-stimulated lipogenesis, and glucose uptake into the adipose tissue [94]. Adipocytes from obese or T2D patients have decreased phosphorylation of IRS1, which appears to be one of the initial changes in the insulin signaling pathway [95]. In one study, chronic treatment of 3T3-L1 pre-adipocytes with 1 nM of BPA during differentiation lead to reduced insulin-stimulated phosphorylation of Akt and glucose uptake [92]. In a different study, 3T3-F442A adipocytes treated with 10 μM of BPA for 24 h had increased glucose uptake and GLUT4 protein levels, therefore emphasizing the different effects that can occur depending on the concentration of BPA and length of exposure [96]. In one of our previous studies, we showed that human whole adipose tissue treated with 10 nM BPA for 24 h leads to a reduction in insulin-stimulated glucose uptake without altering protein phosphorylation of Akt, protein levels of GLUT4, or gene expression of AKT, IRS1, GLUT1, and GLUT4 [97]. Although there were no apparent changes in the insulin signaling pathway to explain the reduction in glucose uptake, it is possible that there were changes in the GLUT4 translocation to the membrane. Furthermore, this coincided with other diabetogenic drugs, such as cyclosporin A, which can reduce glucose uptake without altering the levels of key proteins in the insulin signaling pathway [98]. Often, studies investigating the effects of bisphenols on adipocyte and adipose tissue metabolism have inconsistent results. For instance, 3T3-L1 adipocytes incubated with 105 nM of BPA showed increased glucose uptake and GLUT4 expression at high concentrations, however, there were no effects at environmentally-relevant concentrations (1–104 nM) [96]. In contrast, human adipocytes incubated with 1 and 100 nM of BPA for 8 h had no changes in glucose uptake, whereas longer incubations (24 and 48 h) lead to reduced glucose uptake [97,99]. This reflects an important aspect of exposure to endocrine disruptors: the window of susceptibility and vulnerability. It is evident that adipocytes have divergent effects on factors such as experimental models, incubation times, and concentrations used, all of which make it difficult to draw a single conclusion on the effects of BPA on adipocyte and adipose tissue insulin signaling and glucose metabolism. The length and consistency of BPA exposure (acute or chronic) appears to be an important factor in adverse outcomes [100]. Furthermore, different groups may have varying vulnerability to the adverse effects of BPA. For instance, pregnancy is considered to be a critical window of susceptibility, for both the mother and the offspring, as reviewed by Alonso-Magdalena et al. (2015), and Roto and Soto, 2009 [101,102]. What can be concluded, however, is that it is evident from several studies that BPA exposure can alter glucose metabolism of adipocytes, and chronic exposure may have consequences on glucose metabolism in vivo.
6.3. Body Weight, Adipocyte Size, Lipid Accumulation
It has been shown that fetal exposure to BPA at levels at or lower than the established daily human safe dose (50 μg BPA/kg bw/day) increases body weight and postnatal growth in in vivo studies. In one study, pregnant rodents administered 10 mg/L of BPA orally, had 10–25 ng/g of BPA in tissue, which is comparable to human samples [100]. It was demonstrated that maternal BPA exposure leads to offspring obesity, hypertrophy of adipocytes, and augmented expression of adipogenic and lipogenic factors [100]. This was also shown in in vitro studies where pre-adipocytes from rats incubated with 1–20 μM of BPA for 5 days had an increased number of adipocytes, increased expression of adipogenic transcription factors (PPARγ, C/EBPα), and increased expression of TNFα [103]. In a similar study, rats exposed to 0.5 μg/kg bw/day of BPA orally from gestational day 3.5 until postnatal day 22, which is 8–10 times lower than European Food and Safety Authority daily tolerable dose, had higher plasma triglyceride concentrations and inguinal WAT adipocyte density in offspring [104]. It appeared that there was adipocyte hyperplasia, which is suggested to occur in the early stages of development. Therefore, gestational exposure to BPA can lead to lifelong adverse effects on adipose tissue metabolism in offspring.
Although the effects of BPS on adipocytes are even less understood, BPS has been shown to increase lipid accumulation and gene expression of adipogenic markers in human primary adipocytes [15]. In one study, treatment of 3T3-L1 cells with high concentrations of BPS (10 μM) during differentiation induced lipid accumulation and increased expression of adipogenic markers [15]. Furthermore, they showed that BPA and BPS can activate PPARγ, which is required for BPA- and BPS-induced adipogenesis [15]. Interestingly, BPS binding to the nuclear receptor PPARγ is stronger compared to BPA, thus implicating that BPS may not be a safe alternative for BPA [105]. Similarly, in a separate study, it was shown that BPA and BPS levels in the pM range were able to induce an increase in adipocyte triglyceride levels and a reduction in lipolysis in 3T3-L1 cells [105]. Increased triglyceride levels and reduction in lipolysis are two factors that may promote the development of obesity, highlighting one possible mechanism by which bisphenol exposure can lead to the development of obesity [106].
6.4. Adipokine Signaling
Adipokines are cytokines specifically released from adipose tissue and can have pro- inflammatory or anti-inflammatory effects [106]. Anti-inflammatory adipokines include adiponectin, transforming growth factor β (TGFβ), interleukin-10 (IL-10), and IL-4, whereas pro-inflammatory adipokines include IL-6, IL-1β, and leptin [106]. It has been shown that changes in certain adipokines, such as lower levels of adiponectin, and higher levels of IL-6 and TNFα, are implicated in the development of insulin resistance [107]. Low doses of BPA have been shown to alter adipokine expression in adipocytes, as in the study by Cimmino et al. (2019) that showed that human mature adipocytes and stromal vascular cells (SVFs) from subcutaneous mammary adipose tissue treated with 0.1 nM of BPA had increased levels of IL-6 and MCP1α via GPR30 signaling [108].
Human pre-adipocytes isolated from human adipose tissue explants incubated for 6 h with 0.1–10 nM of BPA have been shown to have reduced adiponectin release [108]. This is important since adiponectin is a critical adipokine involved in insulin sensitivity and inflammation and has been shown to be reduced in T2D [109]. Yamauchi et al., 2001 showed that mice lacking WAT had nearly no circulating adiponectin and were insulin resistant [109]. However, adiponectin administration to these mice led to improved insulin sensitivity and a reduction in muscle and hepatic triglyceride accumulation [4]. This is consistent with a study by Menale et al. (2016) that found a strong inverse association between BPA and adiponectin in children with obesity [4]. Furthermore, they found that resistin, an adipokine which increases TNFα and IL-6 expression, was detected in adipocytes only after BPA treatment [110,111].
In one of our recent studies, we found reduced rather than increased levels of the pro-inflammatory cytokines IL-6, IL-1B, and TNFA following 24 h BPA (1 or 10 nM) exposure [97]. This corresponds to studies by Peshdary et al 2020, who showed that BPS (0.1 µM) reduces IL-6 expression in subcutaneous human adipocytes. It is generally understood that increased IL-6 impairs insulin signaling in adipocytes and adipose tissue. For instance, in human adipose tissue IL-6 has been shown to reduce adiponectin expression, and in 3T3-L1 adipocytes, IRS1 and GLUT4 are reduced [112]. Although adipocytes secrete IL-6, it only accounts for approximately 10% of total tissue production, thus other cells in the adipose tissue may contribute to increased IL-6 production, which might explain the reduced IL-6 levels in adipocytes treated with BPA [113]. Furthermore, IL-1β mediates macrophage-induced impairments in the insulin signaling pathway, such as reduced phosphorylation of Akt in human adipocytes [114]. Although BPA can increase inflammatory markers in adipocytes, it has also been shown to reduce pro-inflammatory cytokine production in macrophages [115]. Interestingly, in a different study BPA was shown to promote macrophage polarization to an M1 pro-inflammatory subtype, which suggests future studies should focus on characterizing the effects of BPA on macrophage-induced inflammation [4,97]. Therefore, it is possible that whole adipose tissue cultured with BPA may have a different inflammatory response than isolated adipocytes due to the presence of the adipose tissue stroma [116]. BPA has also been proposed to elicit inflammatory pathways in adipocytes by activating c-Jun N-terminal kinases/signal transducer and activator of transcription 3 (JNK/STAT3) signaling [99]. Specifically, JNK has been proposed to contribute to metabolic syndrome, obesity, and insulin resistance by different mechanisms, such as by regulating the production of pro-inflammatory cytokines, and apoptosis [117]. Furthermore, it was shown that the inhibition of JNK leads to improved insulin sensitivity and glucose tolerance [118]. In one study, human and 3T3-L1 cells treated with BPA for 24 h had increased phosphorylation of JNK and STAT3 [99]. Additionally, they showed that BPA treatment leads to an increase in nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in nuclear extracts [99]. Interestingly, inhibition of JNK reverted the effects of BPA on glucose utilization and insulin signaling [99]. Other studies have shown similar results on the effects of BPA on JNK/STAT signaling, highlighting the link between BPA exposure and increased inflammation [119,120,121,122].
6.5. Adipose Depot Effects
Studies have shown hormonal heterogeneity between subcutaneous, abdominal, and gluteal adipose tissue [116]. In addition, distinct transcriptomic profiles have been identified in 15 different adipose tissue depots [116]. It is known that the molecular mechanisms of adipocytes in these adipose tissue depots differ. For example, visceral adipose tissue has lower insulin sensitivity, transfers and releases fatty acids more extensively, produces higher levels IL-6, and lower levels of leptin and adiponectin, in comparison to subcutaneous adipose tissue [112,123]. It is possible that BPA and BPS can have distinct effects in different depots. This occurs with other drugs like dexamethasone, which has depot dependent effects [124]. In one study, the effects of environmentally-relevant concentrations of BPA on subcutaneous and visceral breast adipocytes were investigated [108]. Interestingly, they showed that BPA-induced adiponectin decrease in these adipocytes was reversed by an estrogen receptor antagonist in visceral, and not subcutaneous adipose tissue [108]. Peshdary et al. showed that human pre-adipocytes treated with BPS had different levels of certain proteins such as IL-6, angiopoietin-2, and insulin-like growth factor binding protein 6 (IGFBP-6) in subcutaneous vs omental adipose tissue, also highlighting the different response to bisphenols in each depot [125]. Therefore, this highlights the possible differences in the mechanism of action of bisphenols in different adipose depots, and future studies should investigate these possible differences.
Together, these studies have suggested that BPA may not only be obesogenic but also diabetogenic. Understanding the effects of bisphenols on adipocytes and adipose tissue provides insight into its effect on whole-body metabolism, however, discrepancies in results may be due to differences in cell types used in the above discussed studies. Since whole adipose tissue contains pre-adipocytes, fibroblasts, endothelial cells, and immune cells such as macrophages, investigating the effects of bisphenols on whole adipose tissue compared to adipocytes and immune cells alone may provide insight on the effects that occur in vivo. Furthermore, the effects of BPA on other measures of lipid metabolism such as lipogenesis has been seldom reported, and future studies should be conducted.
7. Can Bisphenols Alter on the Cross-Talk between the Adipose Tissue and the Skeletal Muscle?
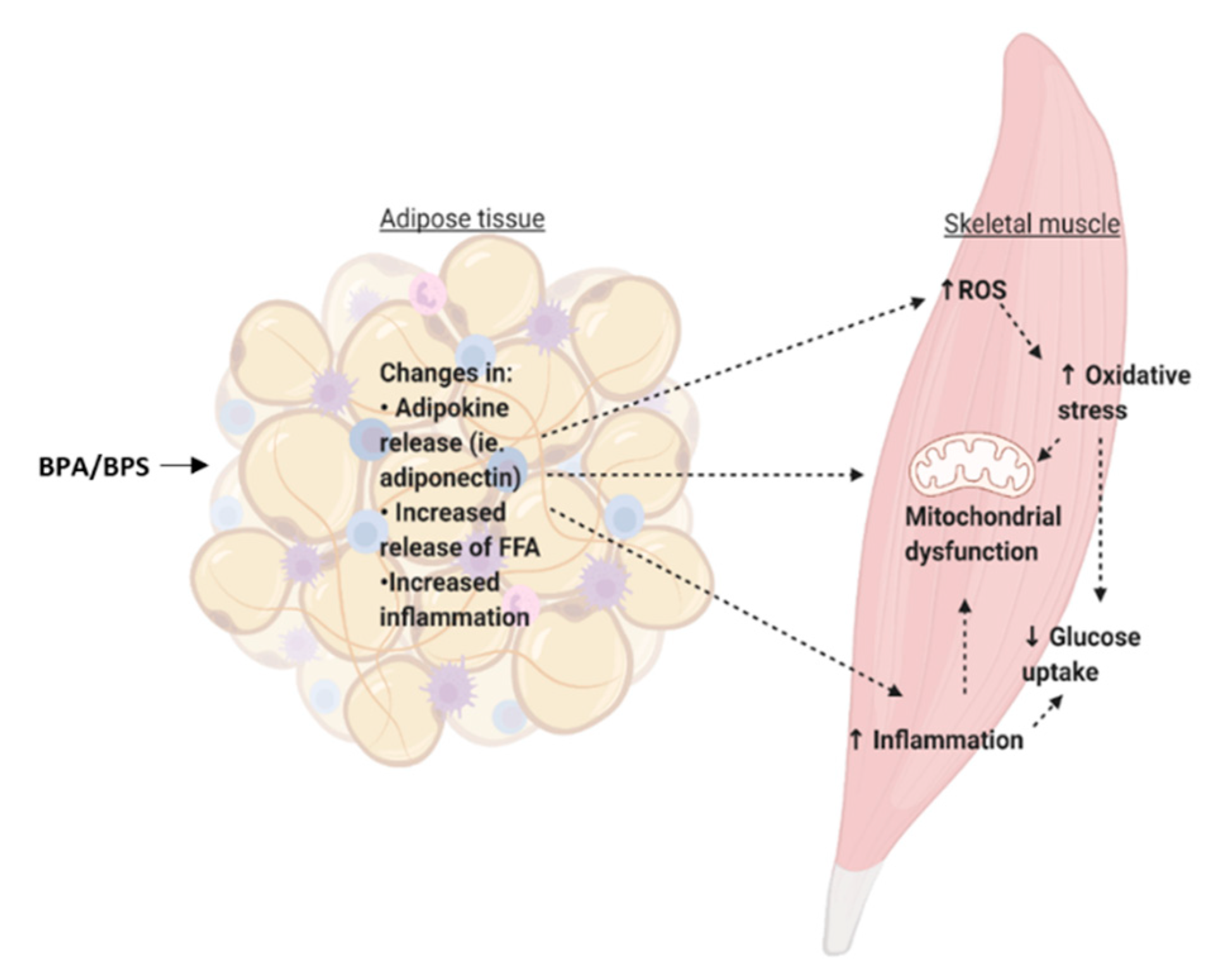
At environmentally-relevant concentrations, BPA exposure in in vivo models has shown impaired glucose metabolism. As mentioned previously, Alonso-Magdalena et al. (2006) showed that mice exposed acutely to a low dose of BPA (10 μg/kg) had a rapid reduction in glycemia that corresponded to an increase in insulin secretion [54]. Adipose tissue incubated for 24 h with BPA had reduced levels of pro-inflammatory cytokine expression, which may contribute to the overall improvement in glucose metabolism of the skeletal muscle [97]. Furthermore, although 24 h exposure leads to reduced glucose uptake in adipose tissue, the skeletal muscle accounts for a great majority (≈80%) of postprandial glucose disposal [126]. Therefore, despite reduced glucose uptake in the adipose tissue that we see in our study, there may be improved insulin sensitivity of the skeletal muscle in response to acute BPA exposure due to the fact that most insulin-stimulated glucose disposal occurs in the muscle. Therefore, future studies should focus on the effects of acute BPA exposure on cross-talk between the adipose tissue and the skeletal muscle to determine if adipokines secreted from adipose tissue explain the improvements in skeletal muscle metabolism.
Metabolic changes in adipose tissue that lead to increased release of free fatty acids (FFA), such as through lipolysis, can alter the metabolism of peripheral tissues and cause an ectopic lipid deposition in organs such as the skeletal muscle [127]. Under normal conditions, insulin inhibits lipolysis [127]. Although it is currently unclear whether BPA alters lipolysis in whole adipose tissue, in 3T3-L1 cells, BPA (1 fM, 1 pM, 1 nM) exposure for ten days has been shown to reduce lipolysis [105]. Therefore, it is possible that BPA does not exert negative effects on adipose-to-peripheral communication through increased lipolysis and increased ectopic lipid deposition. However, this particular study was performed in vitro, and BPA has been shown to induce pancreatic islet cell dysfunction, which includes reduced insulin secretion and increased apoptosis [128]. In vivo, it is possible that BPA-induced changes in pancreatic islet function and reduced insulin release can prevent the inhibition of lipolysis and thus lead to the release of FFA, which may accumulate in organs such as the skeletal muscle. The skeletal muscle can store approximately 0.5% of lipid droplet volume density, however, this can increase to about 1.5% in individuals with obesity [126]. Moreover, lipid accumulation in the muscle is negatively associated with insulin sensitivity [129]. Therefore, future studies should investigate the effects of BPA on islet cell-adipose tissue-muscle communication.
Adipokines play an important role on adipose-to-muscle communication. As previously mentioned, the adipose tissue can function as an endocrine organ and can secrete adipokines such as adiponectin, and IL-6. Adiponectin can bind to the adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2) in the skeletal muscle, leading to the activation of signaling pathways such as AMP-activated protein kinase (AMPK) and PPARα [130]. Adiponectin signaling can increase FA oxidation and glucose uptake in the skeletal muscle. It has been shown in in vitro studies that adiponectin can increase insulin sensitivity in the skeletal muscle. For instance, C2C12 muscle cells incubated with adiponectin for 60 minutes have increased glucose uptake and β-oxidation [131]. Similarly, L6 muscle cells exposed to adiponectin for 4 h were shown to have increased GLUT4 translocation and glucose uptake, which is consistent with increased activation of AMPK [131]. Interestingly, we showed that human adipose tissue treated with BPA in the nanomolar range has reduced expression of adiponectin [97]. This is consistent with a study by Hugo et al. (2008) that showed human pre-adipocytes incubated for 6 h with 0.1, 1, and 10 nM of BPA to have reduced levels of adiponectin [108]. Therefore, in vivo, BPA-induced reduction in adiponectin may lead to reduced insulin sensitivity of the skeletal muscle. BPA has also been shown to alter the expression of pro-inflammatory cytokines such as IL-6 and TNFα. As mentioned in the adipokine signaling section, BPA has been shown to increase expression of pro-inflammatory markers in some studies, whereas their expression was reduced in others, which may be due to factors such as cell type, concentration, and treatment length. Nonetheless, this has implications for altered communication with the skeletal muscle. For instance, palmitate-induced IL-6 production leads to reduced GLUT4 translocation in C2C12 skeletal muscle [132]. Thus, it is possible that elevated FFA levels, such as in obesity, can lead to an increase in IL-6 production, which can then alter skeletal muscle glucose uptake. Likewise, skeletal muscle treated with TNFα resulted in increased phosphorylation of p70 S6 kinase (S6K), extracellular signal-regulated kinases (ERK)-1/2, and JNK and increased phosphorylation of IRS1 at Ser 312, all of which are implicated in the negative regulation of insulin signaling [133]. Moreover, changes in circulating factors such as adipokines may lead to increased oxidative stress in the skeletal muscle. We recently showed that in L6 cells, BPA does not alter levels of oxidative stress markers, however, it may be possible that in vivo, cross-talk with the adipose tissue may lead to increased oxidative stress, potentially leading to a reduction in mitochondrial function. Thus, it is possible that altered adipokine release due to BPA exposure of adipose tissue may lead to impairment in muscle glucose metabolism.
Taken together, impaired adipose tissue metabolism due to BPA may result in changes in communication with the skeletal muscle, through changes in adipokine and lipid release. Future studies should investigate adipose-to-skeletal muscle cross-talk in order to better characterize this communication.
Cross-talk between adipose tissue and skeletal muscle is summarized in Figure 5.
8. Conclusions
It is evident that we are constantly exposed to low levels of BPA and/or its analogs, whether it is from thermal receipts, plastic products, or airborne particles. Several studies have demonstrated that BPA exposure is associated with altered glucose metabolism and insulin resistance, however, the exact mechanisms are not fully understood. In skeletal muscle, it has been shown that BPA can alter mitochondrial function and glucose metabolism. In adipocytes and adipose tissue, it is apparent that BPA exposure can alter differentiation, glucose uptake, insulin signaling, and adipokine levels. Since both tissues play a crucial role in whole body glucose metabolism, chronic exposure to BPA and its analogs are a concern, since it may lead to insulin resistance and the development of T2D. Cross-talk between the skeletal muscle and adipose tissue involves an interplay of adipokines and fatty acids released from the adipose tissue, and myokines released from the skeletal muscle. Whether BPA and its analogs can alter this cross-talk is of interest, and would provide deeper insight into the effects of BPA on the development of metabolic disease, and would then have implications for future manufacturing regulations.
Author Contributions
Conceptualization, F.A., M.J.P. and C.A.; writing—original draft preparation, F.A., M.J.P. and C.A.; writing—review and editing, F.A., M.J.P. and C.A.; visualization, F.A., M.J.P. and C.A.; supervision, C.A. and M.J.P.; project administration, M.J.P. and C.A.; funding acquisition, C.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grants (grant numbers 2015-06263).
Acknowledgments
We acknowledge Léa Garneau for critically reading the manuscript.
Conflicts of Interest
The authors have no competing interest to report and have no potential or real conflicts of interest to declare.
References
- Roglic, G. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Soundararajan, A.; Prabu, P.; Mohan, V.; Gibert, Y.; Balasubramanyam, M. Novel insights of elevated systemic levels of bisphenol-A (BPA) linked to poor glycemic control, accelerated cellular senescence and insulin resistance in patients with type 2 diabetes. Mol. Cell. Biochem. 2019, 458, 171–183. [Google Scholar] [CrossRef]
- Stahlhut, R.W.; Myers, J.P.; Taylor, J.A.; Nadal, A.; Dyer, J.A.; vom Saal, F.S. Experimental BPA exposure and glucose-stimulated insulin response in adult men and women. J. Endocr. Soc. 2018, 2, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Menale, C.; Grandone, A.; Nicolucci, C.; Cirillo, G.; Crispi, S.; di Sessa, A.; Marzuillo, P.; Rossi, S.; Mita, D.G.; Perrone, L.; et al. Bisphenol A is associated with insulin resistance and modulates adiponectin and resistin gene expression in obese children. Pediatric Obes. 2017, 12, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Is the Diabetes Epidemic Primarily Due to Toxins? Integr. Med. 2016, 15, 8–17. [Google Scholar]
- Ben-Jonathan, N.; Hugo, E.R. Bisphenols come in different flavors: Is “S” better than “A”? Endocrinology 2016, 157, 1321–1323. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.E.; Kendig, E.L.; Belcher, S.M. Assessment of bisphenol A released from reusable plastic, aluminium and stainless steel water bottles. Chemosphere 2011, 85, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geens, T.; Goeyens, L.; Covaci, A. Are potential sources for human exposure to bisphenol-A overlooked? Int. J. Hyg. Environ. Health 2011, 214, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Gould, J.C.; Leonard, L.S.; Maness, S.C.; Wagner, B.L.; Conner, K.; Zacharewski, T.; Safe, S.; McDonnell, D.P.; Gaido, K.W. Bisphenol A interacts with the estrogen receptor h in a distinct manner from estradiol. Mol. Cell. Endocrinol. 1998, 142, 203–214. [Google Scholar] [CrossRef]
- Nakagomi, M.; Suzuki, E.; Saito, Y.; Nagao, T. Endocrine disrupting chemicals, 4-nonylphenol, bisphenol A and butyl benzyl phthalate, impair metabolism of estradiol in male and female rats as assessed by levels of 15α-hydroxyestrogens and catechol estrogens in urine. J. Appl. Toxicol. 2018, 38, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Sajiki, J.; Yonekubo, J. Leaching of bisphenol A (BPA) from polycarbonate plastic to water containing amino acids and its degradation by radical oxygen species. Chemosphere 2004, 55, 861–867. [Google Scholar] [CrossRef]
- Vogel, S.A. The Politics of Plastics: The Making and Unmaking of Bisphenol A “Safety”. Am. J. Public Health 2009, 99, S559–S566. [Google Scholar] [CrossRef]
- Kang, J.S.; Choi, J.S.; Kim, W.K.; Lee, Y.J.; Park, J.W. Estrogenic potency of bisphenol, S. polyethersulfone and their metabolites generated by the rat liver S9 fractions on a MVLN cell using a luciferase reporter gene assay. Reprod. Biol. Endocrinol. 2014, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochester, J.R.; Bolden, A.L. Bisphenol S and F: A systematic review and comparison of the hormonal activity of bisphenol a substitutes. Environ. Health Perspect. 2015, 123, 643–650. [Google Scholar] [CrossRef]
- Ahmed, S.; Atlas, E. Bisphenol S- and bisphenol A-induced adipogenesis of murine preadipocytes occurs through direct peroxisome proliferator-activated receptor gamma activation. Int. J. Obes. 2016, 40, 1566–1573. [Google Scholar] [CrossRef]
- Liao, C.; Kannan, K. Concentrations and profiles of bisphenol a and other bisphenol analogues in foodstuffs from the united states and their implications for human exposure. J. Agric. Food Chem. 2013, 61, 4655–4662. [Google Scholar] [CrossRef]
- Caballero-Casero, N.; Lunar, L.; Rubio, S. Analytical methods for the determination of mixtures of bisphenols and derivatives in human and environmental exposure sources and biological fluids. A review. Anal. Chim. Acta 2016, 908, 22–53. [Google Scholar] [CrossRef]
- Chen, D.; Kannan, K.; Tan, H.; Zheng, Z.; Feng, Y.L.; Wu, Y.; Widelka, M. Bisphenol Analogues Other Than BPA: Environmental Occurrence, Human Exposure, and Toxicity—A Review. Environ. Sci. Technol. 2016, 50, 5438–5453. [Google Scholar] [CrossRef] [PubMed]
- Eladak, S.; Grisin, T.; Moison, D.; Guerquin, M.J.; N’Tumba-Byn, T.; Pozzi-Gaudin, S.; Benachi, A.; Livera, G.; Rouiller-Fabre, V.; Habert, R. A new chapter in the bisphenol a story: Bisphenol S and bisphenol F are not safe alternatives to this compound. Fertil. Steril. 2015, 103, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, L.N.; Chahoud, I.; Heindel, J.J.; Padmanabhan, V.; Paumgartten, F.J.R.; Schoenfelder, G. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ. Health Perspect. 2010, 118, 1055–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Department of Health and Human Services. Fourth National Report on Human Exposure to Environmental Chemicals. Updated Tables, January 2019, Volume One, pp. 1–529. Available online: https://www.cdc.gov/exposurereport/pdf/FourthReport_UpdatedTables_Volume1_Jan2019-508.pdf (accessed on 19 April 2021).
- Welshons, W.v.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, 56–69. [Google Scholar] [CrossRef]
- Manukyan, L.; Dunder, L.; Lind, P.M.; Bergsten, P.; Lejonklou, M.H. Developmental exposure to a very low dose of bisphenol A induces persistent islet insulin hypersecretion in Fischer 344 rat offspring. Environ. Res. 2019, 172, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Timms, B.G.; Howdeshell, K.L.; Barton, L.; Bradley, S.; Richter, C.A.; vom Saal, F.S. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc. Natl. Acad. Sci. USA 2005, 102, 7014–7019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonini, C.; Segatto, M.; Gagliardi, S.; Bertoli, S.; Leone, A.; Barberio, L.; Mandalà, M.; Pallottini, V. Maternal dietary exposure to low-dose bisphenol a affects metabolic and signaling pathways in the brain of rat fetuses. Nutrients 2020, 12, 1448. [Google Scholar] [CrossRef] [PubMed]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Furuta, I.; Kato, E.H.; Kataoka, S.; Usuki, Y.; Kobashi, G.; Sata, F.; Kishi, R.; Fujimoto, S. Maternal serum and amniotic fluid bisphenol A concentrations in the early second trimester. Reprod. Toxicol. 2002, 16, 735–739. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tsutsumi, O.; Ikezuki, Y.; Takai, Y.; Taketani, Y. Positive Relationship between Androgen and the Endocrine Disruptor, Bisphenol A, in Normal Women and Women with Ovarian Dysfunction. Endocr. J. 2004, 51, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Le, H.H.; Carlson, E.M.; Chua, J.P.; Belcher, S.M. Bisphenol A is released from polycarbonate drinking bottles and mimics the neurotoxic actions of estrogen in developing cerebellar neurons. Toxicol. Lett. 2008, 176, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Almeida, S.; Raposo, A.; Almeida-González, M.; Carrascosa, C. Bisphenol A: Food Exposure and Impact on Human Health. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1503–1517. [Google Scholar] [CrossRef] [Green Version]
- Bushnik, P.T.; Haines, D.; Levallois, P.; Levesque, J.; Van Oostdom, C.; Viau, C. Lead and bisphenol A concentrations in the Canadian population. Health Rep. 2009, 54, 1547–1554. [Google Scholar]
- VandeVoort, C.A.; Gerona, R.R.; vom Saal, F.S.; Tarantal, A.F.; Hunt, P.A.; Hillenweck, A.; Zalko, D. Maternal and fetal pharmacokinetics of oral radiolabeled and authentic bisphenol a in the rhesus monkey. PLoS ONE 2016, 11, e0165410. [Google Scholar] [CrossRef] [Green Version]
- Schönfelder, G.; Wittfoht, W.; Hopp, H.; Talsness, C.E.; Paul, M.; Chahoud, I. Parent bisphenol a accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 2002, 110, 703–707. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Lemmen, J.G.; Carlsson, B.O.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular mechanisms of action of BPA. Dose-Response 2015, 13, 1559325815610582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heard, D.J.; Norby, P.L.; Holloway, J.; Vissing, H. Human ERRgamm, a Third Member of the Estrogen Receptor-Related Receptor (ERR) Subfamily of Orphan Nuclear Receptors: Tissue-Specific Isoforms Are Expressed during Development and in the Adult. Mol. Endocrinol. 2000, 14, 382–392. [Google Scholar] [PubMed] [Green Version]
- Freitas, J.; Cano, P.; Craig-Veit, C.; Goodson, M.L.; David Furlow, J.; Murk, A.J. Detection of thyroid hormone receptor disruptors by a novel stable in vitro reporter gene assay. Toxicol. Vitr. 2011, 25, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Tagami, T.; Akamizu, T.; Usui, T.; Saijo, M.; Kanamoto, N.; Hataya, Y.; Shimatsu, A.; Kuzuya, H.; Nakao, K. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 2002, 87, 5185–5190. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Park, Y.J. Bisphenols and thyroid hormone. Endocrinol. Metab. 2019, 34, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Bonefeld-Jørgensen, E.C.; Long, M.; Hofmeister, M.v.; Vinggaard, A.M. Endocrine-disrupting potential of Bisphenol A, Bisphenol A dimethacrylate, 4-n-nonylphenol, and 4-n-octylphenol in vitro: New data and a brief review. Environ. Health Perspect. 2007, 115, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Prasanth, G.K.; Divya, L.M.; Sadasivan, C. Bisphenol-A can bind to human glucocorticoid receptor as an agonist: An in silico study. J. Appl. Toxicol. 2010, 30, 769–774. [Google Scholar] [CrossRef]
- Wozniak, A.L.; Bulayeva, N.N.; Watson, C.S. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-α-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ. Health Perspect. 2005, 113, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Alavian-ghavanini, A.; Lin, P.; Lind, P.M.; Rimfors, S.R.; Halin, M.; Dunder, L.; Tang, M.; Lindh, C.; Bornehag, C.-G.; Rüegg, J. Prenatal Bisphenol A Exposure is Linked to Epigenetic Changes in Glutamate Receptor Subunit Gene Grin2b in Female Rats and Humans. Sci. Rep. 2018, 8, 11315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avissar-Whiting, M.; Veiga, K.R.; Uhl, K.M.; Maccani, M.A.; Gagne, L.A.; Moen, E.L.; Marsit, C.J. Bisphenol A exposure leads to specific microRNA alterations in placental cells. Reprod. Toxicol. (Elmsford N. Y.) 2010, 29, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Thakur, M.K. Effect of perinatal exposure to Bisphenol-A on DNA methylation and histone acetylation in cerebral cortex and hippocampus of postnatal male mice. J. Toxicol. Sci. 2017, 42, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Zheng, L.D.; Smith, C.; Luo, J.; Robinson, A.; Almeida, F.A.; Wang, Z.; Olumi, A.F.; Liu, D.; Cheng, Z. Estradiol signaling mediates gender difference in visceral adiposity via autophagy article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Arambula, S.E.; Jima, D.; Patisaul, H.B. Prenatal bisphenol A (BPA) exposure alters the transcriptome of the neonate rat amygdala in a sex-specific manner: A CLARITY-BPA consortium study. Neurotoxicology 2018, 65, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Teppala, S. Relationship between urinary bisphenol A levels and diabetes mellitus. J. Clin. Endocrinol. Metab. 2011, 96, 3822–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadkhaniha, R.; Mansouri, M.; Yunesian, M.; Omidfar, K.; Jeddi, M.Z.; Larijani, B.; Mesdaghinia, A.; Rastkari, N. Association of urinary bisphenol a concentration with type-2 diabetes mellitus. J. Environ. Health Sci. Eng. 2014, 12, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Cornelis, M.C.; Townsend, M.K.; Tobias, D.K.; Heather Eliassen, A.; Franke, A.A.; Hauser, R.; Hu, F.B. Association of urinary concentrations of bisphenol A and phthalate metabolites with risk of type 2 diabetes: A prospective investigation in the nurses’ health study (NHS) and NHSII cohorts. Environ. Health Perspect. 2014, 122, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.K.; O’Neill, M.S.; Sowers, M.F.R.; Park, S.K. Urinary Bisphenol a and type-2 diabetes in U.S. Adults: Data from NHANES 2003–2008. PLoS ONE 2011, 6, e26868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaKind, J.S.; Goodman, M.; Naiman, D.Q. Use of NHANES Data to Link Chemical Exposures to Chronic Diseases: A Cautionary Tale. PLoS ONE 2012, 7, e51086. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol a disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Makaji, E.; Raha, S.; Wade, M.G.; Holloway, A.C. Effect of Environmental Contaminants on Beta Cell Function. Int. J. Toxicol. 2011, 30, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Weldingh, N.M.; Jørgensen-Kaur, L.; Becher, R.; Holme, J.A.; Bodin, J.; Nygaard, U.C.; Bølling, A.K. Bisphenol A Is More Potent than Phthalate Metabolites in Reducing Pancreatic β-Cell Function. BioMed Res. Int. 2017, 2017, 4614379. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Romacho, T.; Elsen, M.; Röhrborn, D.; Eckel, J. Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, R.W.A.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. In Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy; Dove Medical Press Ltd.: Macclesfield, UK, 2014; Volume 7, pp. 55–64. [Google Scholar]
- Storgaard, H.; Song, X.M.; Jensen, C.B.; Madsbad, S.; Bjö, M.; Vaag, A.; Zierath, J.R. Insulin Signal Transduction in Skeletal Muscle From Glucose-Intolerant Relatives With Type 2 Diabetes. Diabetes 2001, 50, 2770–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Neschen, S.; Bilz, S.; Sono, S.; Tsirigotis, D.; Reznick, R.M.; Moore, I.; Nagai, Y.; Samuel, V.; Sebastian, D.; et al. Muscle-Specific IRS-1 Ser 3 Ala Transgenic Mice Are Skeletal Muscle. Diabetes 2008, 57, 2644–2651. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, C.; Tremblay, F.; Asselin, G.; Jacques, H.; Marette, A. Prevention of Skeletal Muscle Insulin Resistance by Dietary Cod Protein in High Fat-Fed Rats. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E62–E71. [Google Scholar] [CrossRef]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jesse, N. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef]
- Mullainadhan, V.; Viswanathan, M.P.; Karundevi, B. Effect of Bisphenol-A (BPA) on insulin signal transduction and GLUT4 translocation in gastrocnemius muscle of adult male albino rat. Int. J. Biochem. Cell Biol. 2017, 90, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Jeong, I.K.; Oh, T.J.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indumathi, D.; Jayashree, S.; Selvaraj, J.; Sathish, S.; Mayilvanan, C.; Akilavalli, N.; Balasubramanian, K. Effect of bisphenol-A on insulin signal transduction and glucose oxidation in skeletal muscle of adult male albino rat. Hum. Exp. Toxicol. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Chehadé, L.; Garneau, L.; Caron, A.; Aguer, C. The effects of acute BPA exposure on skeletal muscle mitochondrial function and glucose metabolism. Mol. Cell. Endocrinol. 2020, 499, 110580. [Google Scholar] [CrossRef]
- Detimary, P.; Gilon, P.; Henquin, J.-C. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: A feedback control mechanism in mouse pancreatic islets. Biochem. J. 1998, 333, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; He, J.; Menshikova, E.v.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, E.M.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmann, Y.W.; Stump, C.S.; Short, K.R.; Coenen-Schimke, J.M.; Guo, Z.K.; Bigelow, M.L.; Nair, K.S. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes 2006, 55, 3309–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; ’t Hart, L.M.; van Essen, E.; Heine, R.J.; Nijpels, G.; Tafrechi, R.S.J.; Raap, A.K.; Janssen, G.M.C.; Lemkes, H.H.P.J. Mitochondrial Diabetes Molecular Mechanisms and Clinical Presentation. Diabetes 2004, 53 (Suppl. 1), S103–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boden, G.; Lebed, B.; Schatz, M.; Homko, C.; Lemieux, S. Effects of Acute Changes of Plasma Free Fatty Acids on Intramyocellular Fat Content and Insulin Resistance in Healthy Subjects. Diabetes 2001, 50, 1612–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Qu, W.; Wang, K.; Chen, S.; Zhang, L.; Wu, D.; Chen, Z. Bisphenol A inhibits mucin 2 secretion in intestinal goblet cells through mitochondrial dysfunction and oxidative stress. Biomed. Pharmacother. 2019, 111, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.C.; et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Aguer, C.; Pasqua, M.; Thrush, A.B.; Moffat, C.; McBurney, M.; Jardine, K.; Zhang, R.; Beauchamp, B.; Dent, R.; McPherson, R.; et al. Increased proton leak and SOD2 expression in myotubes from obese non-diabetic subjects with a family history of type 2 diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1624–1633. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, X.; Qiu, L.; Wei, J.; Huang, Q.; Fang, C.; Ye, T.; Kang, M.; Shen, H.; Dong, S. Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death Dis. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apovian, C.M.; Okemah, J.; O’Neil, P.M. Body Weight Considerations in the Management of Type 2 Diabetes. Adv. Ther. 2019, 36, 44–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes UK. Diabetes: Facts and Stats. Diabetes UK 2016, 8. Available online: https://mrc.ukri.org/documents/pdf/diabetes-uk-facts-and-stats-june-2015/%0Afile:///C:/Users/sfair/OneDrive/HumanBiology3rdyear/ResearchProject/DissertationWriteUp/DiabetesUK_Facts_Stats_Oct16.pdf (accessed on 20 January 2021).
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol a enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef]
- Boucher, J.G.; Boudreau, A.; Ahmed, S.; Atlas, E. In vitro effects of bisphenol A β-D-glucuronide (BPA-G) on adipogenesis in human and murine preadipocytes. Environ. Health Perspect. 2015, 123, 1287–1293. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11β-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef]
- Riu, A.; Grimaldi, M.; le Maire, A.; Bey, G.; Phillips, K.; Boulahtouf, A.; Perdu, E.; Zalko, D.; Bourguet, W.; Balaguer, P. Peroxisome proliferator-activated receptor γ is a target for halogenated analogs of bisphenol A. Environ. Health Perspect. 2011, 119, 1227–1232. [Google Scholar] [CrossRef]
- de Filippis, E.; Li, T.; Rosen, E.D. Exposure of adipocytes to bisphenol-A in vitro interferes with insulin action without enhancing adipogenesis. PLoS ONE 2018, 13, e0201122. [Google Scholar] [CrossRef] [Green Version]
- Chamorro-García, R.; Kirchner, S.; Li, X.; Janesick, A.; Casey, S.C.; Chow, C.; Blumberg, B. Bisphenol A diglycidyl ether induces adipogenic differentiation of multipotent stromal stem cells through a peroxisome proliferator-activated receptor gamma-independent mechanism. Environ. Health Perspect. 2012, 120, 984–989. [Google Scholar] [CrossRef]
- Lonnroth, P.; Digirolamo, M.; Krotkiewski, M.; Smith, U. Jnsulin Binding and Responsiveness in Fat Cells from Patients with Reduced Glucose Tolerance and Type II Diabetes. Diabetes 1983, 32, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, A.; Öst, A.; Nystrom, F.H.; Strålfors, P. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J. Biol. Chem. 2005, 280, 34389–34392. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Kawazuma, M.; Adachi, T.; Harigaya, T.; Saito, Y.; Hashimoto, N.; Mori, C. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br. J. Pharmacol. 2004, 141, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Sarsenbayeva, A.; Katsogiannos, P.; Aguer, C.; Pereira, M.J. The effects of bisphenol A and bisphenol S on adipokine expression and glucose metabolism in human adipose tissue. Toxicology 2020, 445, 152600. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Palming, J.; Rizell, M.; Aureliano, M.; Carvalho, E.; Svensson, M.K.; Eriksson, J.W. Cyclosporine A and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: Increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J. Clin. Endocrinol. Metab. 2014, 99, E1885–E1894. [Google Scholar] [CrossRef] [Green Version]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.; Ferrini, M.G.; Jellyman, J.K.; Han, G.; Ross, M.G. In vivo and in vitro bisphenol A exposure effects on adiposity. J. Dev. Orig. Health Dis. 2018, 9, 678–687. [Google Scholar] [CrossRef]
- Blesson, C.S.; Yallampalli, C. Pregnancy is a new window of susceptibility for bisphenol a exposure. Endocrinology 2015, 156, 1611–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, Á. Prenatal Exposure to BPA and Offspring Outcomes. Dose-Response 2015, 13, 155932581559039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejonklou, M.H.; Dunder, L.; Bladin, E.; Pettersson, V.; Rönn, M.; Lind, L.; Waldén, T.B.; Lind, P.M. Effects of low-dose developmental bisphenol a exposure on metabolic parameters and gene expression in male and female fischer 344 rat offspring. Environ. Health Perspect. 2017, 125, 067018. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Ahmed, S.; Atlas, E. Bisphenol S induces adipogenesis in primary human preadipocytes from female donors. Endocrinol. 2016, 157, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Héliès-Toussaint, C.; Peyre, L.; Costanzo, C.; Chagnon, M.C.; Rahmani, R. Is bisphenol S a safe substitute for bisphenol A in terms of metabolic function? An in vitro study. Toxicol. Appl. Pharmacol. 2014, 280, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, I.; Oriente, F.; D’esposito, V.; Liguoro, D.; Liguoro, P.; Ambrosio, M.R.; Cabaro, S.; D’Andrea, F.; Beguinot, F.; Formisano, P.; et al. Low-dose bisphenol-a regulates inflammatory cytokines through GPR30 in mammary adipose cells. J. Mol. Endocrinol. 2019, 63, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Hugo, E.R.; Brandebourg, T.D.; Woo, J.G.; Loftus, J.; Alexander, J.W.; Ben-Jonathan, N. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ. Health Perspect. 2008, 116, 1642–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Sopasakis, V.R.; Sandqvist, M.; Gustafson, B.; Hammarstedt, A.; Schmelz, M.; Yang, X.; Jansson, P.-A.; Smith, U. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obes. Res. 2004, 12, 454–460. [Google Scholar] [CrossRef]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) Induces Insulin Resistance in 3T3-L1 Adipocytes and Is, Like IL-8 and Tumor Necrosis Factor-α, Overexpressed in Human Fat Cells from Insulin-resistant Subjects. J. Biol. Chem. 2003, 278, 45777–45784. [Google Scholar] [CrossRef] [Green Version]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and Subcutaneous Adipose Tissues of Obese Subjects Release Interleukin-6: Depot Difference and Regulation by Glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1 mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab. 2014, 307, 289–304. [Google Scholar] [CrossRef]
- Yun Pyo, M.; Ju Kim, H.; Kyung Back, S.; Yang, M. Downregulation of Peritoneal Macrophage Activity in Mice Exposed to Bisphenol A During Pregnancy and Lactation. Arch. Pharm. Res. 2007, 30, 1476–1481. [Google Scholar]
- Lu, X.; Li, M.; Wu, C.; Zhou, C.; Zhang, J.; Zhu, Q.; Shen, T. Bisphenol A promotes macrophage proinflammatory subtype polarization via upregulation of IRF5 expression in vitro. Toxicol. Vitro 2019, 60, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Schleinitz, D.; Krause, K.; Wohland, T.; Gebhardt, C.; Linder, N.; Stumvoll, M.; Blüher, M.; Bechmann, I.; Kovacs, P.; Gericke, M.; et al. Identification of distinct transcriptome signatures of human adipose tissue from fifteen depots. Eur. J. Hum. Genet. 2020, 28, 1714–1725. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Hatazaki, M.; Miyatsuka, T.; Matsuoka, T.; Kajimoto, Y.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Modulation of the JNK Pathway in Liver Affects Insulin Resistance Status. J. Biol. Chem. 2004, 279, 45803–45809. [Google Scholar] [CrossRef] [Green Version]
- Vahdati Hassani, F.; Mehri, S.; Abnous, K.; Birner-Gruenberger, R.; Hosseinzadeh, H. Protective effect of crocin on BPA-induced liver toxicity in rats through inhibition of oxidative stress and downregulation of MAPK and MAPKAP signaling pathway and miRNA-122 expression. Food Chem. Toxicol. 2017, 107, 395–405. [Google Scholar] [CrossRef]
- Lan, H.C.; Lin, I.W.; Yang, Z.J.; Lin, J.H. Low-dose bisphenol A activates Cyp11a1 gene expression and corticosterone secretion in adrenal gland via the JNK signaling pathway. Toxicol. Sci. 2015, 148, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Liu, Y.; Ichikawa, H.; Takemura, S.; Minamiyama, Y. Effects of bisphenol a on oxidative stress in the rat brain. Antioxidants 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Wang, S.; Zhu, W.; Xie, C.; Li, X.; Wu, J.; Zhu, J.; Jiang, Y.; Yang, X.; Li, Y.; et al. Curcumin suppresses JNK pathway to attenuate BPA-induced insulin resistance in LO2 cells. Biomed. Pharmacother. 2018, 97, 1538–1543. [Google Scholar] [CrossRef]
- Wajchenberg, B.L.; Gianella-Neto, D.; da Silva, M.E.R.; Santos, R.F. Depot-specific hormonal characteristics of subcutaneous and visceral adipose tissue and their relation to the metabolic syndrome. Horm. Metab. Res. 2002, 34, 616–621, undefined. [Google Scholar] [CrossRef]
- Pickering, R.T.; Lee, M.J.; Karastergiou, K.; Gower, A.; Fried, S.K. Depot dependent effects of dexamethasone on gene expression in human omental and abdominal subcutaneous adipose tissues from obese women. PLoS ONE 2016, 11, e0167337. [Google Scholar] [CrossRef]
- Peshdary, V.; Styles, G.; Gagné, R.; Yauk, C.L.; Sorisky, A.; Atlas, E. Depot-Specific Analysis of Human Adipose Cells and Their Responses to Bisphenol S. Endocrinology 2020, 161, bqaa044. [Google Scholar] [CrossRef]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan metabolic crosstalk in human insulin resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.-K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.-J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin Inhibits Lipolysis in Adipocytes via the Evolutionarily Conserved mTORC1-Egr1-ATGL-Mediated Pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [Green Version]
- Carchia, E.; Porreca, I.; Almeida, P.J.; D’Angelo, F.; Cuomo, D.; Ceccarelli, M.; De Felice, M.; Mallardo, M.; Ambrosino, C. Evaluation of low doses BPA-induced perturbation of glycemia by toxicogenomics points to a primary role of pancreatic islets and to the mechanism of toxicity. Cell Death Disease 2015, 6, e1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almabouada, F.; Diaz-Ruiz, A.; Rabanal-Ruiz, Y.; Peinado, J.R.; Vazquez-Martinez, R.; Malagon, M.M. Adiponectin receptors form homomers and heteromers exhibiting distinct ligand binding and intracellular signaling properties. J. Biol. Chem. 2013, 288, 3112–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, R.B.; Somwar, R.; Maida, A.; Fang, X.; Bikopoulos, G.; Sweeney, G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 2005, 48, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Planavila, A.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate-induced interleukin 6 production is mediated by protein kinase C and nuclear-factor B activation and leads to glucose transporter 4 down-regulation in skeletal muscle cells. Endocrinology 2005, 146, 3087–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Pathophysiology 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Chemical structure of Bisphenol A (BPA), bisphenol S (BPS), 17β-estradiol (E2), and diethylstilbestrol. (A) BPA is a dimethyl derivative and contains two hydroxyphenol groups. (B) BPS is a sulfonyl derivative and contains two hydroxyphenol groups. (C) E2 is a sex hormone that is derived from cholesterol and contains a hydroxyphenol group that interacts with the estrogen receptor (ER). (D) Diethylstilbestrol is a synthetic estrogen that contains two hydroxyphenol groups.
Figure 1.
Chemical structure of Bisphenol A (BPA), bisphenol S (BPS), 17β-estradiol (E2), and diethylstilbestrol. (A) BPA is a dimethyl derivative and contains two hydroxyphenol groups. (B) BPS is a sulfonyl derivative and contains two hydroxyphenol groups. (C) E2 is a sex hormone that is derived from cholesterol and contains a hydroxyphenol group that interacts with the estrogen receptor (ER). (D) Diethylstilbestrol is a synthetic estrogen that contains two hydroxyphenol groups.

Figure 2.
Potential mechanisms of action of BPA and BPS. Bisphenols have been shown to exert both genomic and non-genomic effects. BPA and BPS can bind to the mER-α/β and cytoplasmic ER-α/β, and this receptor-ligand complex can translocate to the nucleus and bind to the promoter of genes to alter expression. Furthermore, BPA can cross the plasma membrane and bind to the glucocorticoid receptor (GR) and androgen receptor (AR) and may act as an agonist or antagonist. In the nucleus, BPA can bind to ERR-γ. Genomic changes may include altered cell proliferation, apoptosis, and differentiation. Binding to mER-α/β can also lead to non-genomic changes, such as altered protein kinase activity, DNA methylation, miRNA, and histone acetylation. Binding of bisphenols to GPR30 can result in rapid non-genomic responses, such as through the influx of Ca2+. Dashed arrows indicate possible mechanisms of action. Image created with BioRender.com.
Figure 2.
Potential mechanisms of action of BPA and BPS. Bisphenols have been shown to exert both genomic and non-genomic effects. BPA and BPS can bind to the mER-α/β and cytoplasmic ER-α/β, and this receptor-ligand complex can translocate to the nucleus and bind to the promoter of genes to alter expression. Furthermore, BPA can cross the plasma membrane and bind to the glucocorticoid receptor (GR) and androgen receptor (AR) and may act as an agonist or antagonist. In the nucleus, BPA can bind to ERR-γ. Genomic changes may include altered cell proliferation, apoptosis, and differentiation. Binding to mER-α/β can also lead to non-genomic changes, such as altered protein kinase activity, DNA methylation, miRNA, and histone acetylation. Binding of bisphenols to GPR30 can result in rapid non-genomic responses, such as through the influx of Ca2+. Dashed arrows indicate possible mechanisms of action. Image created with BioRender.com.
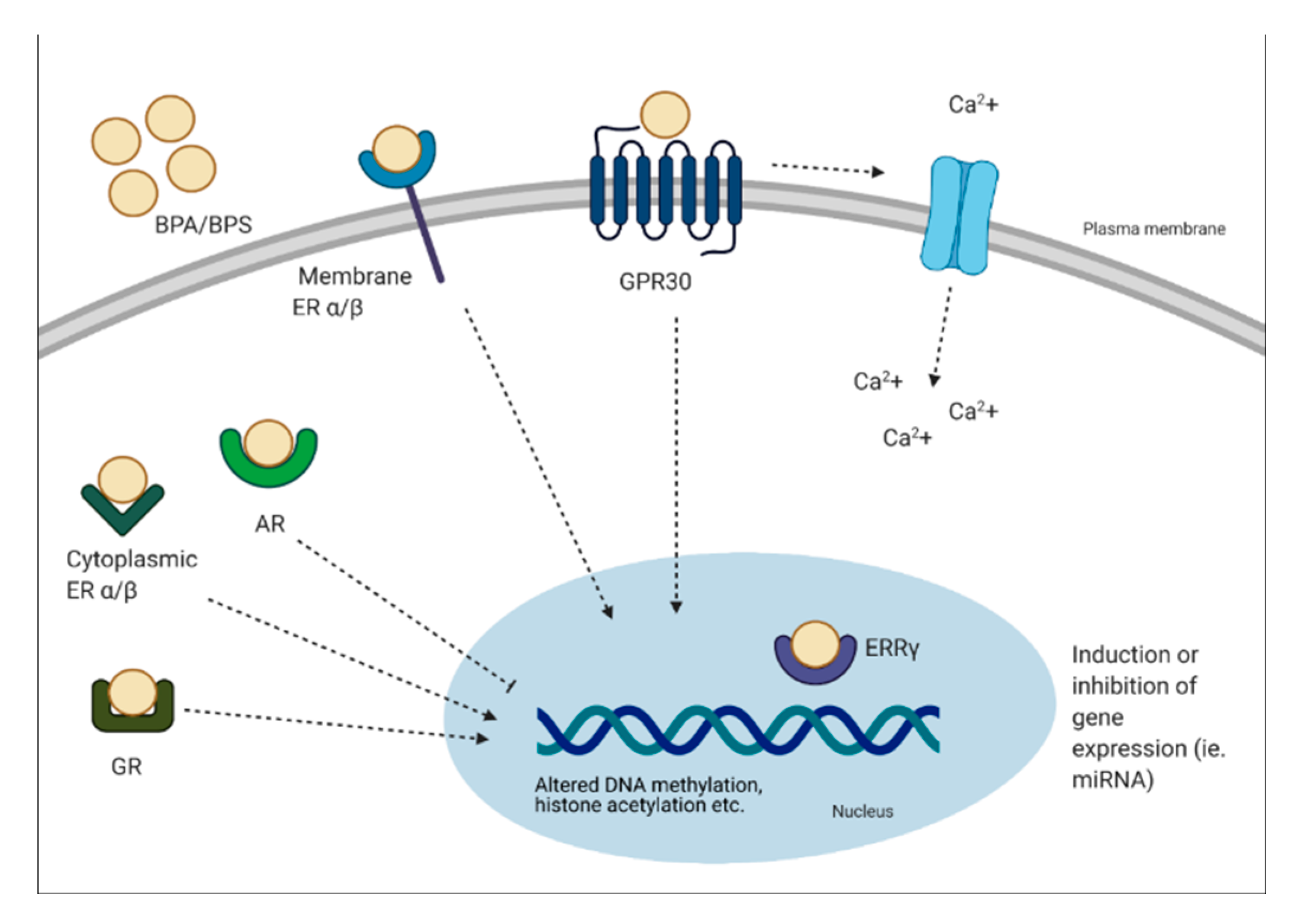
Figure 3.
Mechanism of action of BPA/BPS on glucose metabolism in the skeletal muscle. BPA and BPS can bind to various receptors (as described in Figure 2) and alter gene expression. In muscle cells, it is not completely understood which receptors BPA and BPS bind to and induce metabolic alterations. Changes that have been shown in the skeletal muscle following chronic bisphenol exposure include reduced insulin signaling (reduced insulin receptor substrate 1 (IRS1) and Akt phosphorylation), and reduced mitochondrial function, both of which can lead to reduced glucose uptake. Image created with BioRender.com.
Figure 3.
Mechanism of action of BPA/BPS on glucose metabolism in the skeletal muscle. BPA and BPS can bind to various receptors (as described in Figure 2) and alter gene expression. In muscle cells, it is not completely understood which receptors BPA and BPS bind to and induce metabolic alterations. Changes that have been shown in the skeletal muscle following chronic bisphenol exposure include reduced insulin signaling (reduced insulin receptor substrate 1 (IRS1) and Akt phosphorylation), and reduced mitochondrial function, both of which can lead to reduced glucose uptake. Image created with BioRender.com.
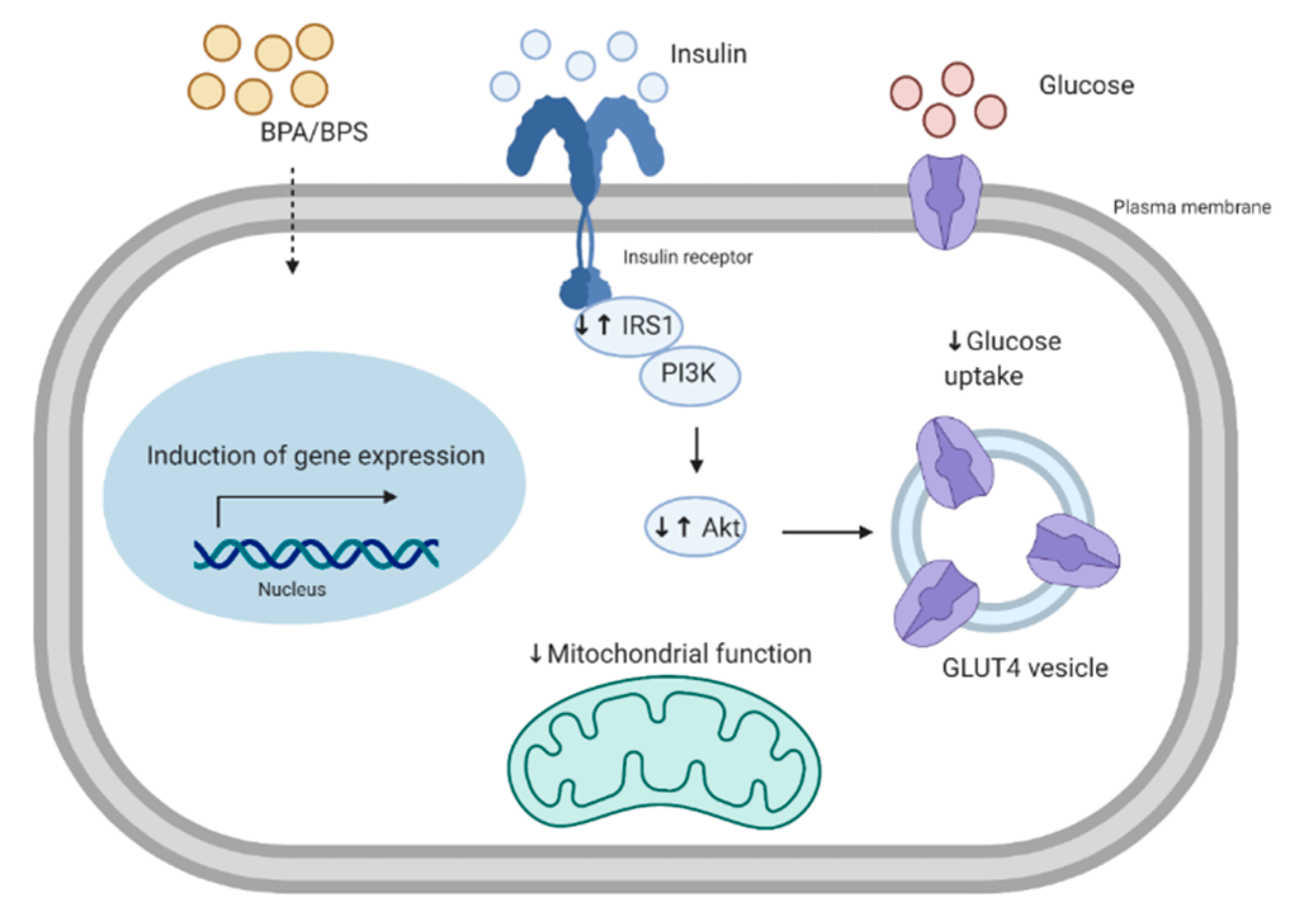
Figure 4.
Mechanism of action of BPA/BPS exposure in adipocytes. In adipocytes it is not completely understood which receptors BPA and BPS bind to and induce metabolic alterations, however, possible receptors are highlighted in Figure 2. This includes altered expression of different factors such as adipogenic and lipogenic markers, inflammation, and adipokines. BPA is known to increase c-Jun N-terminal kinases/signal transducer and activator of transcription 3 (JNK/STAT3), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, which results in an increase in pro-inflammatory cytokines. Lipolysis has been shown to be reduced in 3T3-L1 cells. Bisphenols have also been shown to alter insulin signaling in adipocytes, such as altered phosphorylation of IRS1 and Akt, in addition to increasing lipid accumulation in adipocytes. Furthermore, altered adipokine release, such as adiponectin, has been shown to be a consequence of BPA exposure. Image created with BioRender.com.
Figure 4.
Mechanism of action of BPA/BPS exposure in adipocytes. In adipocytes it is not completely understood which receptors BPA and BPS bind to and induce metabolic alterations, however, possible receptors are highlighted in Figure 2. This includes altered expression of different factors such as adipogenic and lipogenic markers, inflammation, and adipokines. BPA is known to increase c-Jun N-terminal kinases/signal transducer and activator of transcription 3 (JNK/STAT3), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, which results in an increase in pro-inflammatory cytokines. Lipolysis has been shown to be reduced in 3T3-L1 cells. Bisphenols have also been shown to alter insulin signaling in adipocytes, such as altered phosphorylation of IRS1 and Akt, in addition to increasing lipid accumulation in adipocytes. Furthermore, altered adipokine release, such as adiponectin, has been shown to be a consequence of BPA exposure. Image created with BioRender.com.
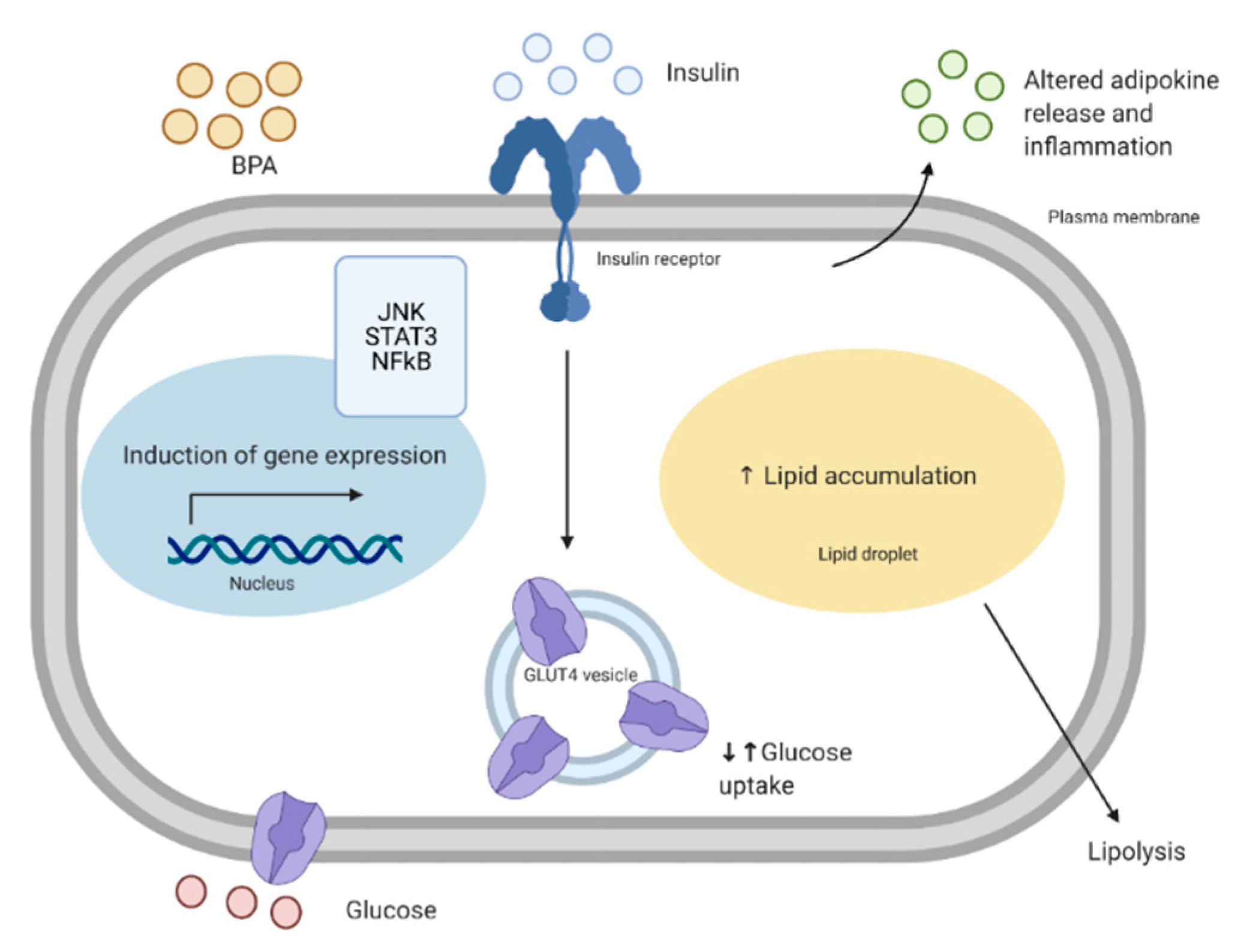
Figure 5.
Possible mechanisms of action of BPA/BPS on adipose tissue-to-skeletal muscle cross-talk. Adipose tissue exposed to BPA has altered adipokine levels and inflammation which may lead to alterations of the skeletal muscle. Some changes may include increased reactive oxygen species (ROS), oxidative stress, and inflammation, which can all induce mitochondrial dysfunction and decrease glucose uptake. Image created with BioRender.com.
Figure 5.
Possible mechanisms of action of BPA/BPS on adipose tissue-to-skeletal muscle cross-talk. Adipose tissue exposed to BPA has altered adipokine levels and inflammation which may lead to alterations of the skeletal muscle. Some changes may include increased reactive oxygen species (ROS), oxidative stress, and inflammation, which can all induce mitochondrial dysfunction and decrease glucose uptake. Image created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ahmed, F.; Pereira, M.J.; Aguer, C. Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue. Environments 2021, 8, 35. https://0-doi-org.brum.beds.ac.uk/10.3390/environments8040035
AMA Style
Ahmed F, Pereira MJ, Aguer C. Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue. Environments. 2021; 8(4):35. https://0-doi-org.brum.beds.ac.uk/10.3390/environments8040035
Chicago/Turabian StyleAhmed, Fozia, Maria João Pereira, and Céline Aguer. 2021. "Bisphenols and the Development of Type 2 Diabetes: The Role of the Skeletal Muscle and Adipose Tissue" Environments 8, no. 4: 35. https://0-doi-org.brum.beds.ac.uk/10.3390/environments8040035
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.