Modal Analysis of the Lysozyme Protein Considering All-Atom and Coarse-Grained Finite Element Models

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Methodology
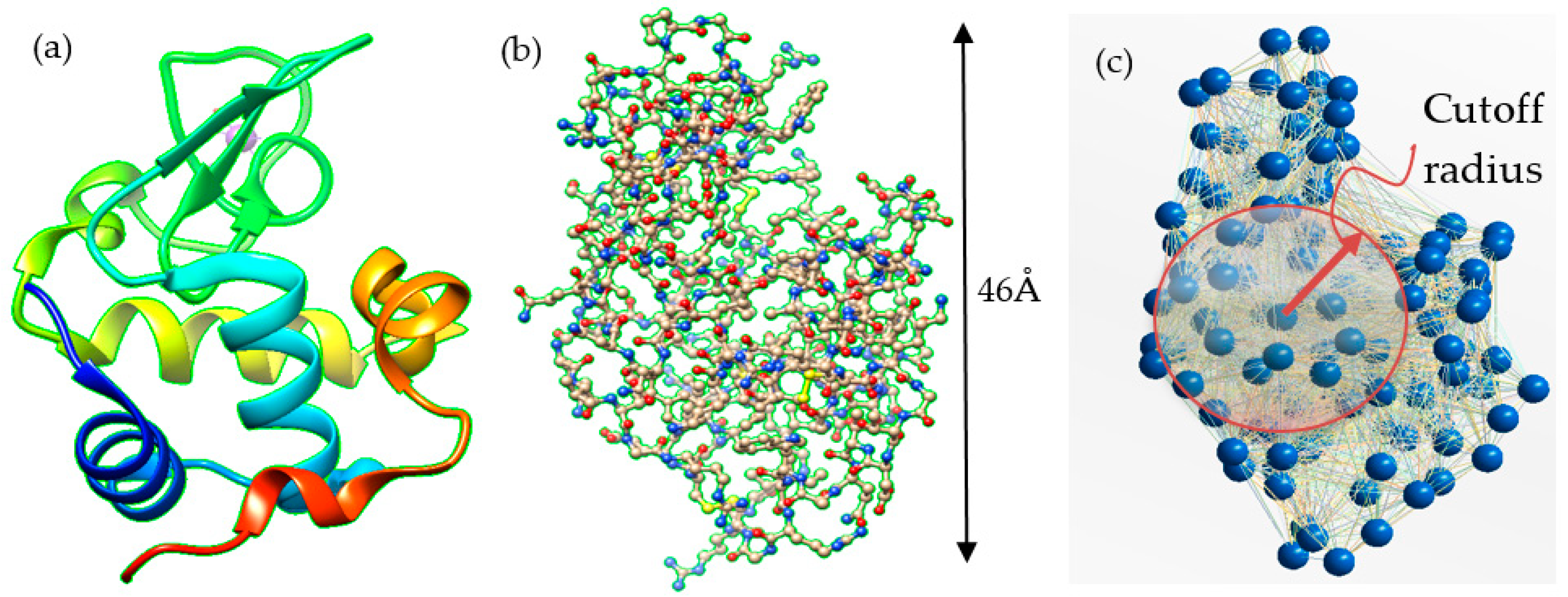
2.1. The Truss-Like Structure, the Connectivity Assumption and Modal Analysis
2.2. Definitions about the All-Atom Model
2.3. Definitions about the Coarse-Grained Model
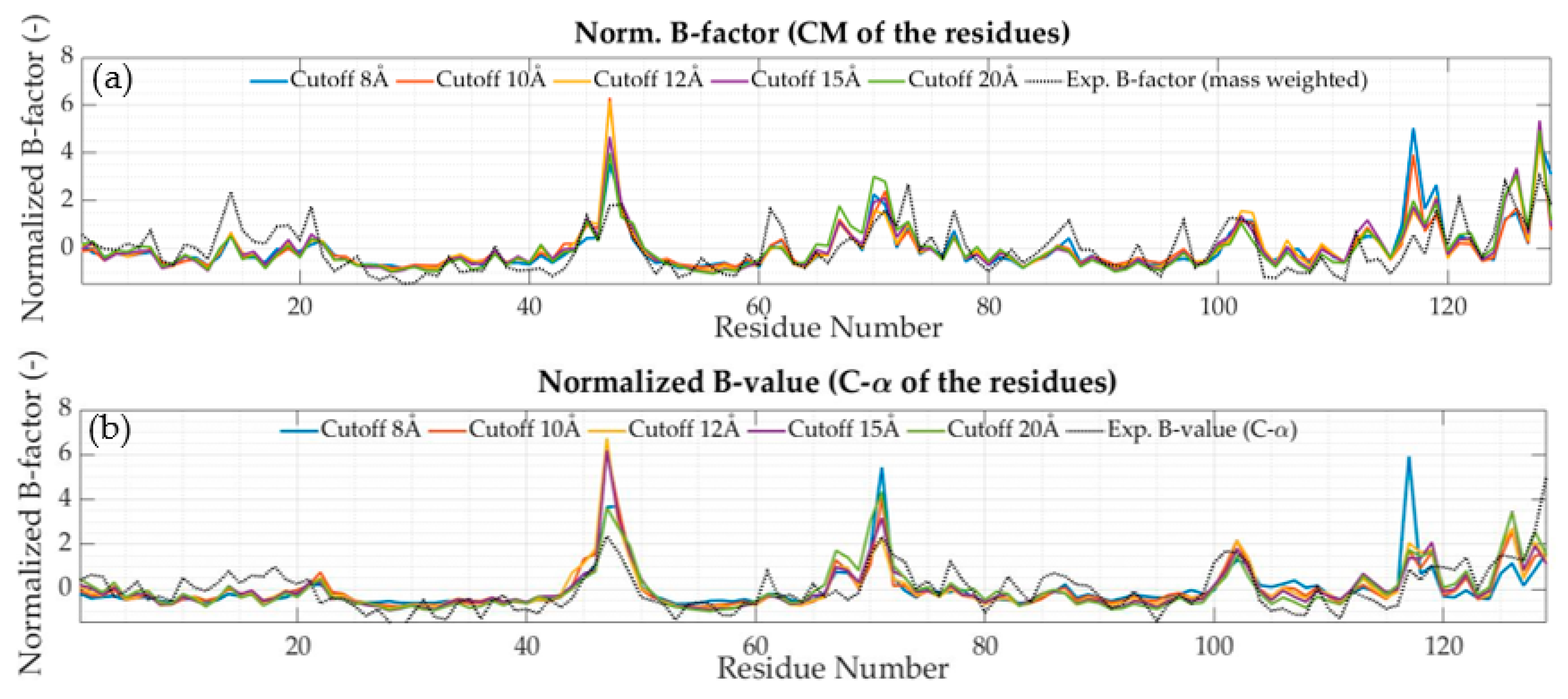
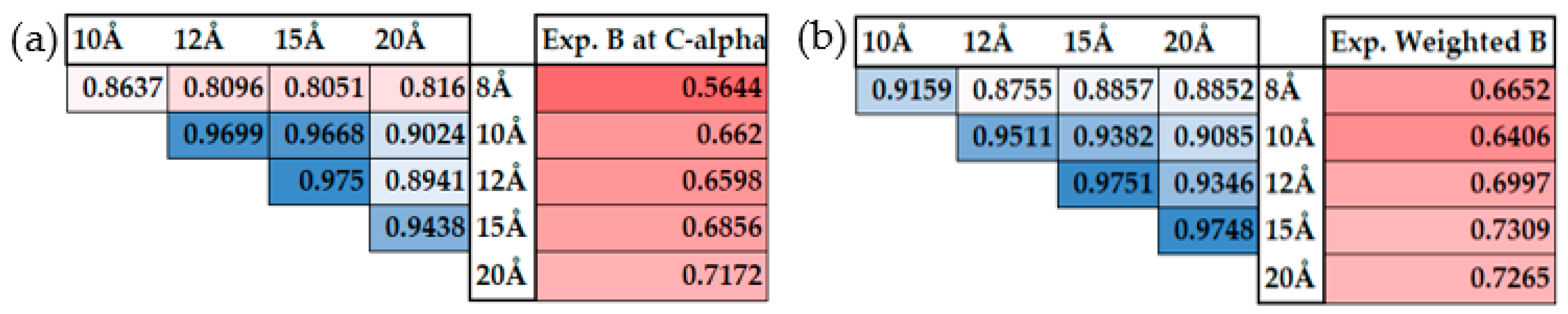
2.4. The Experimental Debye-Waller Factors or B-Factors
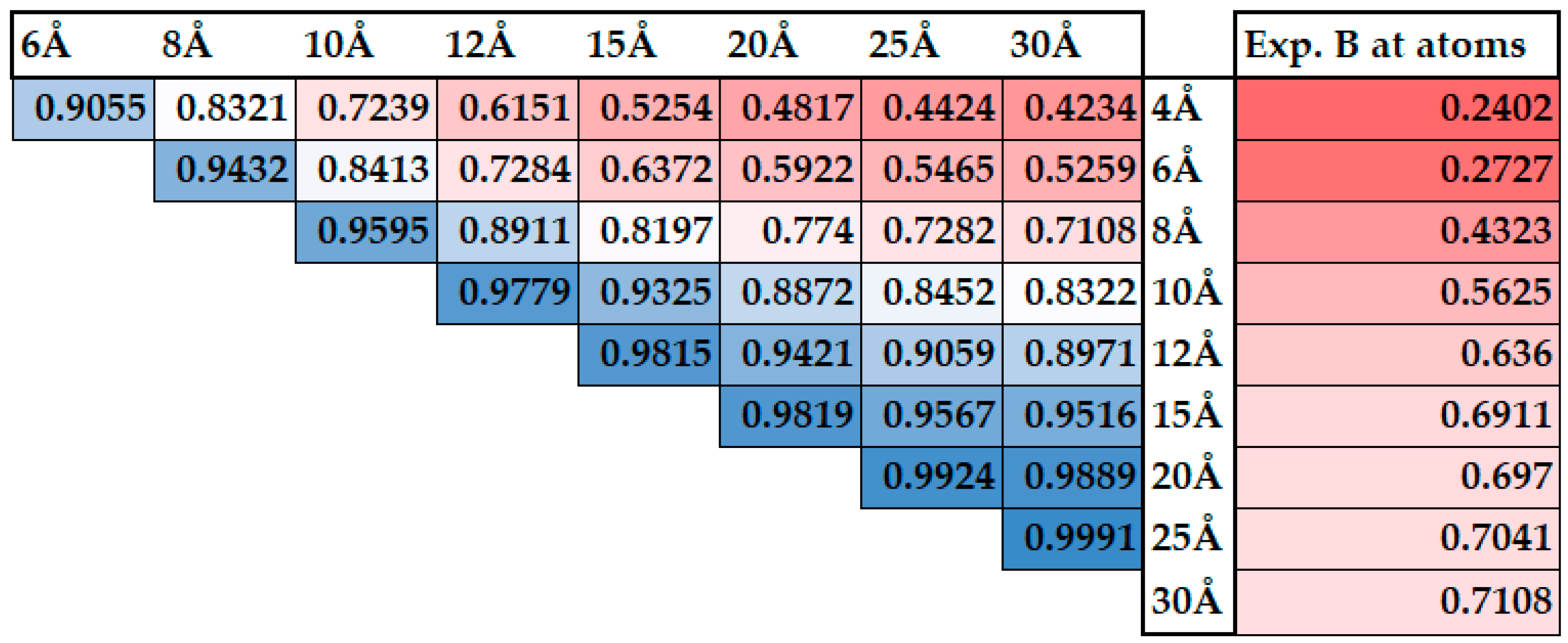
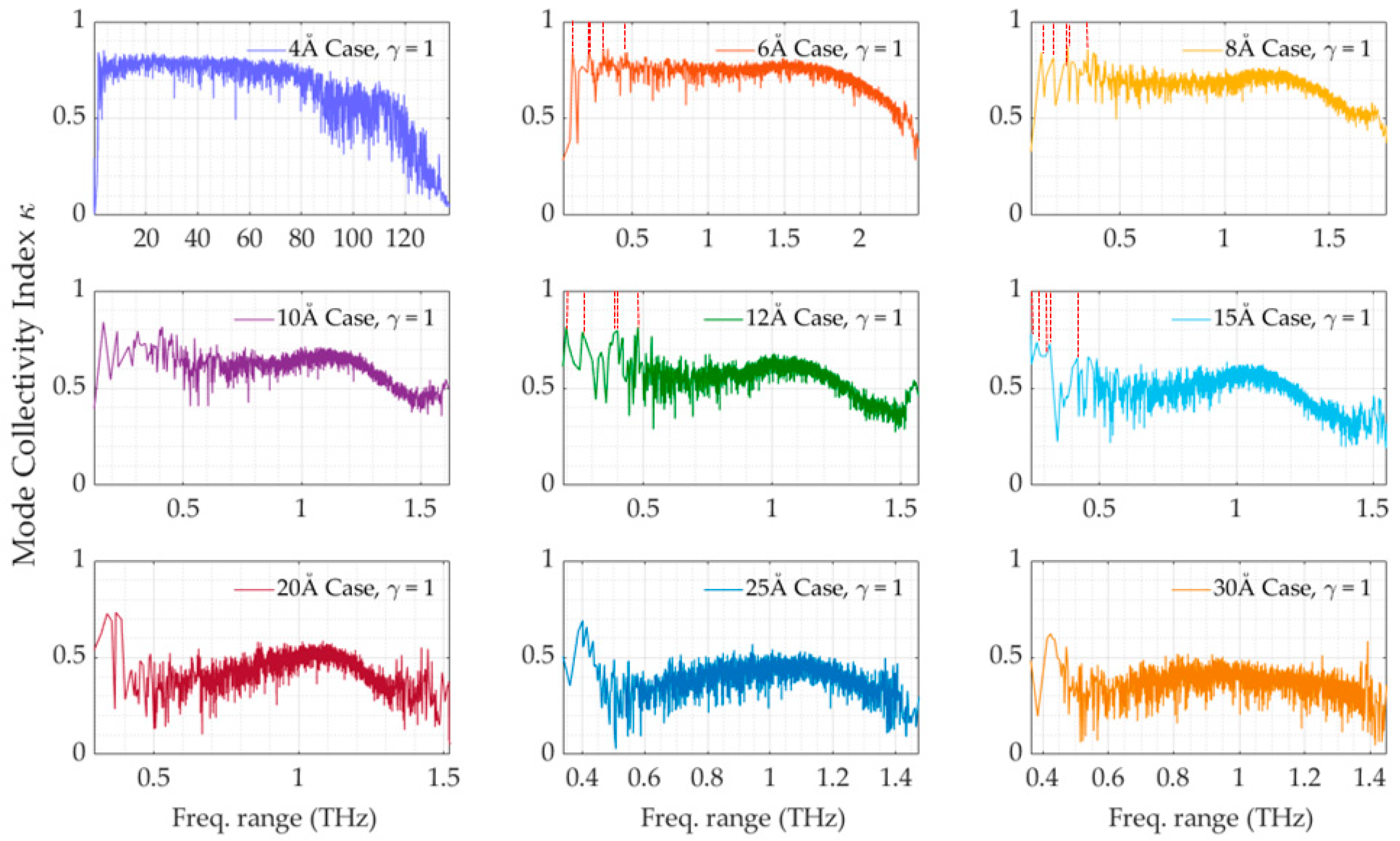
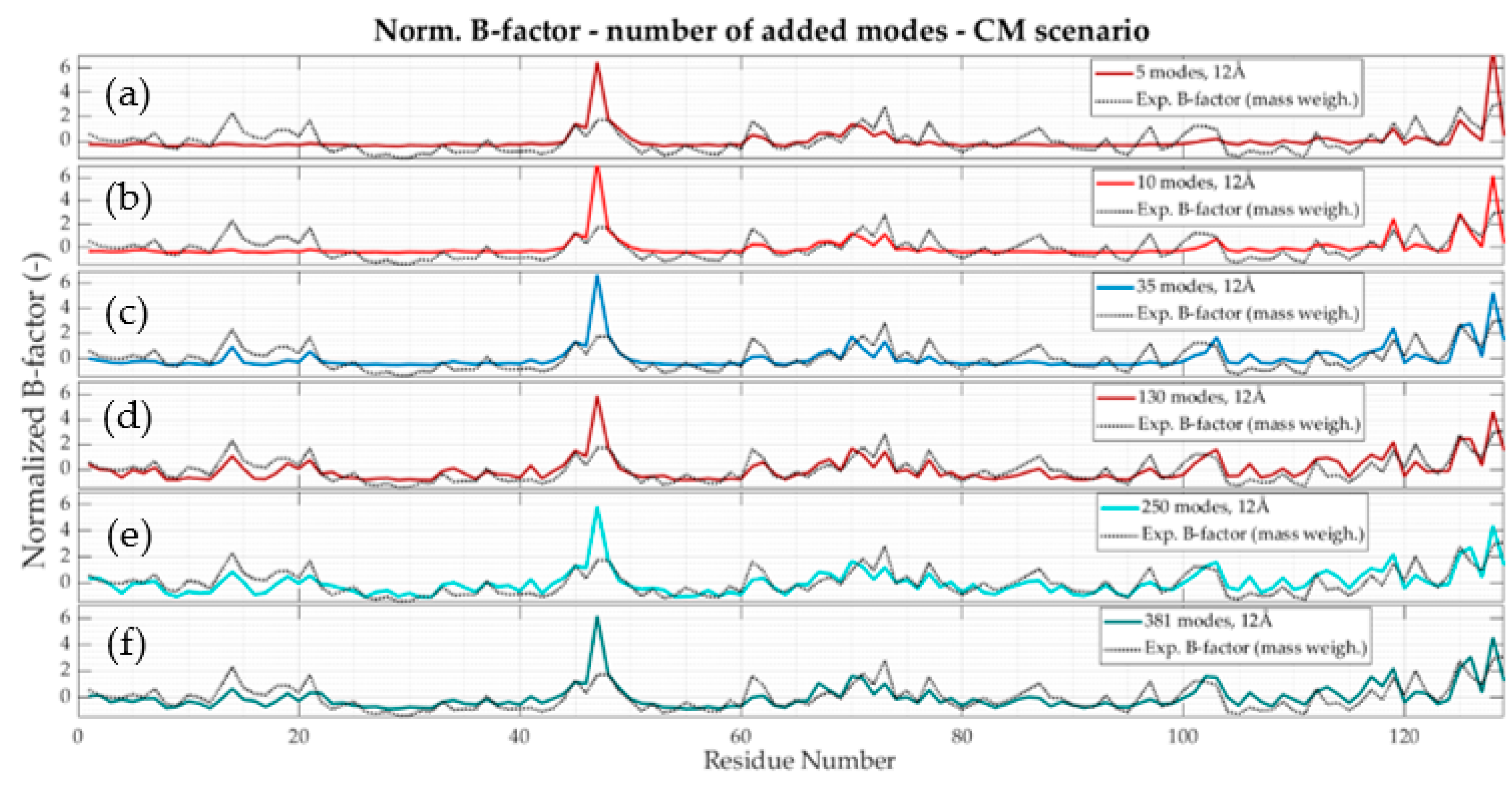
2.5. Collectivity of the Protein Motions
2.6. The Scaling Procedure
3. Results and Discussion
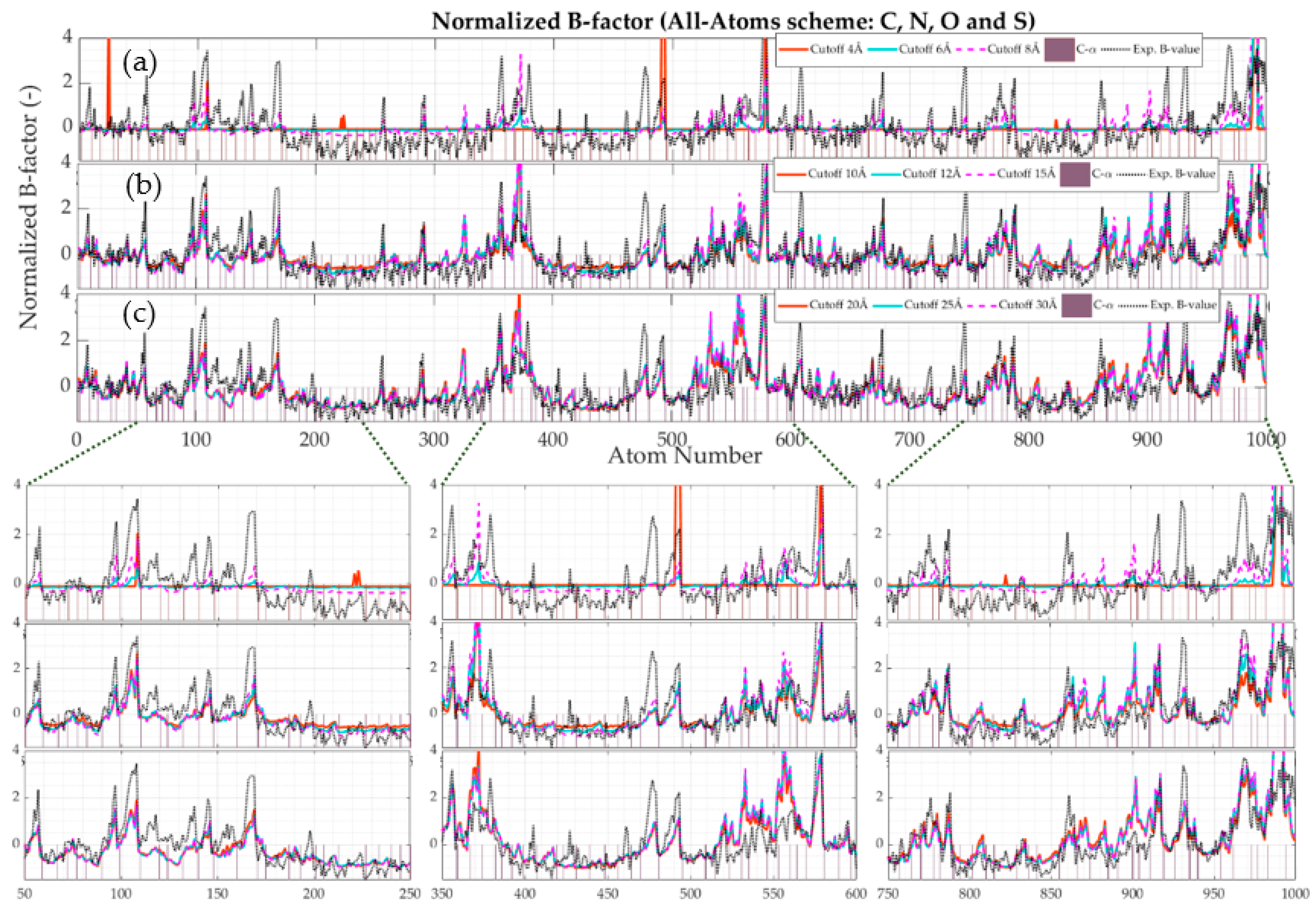
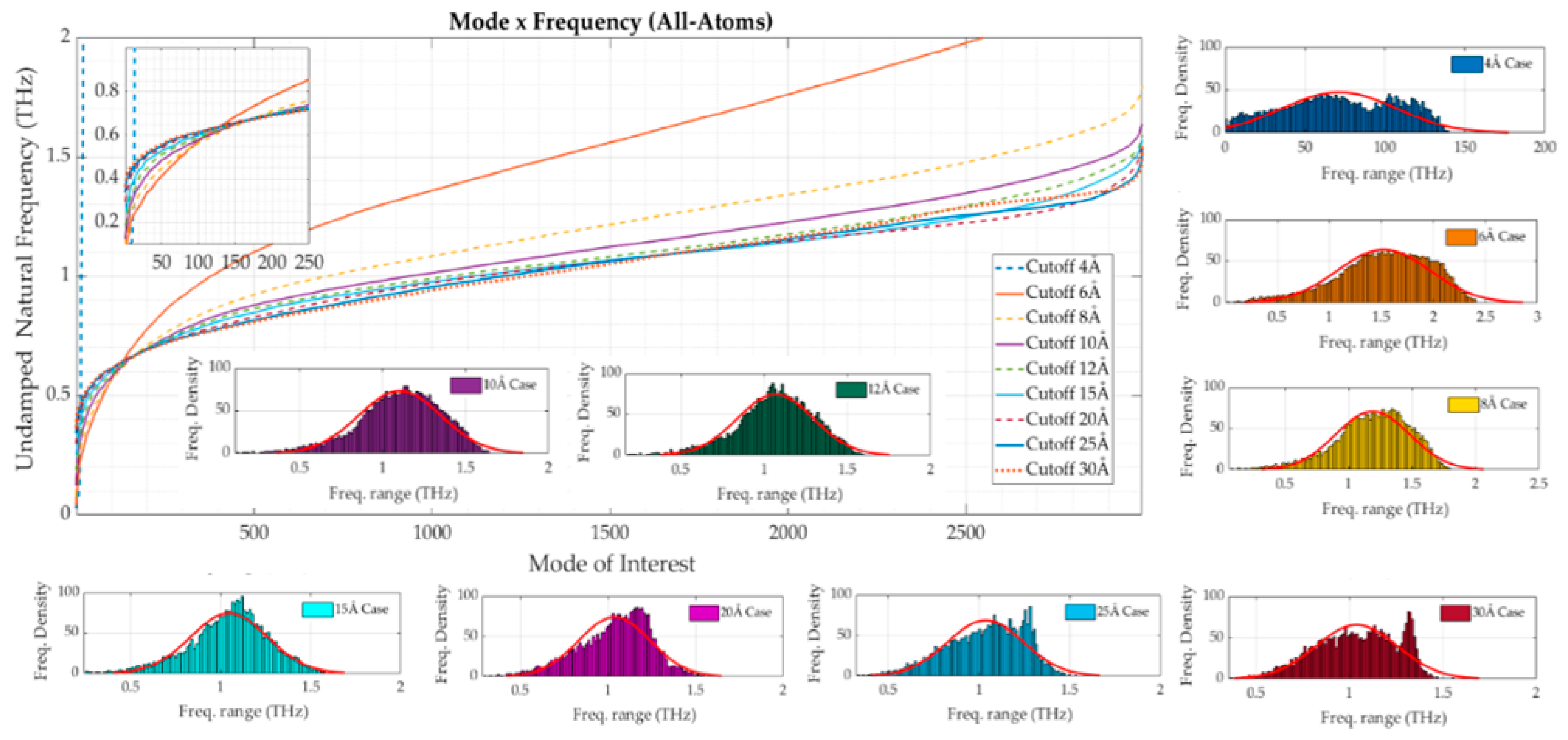
3.1. All-Atom Model
- ○
- in the region 50–250, the major experimental peaks agree with the numerical ones from the model with a cutoff value of 10 Å onwards. For small fluctuations, it is hard to confirm a good representative relationship between the solutions;
- ○
- in the region 350–600, node 379 shows a high experimental B-factor, but none of the numerical solutions provides such increased flexibility. The models are not able to capture this particular behavior. This can either be due to deficiencies in the simplified truss models or measurement errors in the experimental B-factors. Some other smaller peaks have similar patterns. At node 478, the effect that arises from increasing the cutoff value is clearly seen;
- ○
- in the region 750–1000, nodes 991 and 992 present a substantial peak in all cases that go beyond the top of the chart. In that region, a dent extends outside the globular body region, making it similar to a cantilever beam attached to the main structure. This is commonly associated with the first natural mode of numerical solutions and it has high participation in many modes. At first sight, this solution does not present a real behavior according to the experimental B-factors. It is possible that damping related to the surrounding environment may play a role, leading to this discrepancy.
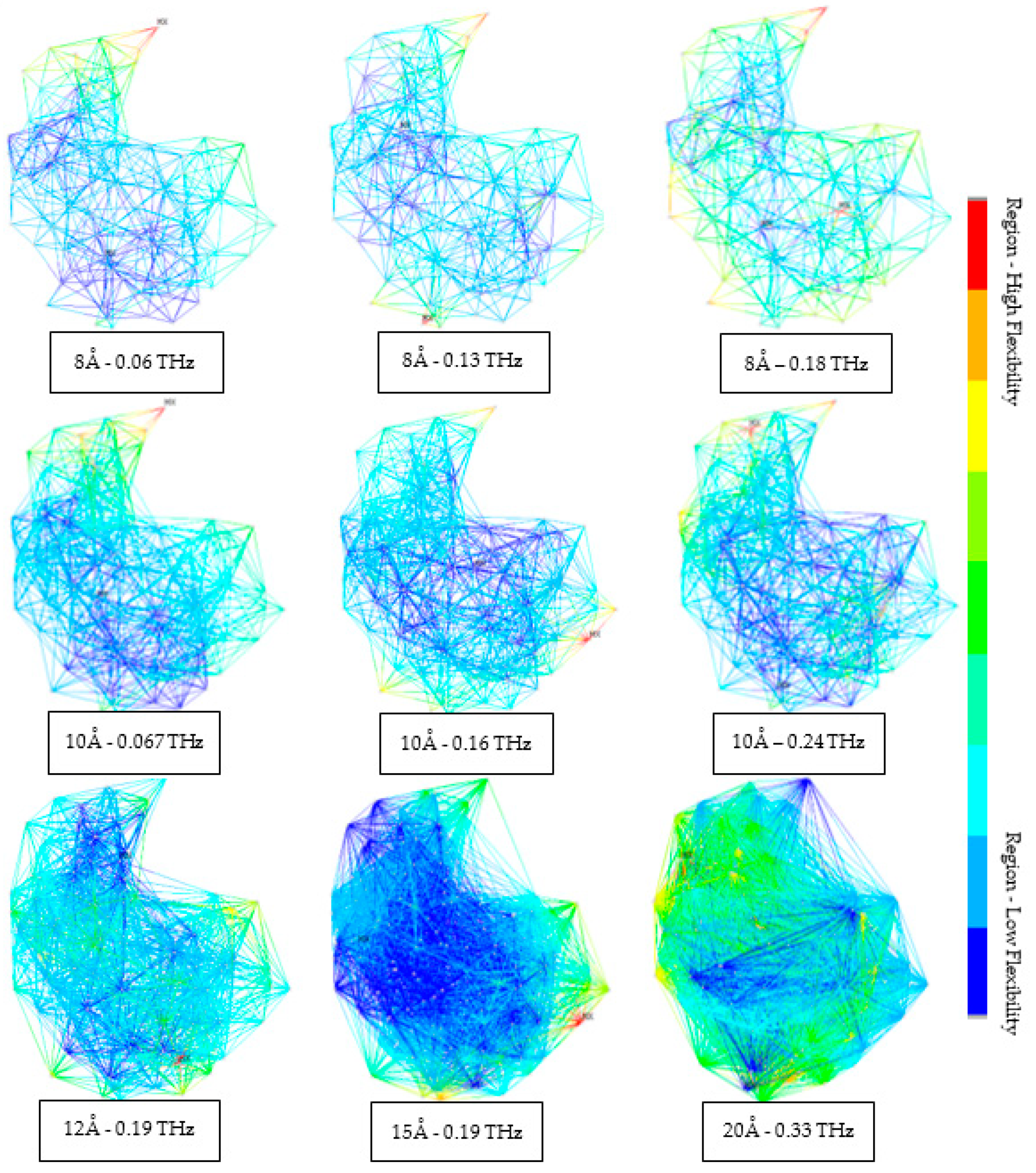
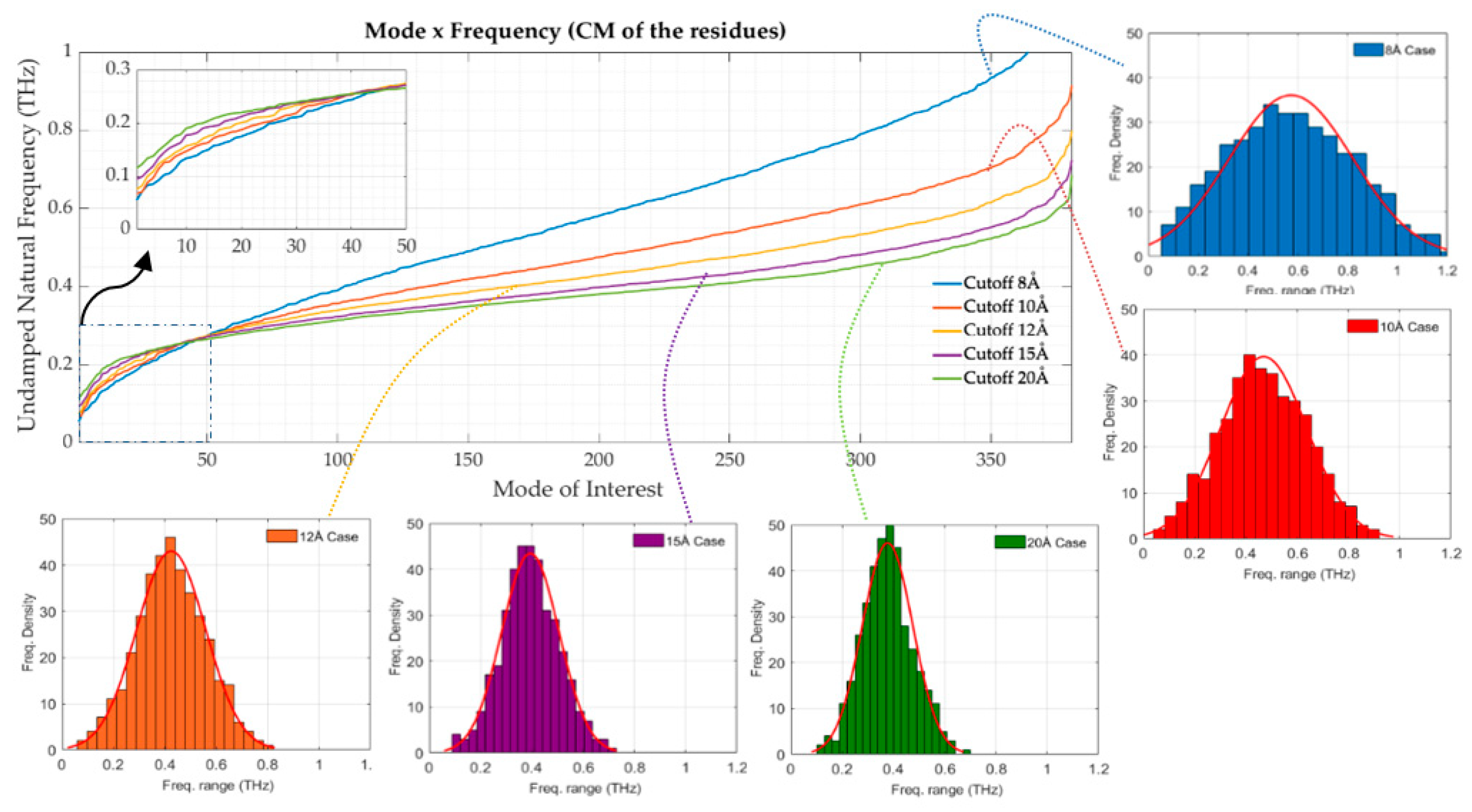
3.2. Coarse-Grained Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bahar, I.; Jernigan, R.L.; Dill, K.A. Protein Actions: Principles & Modeling; Garland Science: New York, NY, USA, 2017. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahar, I.; Rader, A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005, 15, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwo, A. Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-642-28553-0. [Google Scholar]
- Levitt, M.; Sander, C.; Stern, P.S. Protein normal-mode dynamics: Trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985, 181, 423–447. [Google Scholar] [CrossRef]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997, 79, 3090. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar]
- Tama, F.; Gadea, F.X.; Marques, O.; Sanejouand, Y.H. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins Struct. Funct. Genet. 2000, 41, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Song, G.; Jernigan, R.L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. USA 2009, 106, 12347–12352. [Google Scholar] [CrossRef] [Green Version]
- Eyal, E.; Yang, L.W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 22, 2619–2627. [Google Scholar] [CrossRef]
- Hoffmann, A.; Grudinin, S. NOLB: Nonlinear Rigid Block Normal-Mode Analysis Method. J. Chem. Theory Comput. 2017, 13, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Scaramozzino, D.; Lacidogna, G.; Piana, G.; Carpinteri, A. A finite-element-based coarse-grained model for global protein vibration. Meccanica 2019, 54, 1927–1940. [Google Scholar] [CrossRef]
- Song, G.; Jernigan, R.L. An enhanced elastic network model to represent the motions of domain-swapped proteins. Proteins Struct. Funct. Genet. 2006, 63, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Na, H.; Jernigan, R.L.; Song, G. Bridging between NMA and Elastic Network Models: Preserving All-Atom Accuracy in Coarse-Grained Models. PLoS Comput. Biol. 2015, 11, e1004542. [Google Scholar] [CrossRef] [PubMed]
- Tama, F.; Sanejouand, Y.H. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Song, G.; Jernigan, R.L. How well can we understand large-scale protein motions using normal modes of elastic network models? Biophys. J. 2007, 93, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.T.; Kloczkowski, A.; Jernigan, R.L. MAVENs: Motion analysis and visualization of elastic networks and structural ensembles. BMC Bioinform. 2011, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Atilgan, C.; Atilgan, A.R. Perturbation-response scanning reveals ligand entry-exit mechanisms of ferric binding protein. PLoS Comput. Biol. 2009, 5, e1000544. [Google Scholar] [CrossRef] [Green Version]
- Atilgan, C.; Gerek, Z.N.; Ozkan, S.B.; Atilgan, A.R. Manipulation of conformational change in proteins by single-residue perturbations. Biophys. J. 2010, 99, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sankar, K.; Wang, Y.; Jia, K.; Jernigan, R.L. Directional Force Originating from ATP Hydrolysis Drives the GroEL Conformational Change. Biophys. J. 2017, 112, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Scaramozzino, D.; Khade, P.M.; Jernigan, R.L.; Lacidogna, G.; Carpinteri, A. Structural Compliance: A New Metric for Protein Flexibility. Proteins Struct. Funct. Bioinform. 2020, 88, 1482–1492. [Google Scholar]
- Kurkcuoglu, O.; Jernigan, R.L.; Doruker, P. Collective dynamics of large proteins from mixed coarse-grained elastic network model. QSAR Comb. Sci. 2005, 24, 443–448. [Google Scholar] [CrossRef]
- Mishra, S.K.; Jernigan, R.L. Protein dynamic communities from elastic network models align closely to the communities defined by molecular dynamics. PLoS ONE 2018, 13, e0199225. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Song, G.; Carriquiry, A.; Jernigan, R.L. Close Correspondence between the Essential Protein Motions from Principal Component Analysis of Multiple HIV-1 Protease Structures and Elastic Network Modes. Structure 2008, 16, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, K.; Mishra, S.K.; Jernigan, R.L. Comparisons of Protein Dynamics from Experimental Structure Ensembles, Molecular Dynamics Ensembles, and Coarse-Grained Elastic Network Models. J. Phys. Chem. B 2018, 122, 5409–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acbas, G.; Niessen, K.A.; Snell, E.H.; Markelz, A.G. Optical measurements of long-range protein vibrations. Nat. Commun. 2014, 5, 1–7. [Google Scholar]
- Niessen, K.A.; Xu, M.; Paciaroni, A.; Orecchini, A.; Snell, E.H.; Markelz, A.G. Moving in the Right Direction: Protein Vibrational Steering Function. Biophys. J. 2017, 112, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Whitmire, S.E.; Wolpert, D.; Markelz, A.G.; Hillebrecht, J.R.; Galan, J.; Birge, R.R. Protein flexibility and conformational state: A comparison of collective vibrational modes of wild-type and D96N bacteriorhodopsin. Biophys. J. 2003, 85, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Carpinteri, A.; Lacidogna, G.; Piana, G.; Bassani, A. Terahertz mechanical vibrations in lysozyme: Raman spectroscopy vs. modal analysis. J. Mol. Struct. 2017, 1139, 222–230. [Google Scholar] [CrossRef]
- Lacidogna, G.; Piana, G.; Bassani, A.; Carpinteri, A. Raman spectroscopy of Na/K-ATPase with special focus on low-frequency vibrations. Vib. Spectrosc. 2017, 92, 298–301. [Google Scholar]
- Carpinteri, A.; Piana, G.; Bassani, A.; Lacidogna, G. Terahertz vibration modes in Na/K-ATPase. J. Biomol. Struct. Dyn. 2019, 37, 256–264. [Google Scholar] [PubMed]
- Turton, D.A.; Senn, H.M.; Harwood, T.; Lapthorn, A.J.; Ellis, E.M.; Wynne, K. Terahertz underdamped vibrational motion governs protein-ligand binding in solution. Nat. Commun. 2014, 5, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.G.; Erfurth, S.C.; Small, E.W.; Peticolas, W.L. Conformationally dependent low-frequency motions of proteins by laser Raman spectroscopy. Proc. Natl. Acad. Sci. USA 1972, 69, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- Painter, P.C.; Mosher, L.; Rhoads, C. Low-frequency modes in the raman spectra of proteins. Biopolymers 1982, 21, 1469–1472. [Google Scholar]
- Genzel, L.; Keilmann, F.; Martin, T.P.; Wintreling, G.; Yacoby, Y.; Fröhlich, H.; Makinen, M.W. Low-frequency Raman spectra of lysozyme. Biopolymers 1976, 15, 219–225. [Google Scholar] [CrossRef]
- Rygula, A.; Majzner, K.; Marzec, K.M.; Kaczor, A.; Pilarczyk, M.; Baranska, M. Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 2013, 44, 1061–1076. [Google Scholar]
- Thomas, G.J. Raman spectroscopy of protein and nucleic acid assemblies. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 1–27. [Google Scholar]
- Bunaciu, A.A.; Aboul-Enein, H.Y.; Hoang, V.D. Raman spectroscopy for protein analysis. Appl. Spectrosc. Rev. 2015, 50, 377–386. [Google Scholar] [CrossRef]
- Bertoldo Menezes, D.; Reyer, A.; Yüksel, A.; Bertoldo Oliveira, B.; Musso, M. Introduction to Terahertz Raman spectroscopy. Spectrosc. Lett. 2018, 51, 438–445. [Google Scholar]
- Ferreira, A.J.M. Matlab Codes for Finite Element Analysis, Solid and Structures; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Montáns, F.J.; Muñoz, I. Dynamic Analysis of Structures for the Finite Element Method. 2013. [Google Scholar]
- Creighton, T.E. Proteins: Structures and Molecular Properties, 2nd ed.; Freeman, W.H., Ed.; Macmillan: New York, NY, USA, 1993. [Google Scholar]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Karplus, P.A.; Schulz, G.E. Prediction of chain flexibility in proteins—A tool for the selection of peptide antigens. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Schlessinger, A.; Rost, B. Protein flexibility and rigidity predicted from sequence. Proteins Struct. Funct. Bioinform. 2005, 61, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vihinen, M.; Torkkila, E.; Riikonen, P. Accuracy of protein flexibility predictions. Proteins Struct. Funct. Bioinform. 1994, 19, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, K.; Kuriyan, J.; Karplus, M. Temperature dependence of the structure and dynamics of myoglobin. A simulation approach. J. Mol. Biol. 1990, 213, 351–373. [Google Scholar] [CrossRef]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [Green Version]
- Carugo, O. Maximal B-factors in protein crystal structures. BMC Bioinform. 2018, 19, 61. [Google Scholar] [CrossRef]
- Khade, P.; Kumar, A.; Jernigan, R.L. Characterizing and predicting protein hinges for mechanistic insight. J. Mol. Biol. 2020, 432, 508–522. [Google Scholar]
- Dykeman, E.C.; Sankey, O.F. Normal mode analysis and applications in biological physics. J. Phys. Condens. Matter 2010, 22, 423202. [Google Scholar]
- Prompers, J.J.; Lienin, S.F.; Brüschweiler, R. Collective reorientational motion and nuclear spin relaxation in proteins. Pacific Symp. Biocomput. 2001, 88, 79–88. [Google Scholar]
- Brooks, B.; Karplus, M. Harmonic dynamics of proteins: Normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA 1983, 80, 6571–6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Avraham, D. Vibrational normal-mode spectrum of globular proteins. Phys. Rev. B 1993, 47, 14559–14560. [Google Scholar] [CrossRef] [PubMed]
- Russi, S.; Gonzalez, A.; Kenner, L.R.; Keedy, D.A.; Fraser, J.S.; van den Bedem, H. Conformational variation of protein at room is not dominated by radiation damage. J. Syncrothron Radiat. 2017, 24, 73–82. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutoff (Å) | Axial Rigidity EA (N) | Nº of Connections | Average Stiffness k (N/m) |
|---|---|---|---|
| 4 | 3471.7 × 10−10 | 5979 | 1.36 × 103 |
| 6 | 0.6470 × 10−10 | 19,151 | 0.1675 |
| 8 | 0.2436 × 10−10 | 39,456 | 0.0485 |
| 10 | 0.1462 × 10−10 | 69,143 | 0.0236 |
| 12 | 0.1046 × 10−10 | 106,767 | 0.0143 |
| 15 | 0.0739 × 10−10 | 175,875 | 0.0083 |
| 20 | 0.0527 × 10−10 | 301,919 | 0.0047 |
| 25 | 0.0450 × 10−10 | 408,088 | 0.0035 |
| 30 | 0.0424 × 10−10 | 46,6426 | 0.0031 |
| Cutoff (Å) | Axial Rigidity EA-CM (N) | Nº of Connections | Average Stiffness k (N/m) | Axial Rigidity EA-Cα (N) [15] |
|---|---|---|---|---|
| 8 | 4.56 × 10−10 | 627 | 0.783 | 8.31 × 10−10 |
| 10 | 1.95 × 10−10 | 1109 | 0.283 | 2.35 × 10−10 |
| 12 | 1.13 × 10−10 | 1771 | 0.142 | 1.24 × 10−10 |
| 15 | 0.69 × 10−10 | 2938 | 0.0723 | 0.71 × 10−10 |
| 20 | 0.45 × 10−10 | 5043 | 0.0382 | 0.45 × 10−10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordani, G.; Scaramozzino, D.; Iturrioz, I.; Lacidogna, G.; Carpinteri, A. Modal Analysis of the Lysozyme Protein Considering All-Atom and Coarse-Grained Finite Element Models. Appl. Sci. 2021, 11, 547. https://0-doi-org.brum.beds.ac.uk/10.3390/app11020547
Giordani G, Scaramozzino D, Iturrioz I, Lacidogna G, Carpinteri A. Modal Analysis of the Lysozyme Protein Considering All-Atom and Coarse-Grained Finite Element Models. Applied Sciences. 2021; 11(2):547. https://0-doi-org.brum.beds.ac.uk/10.3390/app11020547
Chicago/Turabian StyleGiordani, Gustavo, Domenico Scaramozzino, Ignacio Iturrioz, Giuseppe Lacidogna, and Alberto Carpinteri. 2021. "Modal Analysis of the Lysozyme Protein Considering All-Atom and Coarse-Grained Finite Element Models" Applied Sciences 11, no. 2: 547. https://0-doi-org.brum.beds.ac.uk/10.3390/app11020547