
Electronic Band Structure of Transition Metal Dichalcogenides from Ab Initio and Slater–Koster Tight-Binding Model


Abstract
:1. Introduction
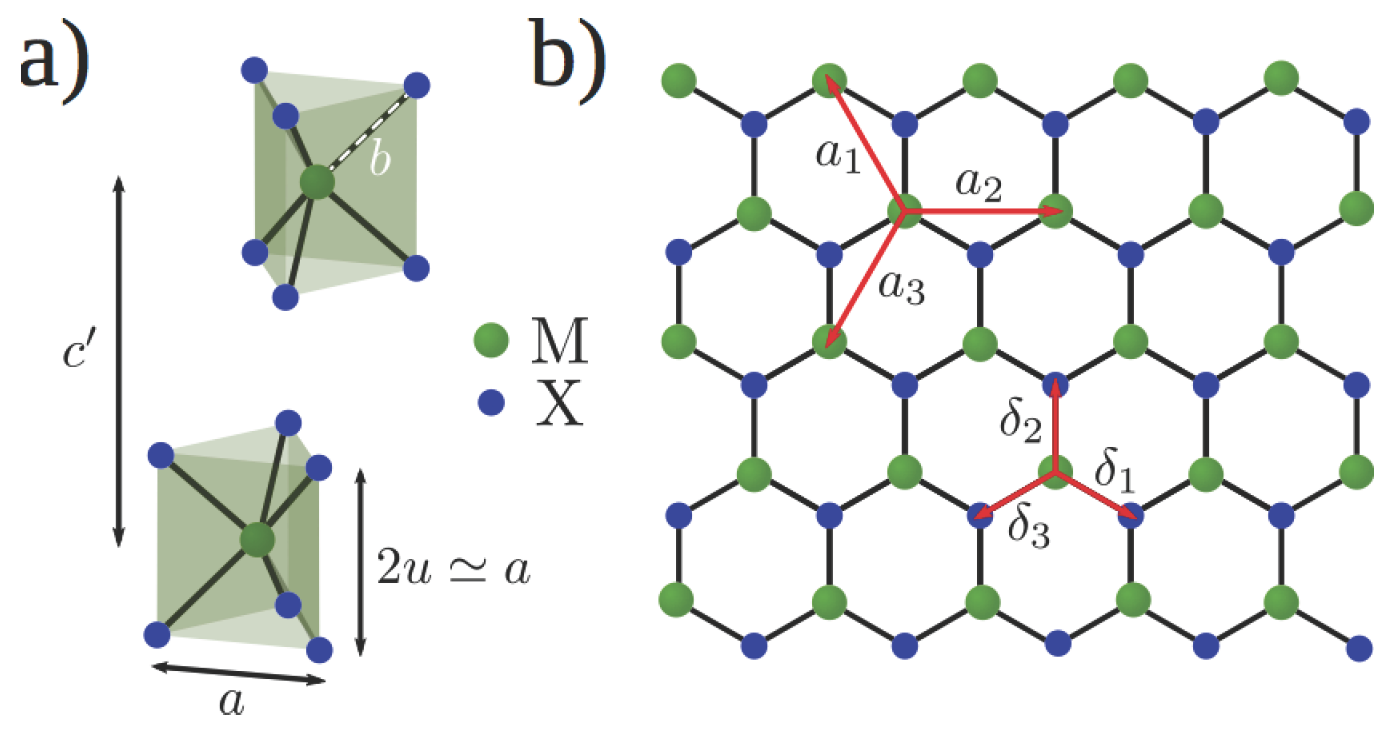
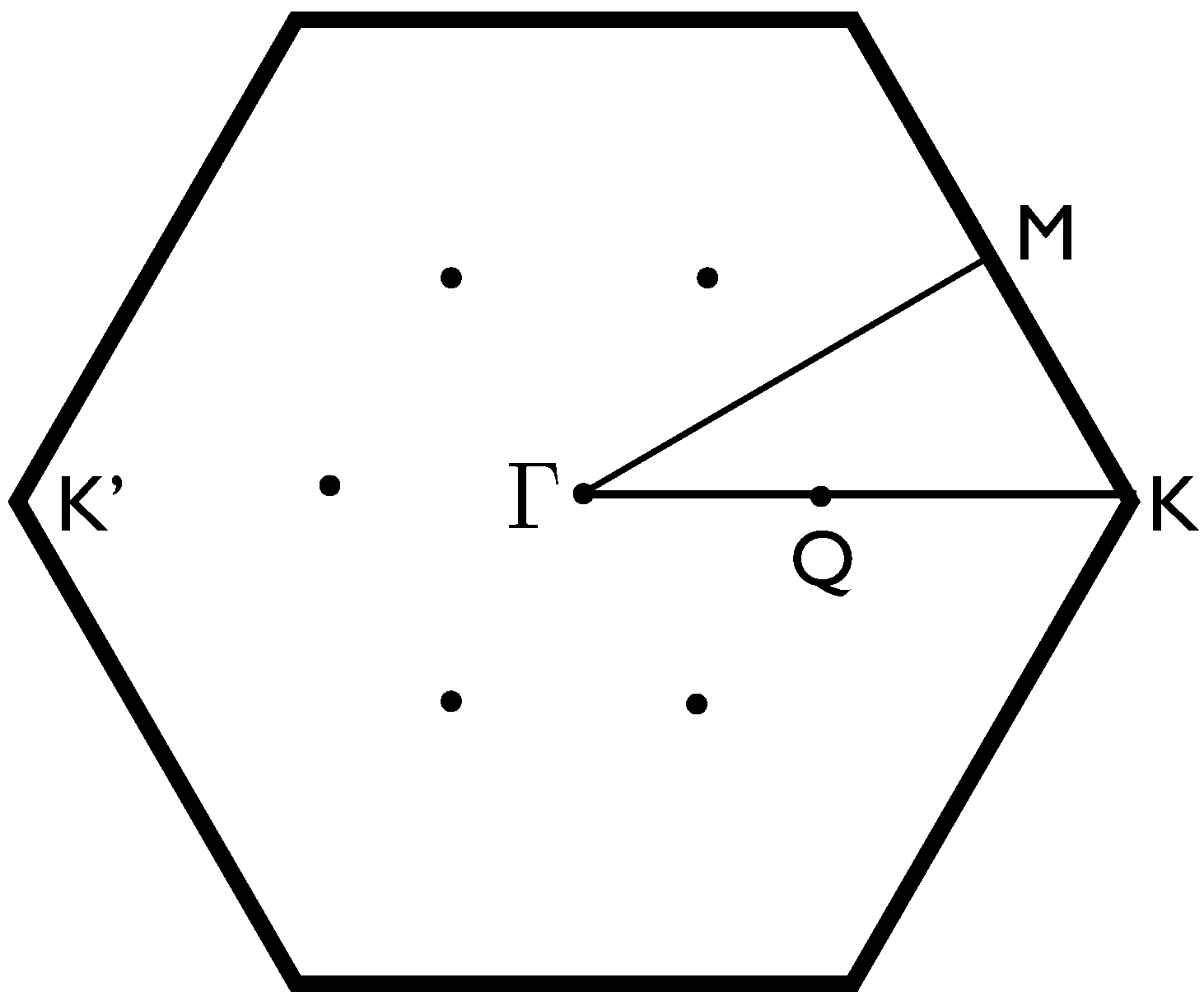
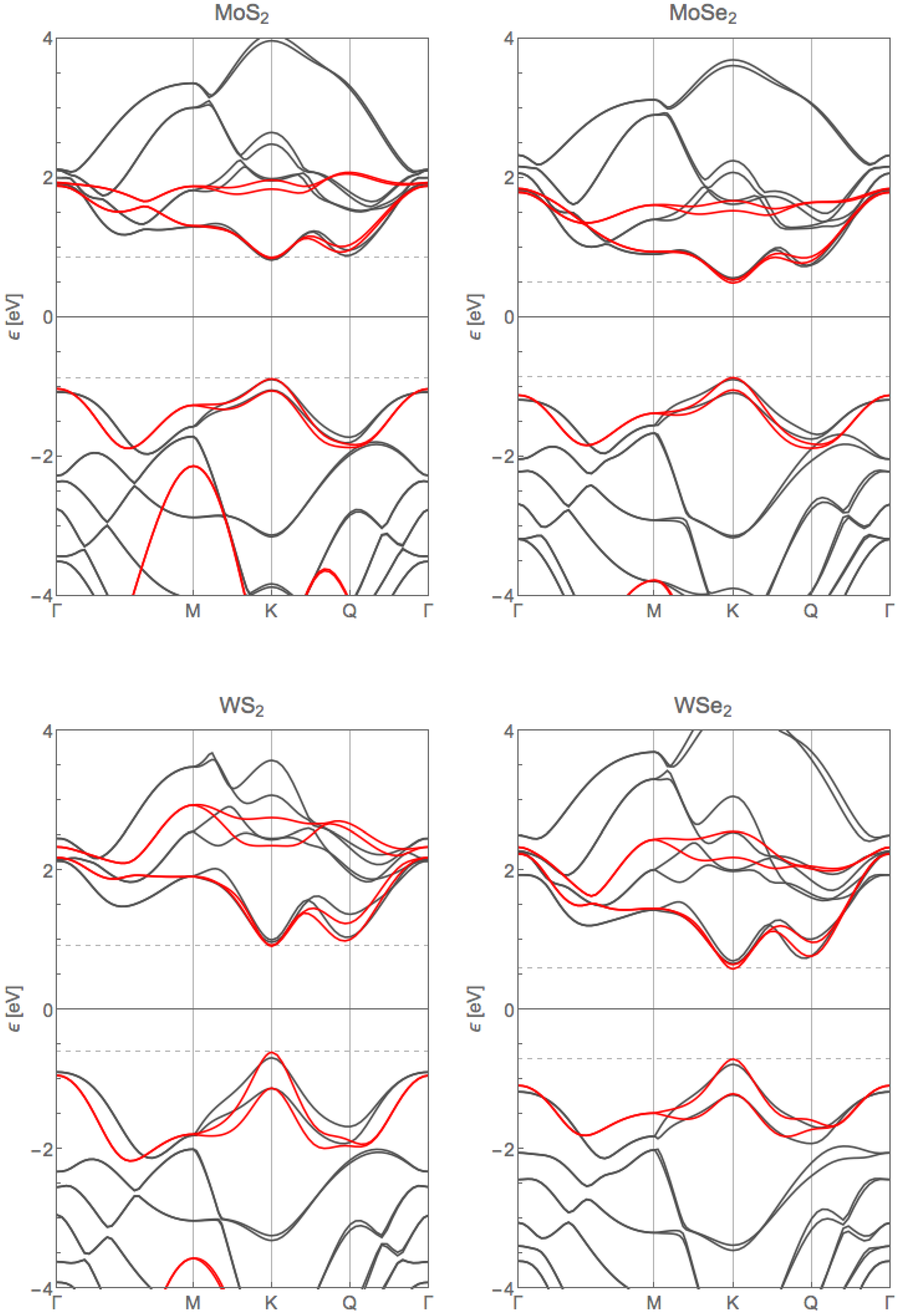
2. Electronic Band Structure
3. Tight-Binding Model
4. Slater–Koster Parameters from Fitting to Ab Initio Calculations
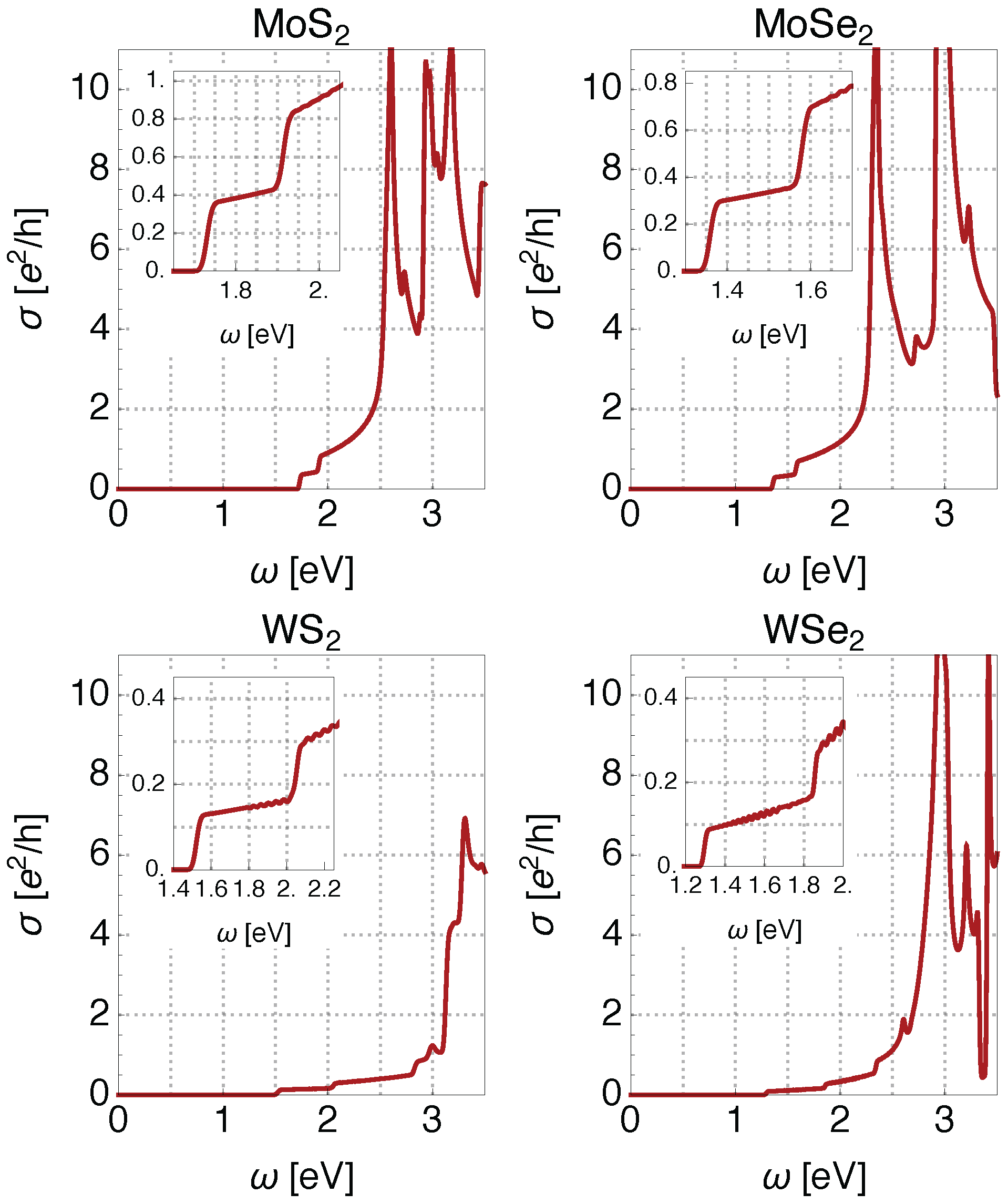
5. Optical Conductivity
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. On-Site and Hopping Matrices of the 6×6 Block
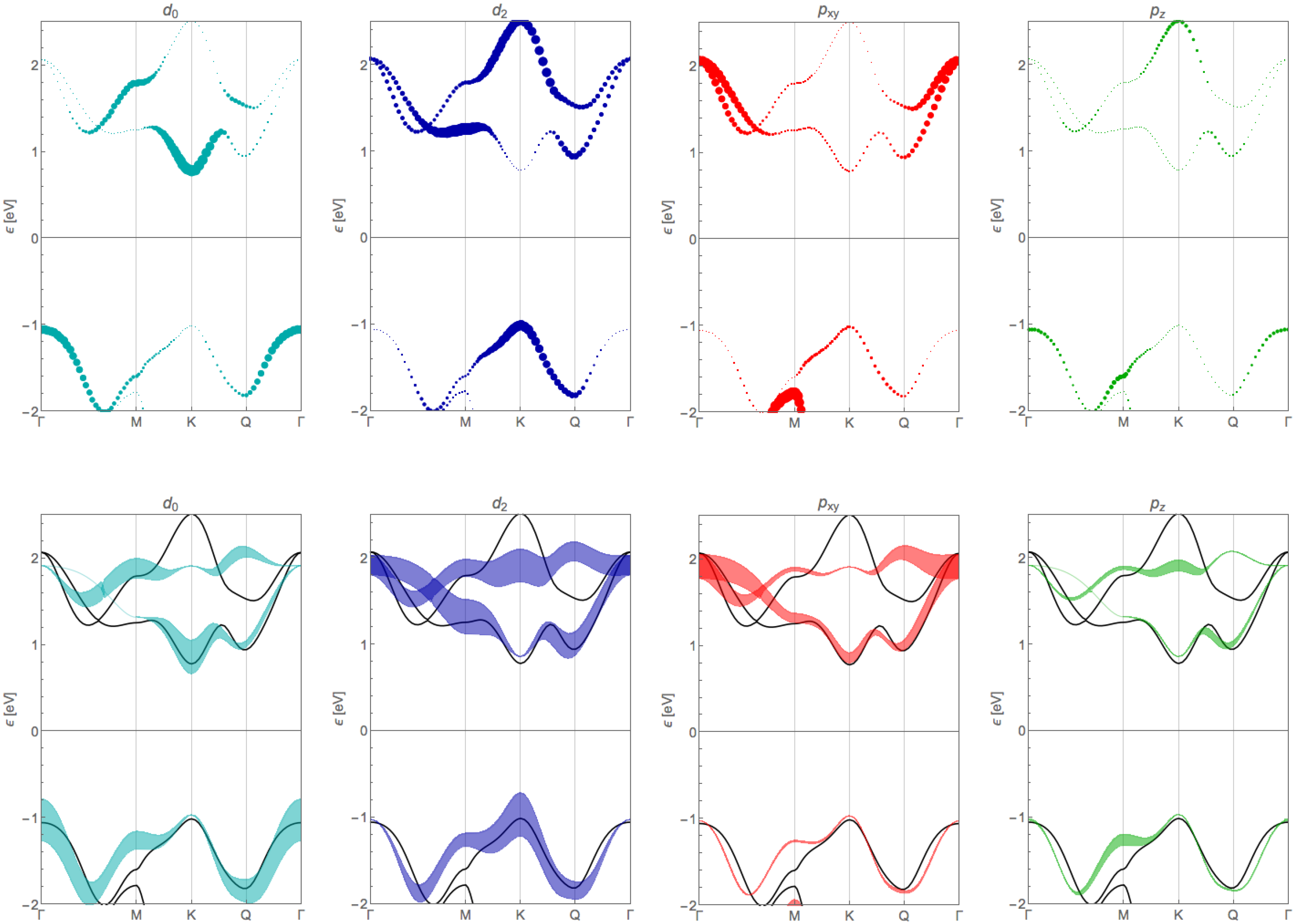
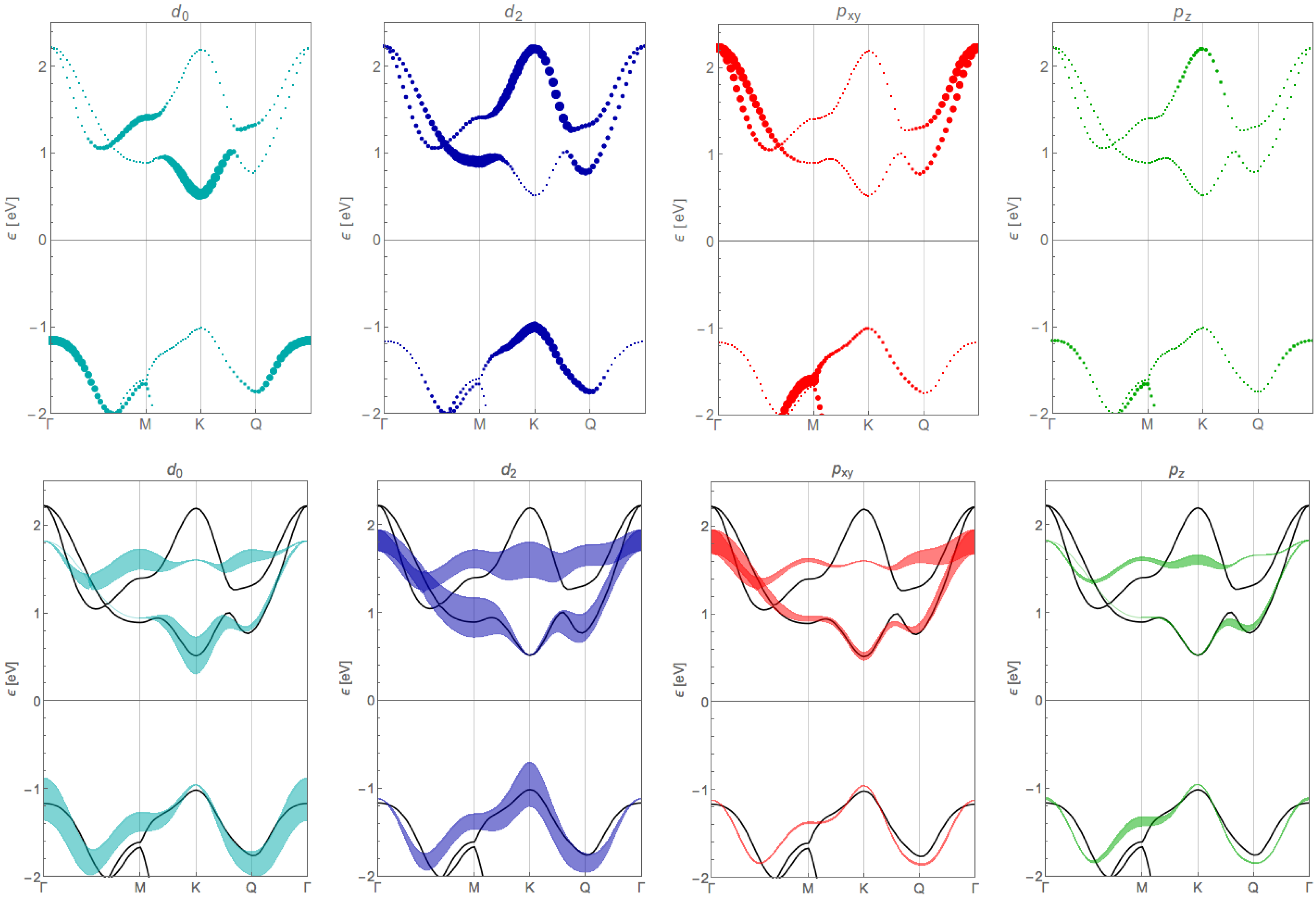
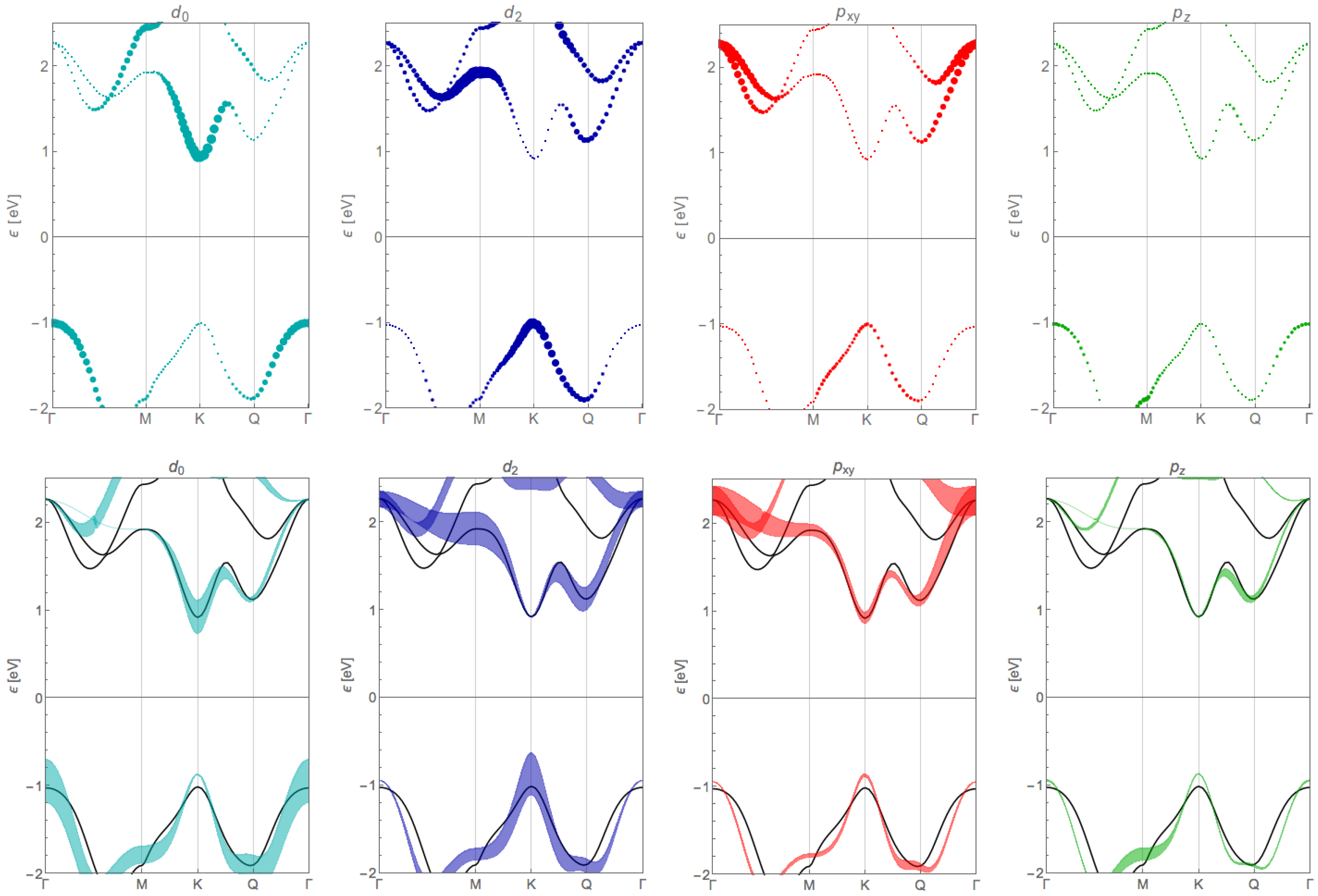
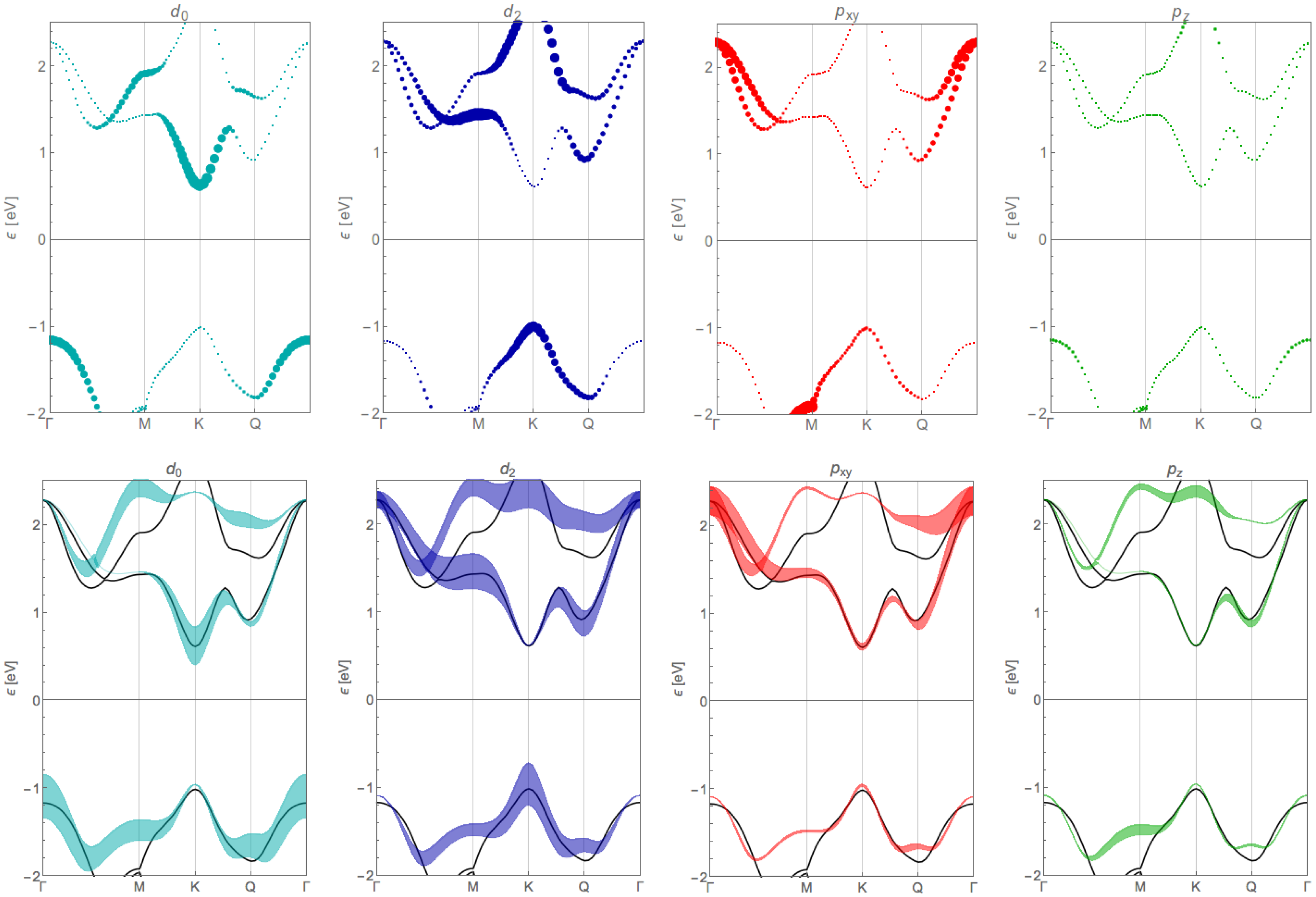
Appendix B. Orbital Contribution of the Tight-Binding Bands



References
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nature Nanotech. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.J.; Marks, T.J.; Hersam, M.C. Emerging Device Applications for Semiconducting Two-Dimensional Transition Metal Dichalcogenides. ACS Nano 2014, 8, 1102–1120. [Google Scholar] [CrossRef] [PubMed]
- Ganatra, R.; Zhang, Q. Few-Layer MoS2: A Promising Layered Semiconductor. ACS Nano 2014, 8, 4074–4099. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Qian, X.; Huang, C.W.; Li, J. Strain-engineered artificial atom as a broad-spectrum solar energy funnel. Nat. Photonics 2012, 6, 866–872. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Y.W. Tuning the electronic and magnetic properties of MoS2 nanoribbons by strain engineering. J. Phys. Chem. C 2012, 116, 11752–11757. [Google Scholar] [CrossRef]
- Peelaers, H.; Van de Walle, C.G. Effects of strain on band structure and effective masses in MoS2. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Scalise, E.; Houssa, M.; Pourtois, G.; Afanas?ev, V.; Stesmans, A. First-principles study of strained 2D MoS2. Phys. E Low-Dimens. Syst. Nanostruct. 2014, 56, 416–421. [Google Scholar] [CrossRef]
- Molina-Sánchez, A.; Hummer, K.; Wirtz, L. Vibrational and optical properties of MoS2: From monolayer to bulk. Surf. Sci. Rep. 2015, 70, 554–586. [Google Scholar] [CrossRef]
- Brumme, T.; Calandra, M.; Mauri, F. First-principles theory of field-effect doping in transition-metal dichalcogenides: Structural properties, electronic structure, Hall coefficient, and electrical conductivity. Phys. Rev. B 2015, 91. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Cheng, Y.C.; Schwingenschlögl, U. Giant spin-orbit-induced spin splitting in two-dimensional transition-metal dichalcogenide semiconductors. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef]
- Xiao, D.; Liu, G.B.; Feng, W.; Xu, X.; Yao, W. Coupled Spin and Valley Physics in Monolayers of MoS2 and Other Group-VI Dichalcogenides. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yao, W.; Xiao, D.; Heinz, T.F. Spin and pseudospins in layered transition metal dichalcogenides. Nat. Phys. 2014, 10, 343–350. [Google Scholar] [CrossRef]
- Babaee Touski, S.; Pourfath, M.; Roldán, R.; López-Sancho, M.P. Enhanced Spin-Flip Scattering by Surface Roughness in WS2 and MoS2 Nanoribbons. 2016; ArXiv e-prints. Available online: http://xxx.lanl.gov/abs/1606.03339 (accessed on 10 June 2016). [Google Scholar]
- Zeng, H.; Dai, J.; Yao, W.; Xiao, D.; Cui, X. Valley polarization in MoS2 monolayers by optical pumping. Nat. Nanotech. 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.F.; He, K.; Sahn, J.; Heinz, T.F. Control of valley polarization in monolayer MoS2 by optical helicity. Nat. Nanotech. 2012, 7, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Wang, G.; Han, W.; Ye, H.; Zhu, C.; Shi, J.; Niu, Q.; Tan, P.; Wang, E.; Liu, B.; et al. Valley-selective circular dichroism of monolayer molybdenum disulphide. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Liu, G.B.; Dai, J.; Yan, Y.; Zhu, B.; He, R.; Xie, L.; Xu, S.; Chen, X.; Yao, W.; et al. Optical signature of symmetry variations and spin-valley coupling in atomically thin tungsten dichalcogenides. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Ross, J.S.; Liu, G.B.; Aivazian, G.; Jones, A.; Fei, Z.; Zhu, W.; Xiao, D.; Yao, W.; Cobden, D.; et al. Electrical tuning of valley magnetic moment through symmetry control in bilayer MoS2. Nat. Phys. 2013, 9, 149–153. [Google Scholar] [CrossRef]
- Wang, Q.; Ge, S.; Li, X.; Qiu, J.; Ji, Y.; Feng, J.; Sun, D. Valley Carrier Dynamics in Monolayer Molybdenum Disulfide from Helicity-Resolved Ultrafast Pump–Probe Spectroscopy. ACS Nano 2013, 7, 11087–11093. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.F.; He, K.; Lee, C.; Lee, G.H.; Hone, J.; Heinz, T.F.; Shan, J. Tightly bound trions in monolayer MoS2. Nat. Mat. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Sallen, G.; Bouet, L.; Marie, X.; Wang, G.; Zhu, C.R.; Han, W.P.; Lu, Y.; Tan, P.H.; Amand, T.; Liu, B.L.; et al. Robust optical emission polarization in MoS2 monolayers through selective valley excitation. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef]
- Kormányos, A.; Zólyomi, V.; Drummond, N.D.; Burkard, G. Spin-Orbit Coupling, Quantum Dots, and Qubits in Monolayer Transition Metal Dichalcogenides. Phys. Rev. X 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Kormányos, A.; Burkard, G.; Gmitra, M.; Fabian, J.; Zólyomi, V.; Drummond, N.D.; Fal’ko, V. k * p theory for two-dimensional transition metal dichalcogenide semiconductors. 2D Mater. 2015, 2. [Google Scholar] [CrossRef]
- Roldán, R.; Castellanos-Gomez, A.; Cappelluti, E.; Guinea, F. Strain engineering in semiconducting two-dimensional crystals. J. Phys. Condens. Matter 2015, 27. [Google Scholar] [CrossRef] [PubMed]
- San-Jose, P.; Parente, V.; Guinea, F.; Roldán, R.; Prada, E. Inverse Funnel Effect of Excitons in Strained Black Phosphorus. Phys. Rev. X 2016, 6. [Google Scholar] [CrossRef]
- Wu, W.; Wang, L.; Li, Y.; Zhang, F.; Lin, L.; Niu, S.; Chenet, D.; Zhang, X.; Hao, Y.; Heinz, T.F.; et al. Piezoelectricity of single-atomic-layer MoS2 for energy conversion and piezotronics. Nature 2014, 514, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Roldán, R.; Silva-Guillén, J.A.; López-Sancho, M.P.; Guinea, F.; Cappelluti, E.; Ordejón, P. Electronic properties of single-layer and multilayer transition metal dichalcogenides MX2 (M = Mo, W and X = S, Se). Ann. Phys. 2014, 526, 347–357. [Google Scholar] [CrossRef]
- Yuan, S.; Roldán, R.; Katsnelson, M.I.; Guinea, F. Effect of point defects on the optical and transport properties of MoS2 and WS2. Phys. Rev. B 2014, 90. [Google Scholar] [CrossRef]
- Rostami, H.; Roldán, R.; Cappelluti, E.; Asgari, R.; Guinea, F. Theory of strain in single-layer transition metal dichalcogenides. Phys. Rev. B 2015, 92. [Google Scholar] [CrossRef]
- Pearce, A.J.; Burkard, G. A Tight Binding Approach to Strain and Curvature in Monolayer Transition-Metal Dichalcogenides. 2015; ArXiv e-prints. Available online: http://xxx.lanl.gov/abs/1511.06254 (accessed on 22 November 2015). [Google Scholar]
- Rostami, H.; Asgari, R.; Guinea, F. Edge modes in zigzag and armchair ribbons of monolayer MoS2. 2015; ArXiv e-prints. Available online: http://xxx.lanl.gov/abs/1511.07003 (accessed on 22 November 2015). [Google Scholar]
- Farmanbar, M.; Amlaki, T.; Brocks, G. Green’s function approach to edge states in transition metal dichalcogenides. Phys. Rev. B 2016, 93. [Google Scholar] [CrossRef]
- Castellanos-Gomez, A.; Roldán, R.; Cappelluti, E.; Buscema, M.; Guinea, F.; van der Zant, H.S.J.; Steele, G.A. Local Strain Engineering in Atomically Thin MoS2. Nano Letters 2013, 13, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Cappelluti, E.; Roldán, R.; Silva-Guillén, J.A.; Ordejón, P.; Guinea, F. Tight-binding model and direct-gap/indirect-gap transition in single-layer and multilayer MoS2. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef]
- Liu, G.B.; Shan, W.Y.; Yao, Y.; Yao, W.; Xiao, D. Three-band tight-binding model for monolayers of group-VIB transition metal dichalcogenides. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef]
- Kośmider, K.; González, J.W.; Fernández-Rossier, J. Large spin splitting in the conduction band of transition metal dichalcogenide monolayers. Phys. Rev. B 2013, 88. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ahluwalia, P. Electronic structure of transition metal dichalcogenides monolayers 1H-MX2 (M= Mo, W; X= S, Se, Te) from ab-initio theory: new direct band gap semiconductors. Eur. Phys. J. B 2012, 85, 1–7. [Google Scholar] [CrossRef]
- Soler, J.; Artacho, E.; Gale, J.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- Artacho, E.; Anglada, E.; Dieguez, O.; Gale, J.; García, A.; Junquera, J.; Martin, R.; Ordejón, P.; Pruneda, J.M.; Sánchez-Portal, D.; Soler, J. The SIESTA method; developments and applicability. J. Phys. Condens. Matter 2008, 20. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45. [Google Scholar] [CrossRef]
- Perdew, P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23. [Google Scholar] [CrossRef]
- Artacho, E.; Sánchez-Portal, D.; Ordejón, P.; García, A.; Soler, J. Linear-Scaling ab-initio Calculations for Large and Complex Systems. Phys. Status Solidi B Basic Res. 1999, 215, 809–817. [Google Scholar] [CrossRef]
- Fernández-Seivane, L.; Oliveira, M.; Sanvito, S.; Ferrer, J. On-site approximation for spin-orbit coupling in linear combination of atomic orbitals density functional methods. J. Phys. Condens. Matter 2006, 18. [Google Scholar] [CrossRef]
- Bromley, R.A.; Murray, R.B.; Yoffe, A.D. The band structures of some transition metal dichalcogenides. III. Group VIA: trigonal prism materials. J. Phys. C Solid State Phys. 1972, 5. [Google Scholar] [CrossRef]
- Schutte, W.; Boer, J.D.; Jellinek, F. Crystal structures of tungsten disulfide and diselenide. J. Solid State Chem. 1987, 70, 207–209. [Google Scholar] [CrossRef]
- Kumar, A.; Ahluwalia, P.K. Electronic structure of transition metal dichalcogenides monolayers 1H-MX2 (M = Mo, W; X = S, Se, Te) from ab-initio theory: new direct band gap semiconductors. Eur. Phys. J. B 2012, 85. [Google Scholar] [CrossRef]
- Roldán, R.; López-Sancho, M.P.; Guinea, F.; Cappelluti, E.; Silva-Guillén, J.A.; Ordejón, P. Momentum dependence of spin–orbit interaction effects in single-layer and multi-layer transition metal dichalcogenides. 2D Mater. 2014, 1. [Google Scholar] [CrossRef]
- Slater, J.C.; Koster, G.F. Simplified LCAO Method for the Periodic Potential Problem. Phys. Rev. 1954, 94, 1498–1524. [Google Scholar] [CrossRef]
- Ridolfi, E.; Le, D.; Rahman, T.S.; Mucciolo, E.R.; Lewenkopf, C.H. A tight-binding model for MoS2 monolayers. J. Phys. Condens. Matter 2015, 27. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Kuate Defo, R.; Shirodkar, S.N.; Lieu, S.; Tritsaris, G.A.; Kaxiras, E. Ab initio tight-binding Hamiltonian for transition metal dichalcogenides. Phys. Rev. B 2015, 92. [Google Scholar] [CrossRef]
- Lado, J.L.; Fernandez-Rossier, J. Landau Levels in two dimensional materials from first principles. 2016. ArXiv e-prints. [Google Scholar] [CrossRef]
- Mak, K.F.; Lee, C.; Hone, J.; Shan, J.; Heinz, T.F. Atomically Thin MoS2: A New Direct-Gap Semiconductor. Phys. Rev. Lett. 2010, 105. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | u | ||
|---|---|---|---|
| MoS | |||
| MoSe | |||
| WS | |||
| WSe |
| MoS | MoSe | WS | WSe | ||
|---|---|---|---|---|---|
| SOC | 0.086 | 0.089 | 0.271 | 0.251 | |
| 0.052 | 0.256 | 0.057 | 0.439 | ||
| Crystal Fields | −1.094 | −1.144 | −1.155 | −0.935 | |
| −0.050 | −0.250 | −0.650 | −1.250 | ||
| −1.511 | −1.488 | −2.279 | −2.321 | ||
| −3.559 | −4.931 | −3.864 | −5.629 | ||
| −6.886 | −7.503 | −7.327 | −6.759 | ||
| M–X | 3.689 | 3.728 | 7.911 | 5.803 | |
| −1.241 | −1.222 | −1.220 | −1.081 | ||
| M–M | −0.895 | −0.823 | −1.328 | −1.129 | |
| 0.252 | 0.215 | 0.121 | 0.094 | ||
| 0.228 | 0.192 | 0.442 | 0.317 | ||
| X–X | 1.225 | 1.256 | 1.178 | 1.530 | |
| −0.467 | −0.205 | −0.273 | −0.123 |
| K | K | ||||||
|---|---|---|---|---|---|---|---|
| DFT | TB | DFT | TB | DFT | TB | ||
| MoS | 0.0 | 0.0 | 0.82 | 0.77 | 0.66 | 0.96 | |
| 0.76 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.20 | 0.0 | 0.12 | 0.23 | 0.0 | 0.0 | ||
| 0.0 | 0.0 | 0.0 | 0.00 | 0.28 | 0.04 | ||
| MoSe | 0.0 | 0.0 | 0.83 | 0.83 | 0.71 | 0.96 | |
| 0.78 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.18 | 0.0 | 0.10 | 0.17 | 0.0 | 0.0 | ||
| 0.0 | 0.0 | 0.0 | 0.0 | 0.23 | 0.04 | ||
| WS | 0.0 | 0.0 | 0.80 | 0.76 | 0.64 | 0.98 | |
| 0.74 | 0.94 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.21 | 0.06 | 0.07 | 0.24 | 0.0 | 0.0 | ||
| 0.0 | 0.0 | 0.0 | 0.0 | 0.28 | 0.02 | ||
| WSe | 0.0 | 0.0 | 0.82 | 0.86 | 0.69 | 0.99 | |
| 0.73 | 0.95 | 0.0 | 0.0 | 0.0 | 0.0 | ||
| 0.20 | 0.05 | 0.05 | 0.14 | 0.0 | 0.0 | ||
| 0.0 | 0.0 | 0.0 | 0.0 | 0.23 | 0.01 | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Guillén, J.Á.; San-Jose, P.; Roldán, R. Electronic Band Structure of Transition Metal Dichalcogenides from Ab Initio and Slater–Koster Tight-Binding Model. Appl. Sci. 2016, 6, 284. https://0-doi-org.brum.beds.ac.uk/10.3390/app6100284
Silva-Guillén JÁ, San-Jose P, Roldán R. Electronic Band Structure of Transition Metal Dichalcogenides from Ab Initio and Slater–Koster Tight-Binding Model. Applied Sciences. 2016; 6(10):284. https://0-doi-org.brum.beds.ac.uk/10.3390/app6100284
Chicago/Turabian StyleSilva-Guillén, Jose Ángel, Pablo San-Jose, and Rafael Roldán. 2016. "Electronic Band Structure of Transition Metal Dichalcogenides from Ab Initio and Slater–Koster Tight-Binding Model" Applied Sciences 6, no. 10: 284. https://0-doi-org.brum.beds.ac.uk/10.3390/app6100284