Combined Effects of Repetitive Mild Traumatic Brain Injury and Alcohol Drinking on the Neuroinflammatory Cytokine Response and Cognitive Behavioral Outcomes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
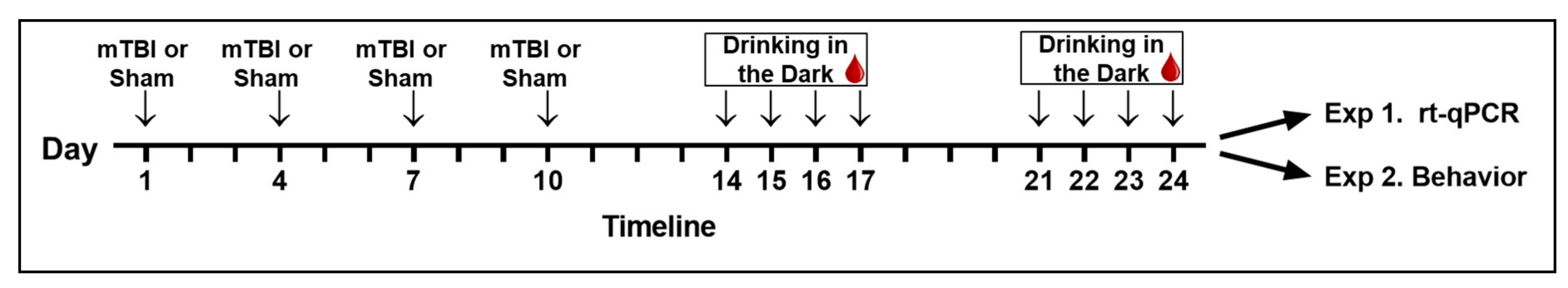
2.2. Repetitive Mild Traumatic Brain Injury
2.3. Voluntary Alcohol Consumption
2.4. Blood Alcohol Concentration
2.5. Experiment 1: Neuronal Cytokine Expression following rmTBI and Alcohol Drinking
2.5.1. RNA Isolation
2.5.2. Complementary DNA (cDNA) Synthesis
2.5.3. Real-Time Polymerase Chain Reaction Assay
2.6. Experiment 2: Cognitive Behavioral Performance following rmTBI and Alcohol Drinking
2.6.1. Serial Spatial Discrimination Reversal Learning
2.6.2. Open Field with Novelty Preference Task
2.7. Analyses
3. Results
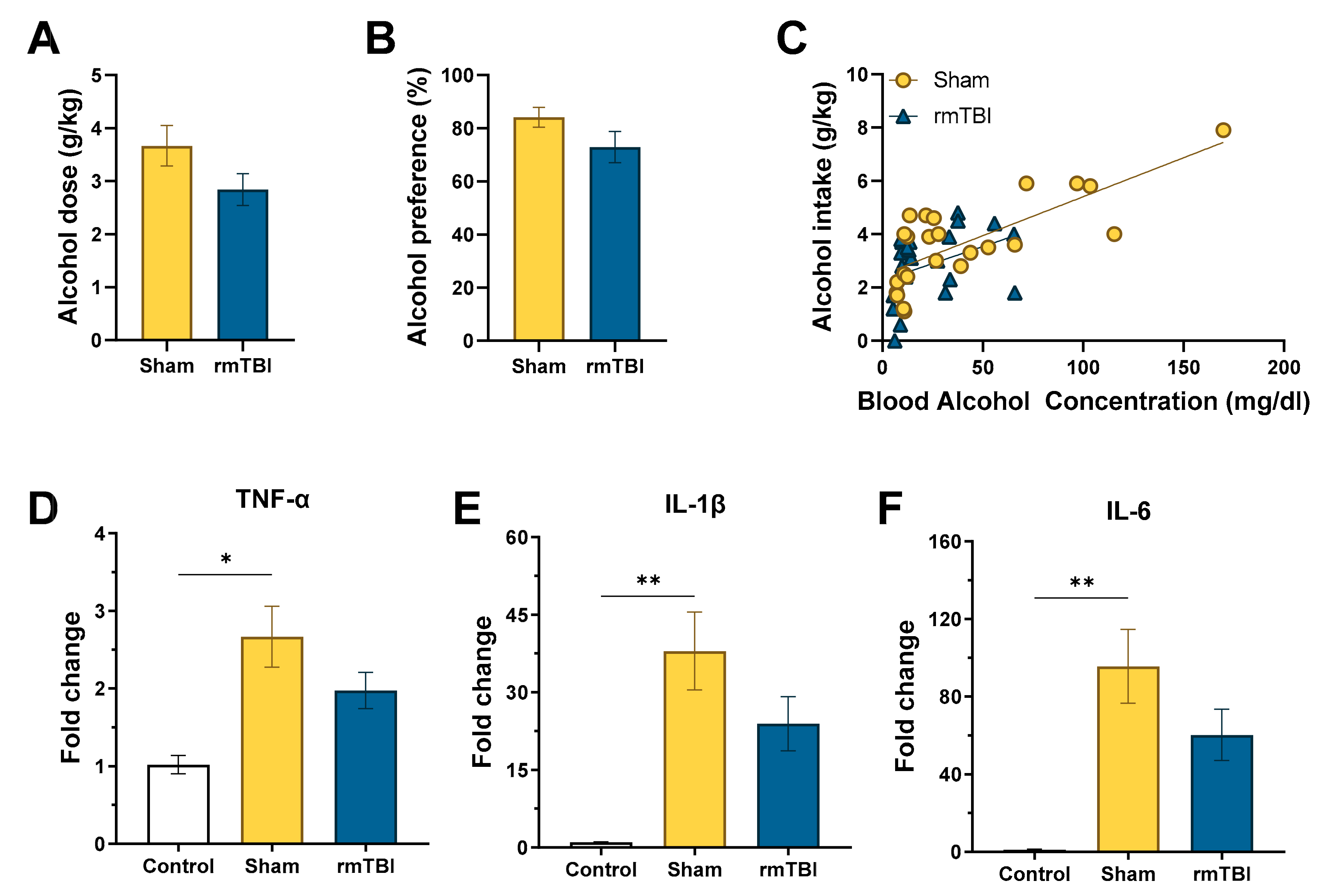
3.1. Experiment 1: rmTBI on Voluntary EtOH Intake and Combined Effects on Neurocytokine Expression
3.1.1. Voluntary Alcohol Consumption
3.1.2. RT-qPCR
3.2. Experiment 2: rmTBI on Voluntary EtOH Intake and Combined Effects on Cognitive Behavioral Performance
3.2.1. Voluntary Alcohol Consumption
3.2.2. Cognitive Behavioral Assessment
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Results from the 2006 National Survey on Drug Use and Health. In PsycEXTRA Dataset; American Psychological Association (APA): Rockville, MD, USA, 2013.
- Langlois, J.A.; Rutland-Brown, W.; Thomas, K.E. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. PsycEXTRA Dataset 2013. [Google Scholar] [CrossRef] [Green Version]
- Peterson, A.B.; Xu, L.; Daugherty, J.; Breiding, M.J. Surveillance Report of Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths, United States, 2014; Centers for Disease Control and Prevention, Department of Health and Human Services: Atlanta, GA, USA, 2019.
- Dikmen, S.S.; E Machamer, J.; Donovan, D.; Winn, H.; Temkin, N. Alcohol Use Before and After Traumatic Head Injury. Ann. Emerg. Med. 1995, 26, 167–176. [Google Scholar] [CrossRef]
- Kraus, J.F.; Morgenstern, H.; Fife, D.; Conroy, C.; Nourjah, P. Blood alcohol tests, prevalence of involvement, and outcomes following brain injury. Am. J. Public Health 1989, 79, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, J.D. Substance abuse as a mediating factor in outcome from traumatic brain injury. Arch. Phys. Med. Rehabil. 1995, 76, 302–309. [Google Scholar] [CrossRef]
- Hibbard, M.R.; Uysal, S.; Kepler, K.; Bogdany, J.; Silver, J. Axis I Psychopathology in Individuals with Traumatic Brain Injury. J. Head Trauma Rehabil. 1998, 13, 24–39. [Google Scholar] [CrossRef]
- Silver, J.M.; Kramer, R.; Greenwald, S.; Weissman, M. The association between head injuries and psychiatric disorders: findings from the New Haven NIMH Epidemiologic Catchment Area Study. Brain Inj. 2001, 15, 935–945. [Google Scholar] [CrossRef]
- Weil, Z.M.; Corrigan, J.D.; Karelina, K. Alcohol abuse after traumatic brain injury: Experimental and clinical evidence. Neurosci. Biobehav. Rev. 2016, 62, 89–99. [Google Scholar] [CrossRef]
- Fann, J.R.; E Burington, B.; Leonetti, A.; Jaffe, K.M.; Katon, W.J.; Thompson, R.S. Psychiatric Illness Following Traumatic Brain Injury in an Adult HealthMaintenance Organization Population. Arch. Gen. Psychiatry 2004, 61, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, J.D.; Bogner, J.; Mellick, D.; Bushnik, T.; Dams-O’Connor, K.; Hammond, F.M.; Hart, T.; Kolakowsky-Hayner, S. Prior History of Traumatic Brain Injury Among Persons in the Traumatic Brain Injury Model Systems National Database. Arch. Phys. Med. Rehabil. 2013, 94, 1940–1950. [Google Scholar] [CrossRef]
- US Department of Health & Human Services; Centers for Disease Control (CDC). National Center for Injury Prevention and Control Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. PsycEXTRA Dataset 2013. [Google Scholar] [CrossRef]
- Holm, L.; Cassidy, J.D.; Carroll, L.J.; Borg, J. Summary of the WHO collaborating centre for neurotrauma task force on mild traumatic brain injury. J. Rehabil. Med. 2005, 37, 137–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delouche, A.; Attyé, A.; Heck, O.; Grand, S.; Kastler, A.; Lamalle, L.; Renard, F.; Krainik, A. Diffusion MRI: Pitfalls, literature review and future directions of research in mild traumatic brain injury. Eur. J. Radiol. 2016, 85, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Honce, J.M.; Nyberg, E.; Jones, I.; Nagae, L. Neuroimaging of Concussion. Phys. Med. Rehabil. Clin. N. Am. 2016, 27, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Morganti-Kossmann, M.C.; Satgunaseelan, L.; Bye, N.; Kossmann, T. Modulation of immune response by head injury. Injury 2007, 38, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury—An inflammatory disease? Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef]
- Ghirnikar, R.S.; Lee, Y.L.; Eng, L.F. Inflammation in Traumatic Brain Injury: Role of Cytokines and Chemokines. Neurochem. Res. 1998, 23, 329–340. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Morganti-Kossmann, M.C. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010, 7, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.A.; Corso, T.D.; Neafsey, E.J. Neuronal Degeneration in Rat Cerebrocortical and Olfactory Regions During Subchronic "Binge" Intoxication with Ethanol: Possible Explanation for Olfactory Deficits in Alcoholics. Alcohol. Clin. Exp. Res. 1996, 20, 284–292. [Google Scholar] [CrossRef]
- Crews, F. Alcohol and Neurodegeneration. CNS Drug Rev. 2006, 5, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Crews, F.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and Alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef]
- Obernier, J.A.; Bouldin, T.W.; Crews, F.T. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin. Exp. Res. 2002, 26, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Crews, F.T. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 2008, 210, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Crews, F. Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J. Neuroinflamm. 2012, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wang, P.; Morgan, D.; Bruijnzeel, A.W.; Lin, D.; Pan, J.; Lin, F.; Strang, K.H.; Selig, T.M.; Perez, P.D.; et al. Temporal MRI characterization, neurobiochemical and neurobehavioral changes in a mouse repetitive concussive head injury model. Sci. Rep. 2015, 5, 11178. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.S.; Best, K.; Belknap, J.K.; Finn, D.A.; Crabbe, J.C. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol. Behav. 2005, 84, 53–63. [Google Scholar] [CrossRef]
- Thiele, T.E.; Crabbe, J.C.; Ii, S.L.B. “Drinking in the Dark” (DID): A Simple Mouse Model of Binge-Like Alcohol Intake. Curr. Protoc. Neurosci. 2014, 68, 9.49.1–9.49.12. [Google Scholar] [CrossRef] [Green Version]
- Thiele, T.E.; Navarro, M. “Drinking in the dark” (DID) procedures: A model of binge-like ethanol drinking in non-dependent mice. Alcohol 2014, 48, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Idrus, N.M.; McGough, N.N.H.; Riley, E.P.; Thomas, J.D. Administration of memantine during withdrawal mitigates overactivity and spatial learning impairments associated with neonatal alcohol exposure in rats. Alcohol. Clin. Exp. Res. 2014, 38, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Idrus, N.M.; McGough, N.N.; Spinetta, M.J.; Thomas, J.D.; Riley, E.P. The effects of a single memantine treatment on behavioral alterations associated with binge alcohol exposure in neonatal rats. Neurotoxicol. Teratol. 2011, 33, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.D.; Fleming, S.; Riley, E. MK-801 Can Exacerbate or Attenuate Behavioral Alterations Associated With Neonatal Alcohol Exposure in the Rat, Depending on the Timing of Administration. Alcohol. Clin. Exp. Res. 2001, 25, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Weinert, S.P.; Sharif, S.; Riley, E.P. MK-801 Administration during Ethanol Withdrawal in Neonatal Rat Pups Attenuates Ethanol-Induced Behavioral Deficits. Alcohol. Clin. Exp. Res. 1997, 21, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, J.C.; Metten, P.; Rhodes, J.S.; Yu, C.-H.; Brown, L.L.; Phillips, T.J.; Finn, D.A. A Line of Mice Selected for High Blood Ethanol Concentrations Shows Drinking in the Dark to Intoxication. Biol. Psychiatry 2009, 65, 662–670. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Coehlo, M.A.; Solton, N.R.; Szumlinski, K.K. Negative Affect and Excessive Alcohol Intake Incubate during Protracted Withdrawal from Binge-Drinking in Adolescent, But Not Adult, Mice. Front. Psychol. 2017, 8, 1128. [Google Scholar] [CrossRef] [Green Version]
- Marianno, P.; Abrahao, K.P.; Camarini, R. Environmental Enrichment Blunts Ethanol Consumption after Restraint Stress in C57BL/6 Mice. PLoS ONE 2017, 12, e0170317. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.S.; Ford, M.M.; Yu, C.-H.; Brown, L.L.; Finn, D.A.; Garland, T.; Crabbe, J.C. Mouse inbred strain differences in ethanol drinking to intoxication. Genes Brain Behav. 2007, 6, 1–18. [Google Scholar] [CrossRef]
- Lim, Y.W.; Meyer, N.P.; Shah, A.S.; Budde, M.D.; Stemper, B.D.; Olsen, C.M. Voluntary Alcohol Intake following Blast Exposure in a Rat Model of Mild Traumatic Brain Injury. PLoS ONE 2015, 10, e0125130. [Google Scholar] [CrossRef] [Green Version]
- Weil, Z.M.; Gaier, K.R.; Karelina, K. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol. Dis. 2014, 70, 108–116. [Google Scholar] [CrossRef]
- Belknap, J.K.; Crabbe, J.C.; Young, E.R. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology 1993, 112, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.A.; McClearn, G.E. Mouse Strain Differences in Preference for Various Concentrations of Alcohol. Q. J. Stud. Alcohol 1962, 23, 26–33. [Google Scholar] [CrossRef] [PubMed]
- York, J.L. Consumption of intoxicating beverages by rats and mice exhibiting high and low preferences for ethanol. Pharmacol. Biochem. Behav. 1981, 15, 207–214. [Google Scholar] [CrossRef]
- Koechling, U.; Amit, Z. Effects of 3-amino-1,2,4-triazole on brain catalase in the mediation of ethanol consumption in mice. Alcohol 1994, 11, 235–239. [Google Scholar] [CrossRef]
- Fritz, B.M.; Ii, S.L.B. Rodent models and mechanisms of voluntary binge-like ethanol consumption: Examples, opportunities, and strategies for preclinical research. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombardier, C.H.; Temkin, N.R.; Machamer, J.; Dikmen, S.S. The natural history of drinking and alcohol-related problems after traumatic brain injury. Arch. Phys. Med. Rehabil. 2003, 84, 185–191. [Google Scholar] [CrossRef]
- Horner, M.D.; Ferguson, P.L.; Selassie, A.W.; Labbate, L.A.; Kniele, K.; Corrigan, J.D. Patterns of alcohol use 1 year after traumatic brain injury: A population-based, epidemiological study. J. Int. Neuropsychol. Soc. 2005, 11, 322–330. [Google Scholar] [CrossRef]
- Crews, F.T.; Sarkar, D.K.; Qin, L.; Zou, J.; Boyadjieva, N.; Vetreno, R.P. Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Res. Curr. Rev. 2015, 37, 331–351. [Google Scholar]
- Chao, C.; Hu, S.; Ehrlich, L.; Peterson, P. Interleukin-1 and Tumor Necrosis Factor-α Synergistically Mediate Neurotoxicity: Involvement of Nitric Oxide and of N-Methyl-D-aspartate Receptors. Brain Behav. Immun. 1995, 9, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Briones, T.L.; Woods, J.; Rogozinska, M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol. Commun. 2013, 1, 57. [Google Scholar] [CrossRef] [Green Version]
- Kremlev, S.G.; Palmer, C. Interleukin-10 inhibits endotoxin-induced pro-inflammatory cytokines in microglial cell cultures. J. Neuroimmunol. 2005, 162, 71–80. [Google Scholar] [CrossRef]
- Penkowa, M.; Camats, J.; Hadberg, H.; Quintana, A.; Rojas, S.; Giralt, M.; Molinero, A.; Campbell, I.L.; Hidalgo, J. Astrocyte-targeted expression of interleukin-6 protects the central nervous system during neuroglial degeneration induced by 6-aminonicotinamide. J. Neurosci. Res. 2003, 73, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.C.B.; Regner, A.; Miotto, K.D.L.; De Moura, S.; Ikuta, N.; Vargas, A.E.; Chies, J.A.B.; Simon, D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014, 28, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.M.S.; De Souza, N.M.; Schwarzbold, M.L.; Diaz, A.P.; Nunes, J.C.; Hohl, A.; Da Silva, P.N.A.; Vieira, J.; De Souza, R.L.; Bertotti, M.M.; et al. Interleukin-10 Is an Independent Biomarker of Severe Traumatic Brain Injury Prognosis. Neuroimmunomodulation 2012, 19, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.D.; Makley, A.T.; Campion, E.M.; Friend, L.A.W.; Lentsch, A.B.; Pritts, T.A. Preinjury alcohol exposure attenuates the neuroinflammatory response to traumatic brain injury. J. Surg. Res. 2013, 184, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiffenstein, P.; Mathis, K.W.; Stouwe, C.V.; Molina, P.E. Alcohol Binge Before Trauma/Hemorrhage Impairs Integrity of Host Defense Mechanisms during Recovery. Alcohol. Clin. Exp. Res. 2007, 31, 704–715. [Google Scholar] [CrossRef]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol 2004, 33, 147–155. [Google Scholar] [CrossRef]
- Crews, F.T.; Qin, L.; Sheedy, D.; Vetreno, R.P.; Zou, J. High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol. Psychiatry 2013, 73, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; He, J.; Hanes, R.N.; Pluzarev, O.; Hong, J.-S.; Crews, F. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflamm. 2008, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.Y.; Crews, F. Release of Neuronal HMGB1 by Ethanol through Decreased HDAC Activity Activates Brain Neuroimmune Signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [Green Version]
- Teng, S.X.; Molina, P.E. Acute Alcohol Intoxication Prolongs Neuroinflammation without Exacerbating Neurobehavioral Dysfunction following Mild Traumatic Brain Injury. J. Neurotrauma 2014, 31, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 Mediates Neuroinflammation and Motor Coordination Deficits after Mild Traumatic Brain Injury and Brief Hypoxia in Mice. Shock 2013, 40, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Bai, L.; Niu, X.; Wang, Z.; Yin, B.; Bai, G.; Zhang, D.; Gan, S.; Sun, C.; Wang, S.; et al. Elevated Serum Levels of Inflammation-Related Cytokines in Mild Traumatic Brain Injury Are Associated With Cognitive Performance. Front. Neurol. 2019, 10, 1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, M.; Hales, A.; Reger, M.; Giza, C.; Hovda, D. Repeat Traumatic Brain Injury in the Juvenile Rat Is Associated with Increased Axonal Injury and Cognitive Impairments. Dev. Neurosci. 2010, 32, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrichs, S.; Stenzel-Poore, M.; Gold, L.; Battenberg, E.; Bloom, F.; Koob, G.; Vale, W.; Pich, E.M. Learning impairment in transgenic mice with central overexpression of corticotropin-releasing factor. Neuroscience 1996, 74, 303–311. [Google Scholar] [CrossRef]
- Vaagenes, I.C.; Tsai, S.-Y.; Ton, S.T.; Husak, V.A.; McGuire, S.O.; O’Brien, T.E.; Kartje, G.L. Binge Ethanol Prior to Traumatic Brain Injury Worsens Sensorimotor Functional Recovery in Rats. PLoS ONE 2015, 10, e0120356. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, J.P.; Teng, S.X.; Katz, P.S.; Gilpin, N.W.; Molina, P.E. Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav. Brain Res. 2015, 279, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Teng, S.X.; Katz, P.S.; Maxi, J.K.; Mayeux, J.P.; Gilpin, N.W.; Molina, P.E. Alcohol exposure after mild focal traumatic brain injury impairs neurological recovery and exacerbates localized neuroinflammation. Brain Behav. Immun. 2015, 45, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Badanich, K.A.; Becker, H.C.; Woodward, J.J. Effects of chronic intermittent ethanol exposure on orbitofrontal and medial prefrontal cortex-dependent behaviors in mice. Behav. Neurosci. 2011, 125, 879–891. [Google Scholar] [CrossRef]
- Milman, A.; Rosenberg, A.; Weizman, R.; Pick, C.G. Mild Traumatic Brain Injury Induces Persistent Cognitive Deficits and Behavioral Disturbances in Mice. J. Neurotrauma 2005, 22, 1003–1010. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cognitive Behavioral Battery Schedule | |
|---|---|
| Day 1 | SSDRL: pretrial training (10 trials) |
| Day 2 | SSDRL: test day (30 trials) |
| Day 3 | SSDRL: test day (30 trials) |
| Day 4 | SSDRL: test day (30 trials) |
| Day 5 | Open field: habituation, (trial 1); 30 min delay; locomotor, (trial 2). |
| Day 6 | Novelty preference: open field + novel object (trial 3); 30 min delay; familiar object + novel object (trial 4). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffman, J.; Yu, J.; Kirstein, C.; Kindy, M.S. Combined Effects of Repetitive Mild Traumatic Brain Injury and Alcohol Drinking on the Neuroinflammatory Cytokine Response and Cognitive Behavioral Outcomes. Brain Sci. 2020, 10, 876. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110876
Hoffman J, Yu J, Kirstein C, Kindy MS. Combined Effects of Repetitive Mild Traumatic Brain Injury and Alcohol Drinking on the Neuroinflammatory Cytokine Response and Cognitive Behavioral Outcomes. Brain Sciences. 2020; 10(11):876. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110876
Chicago/Turabian StyleHoffman, Jessica, Jin Yu, Cheryl Kirstein, and Mark S. Kindy. 2020. "Combined Effects of Repetitive Mild Traumatic Brain Injury and Alcohol Drinking on the Neuroinflammatory Cytokine Response and Cognitive Behavioral Outcomes" Brain Sciences 10, no. 11: 876. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10110876