
Genotyping and Plasma/Cerebrospinal Fluid Profiling of a Cohort of Frontotemporal Dementia–Amyotrophic Lateral Sclerosis Patients


 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
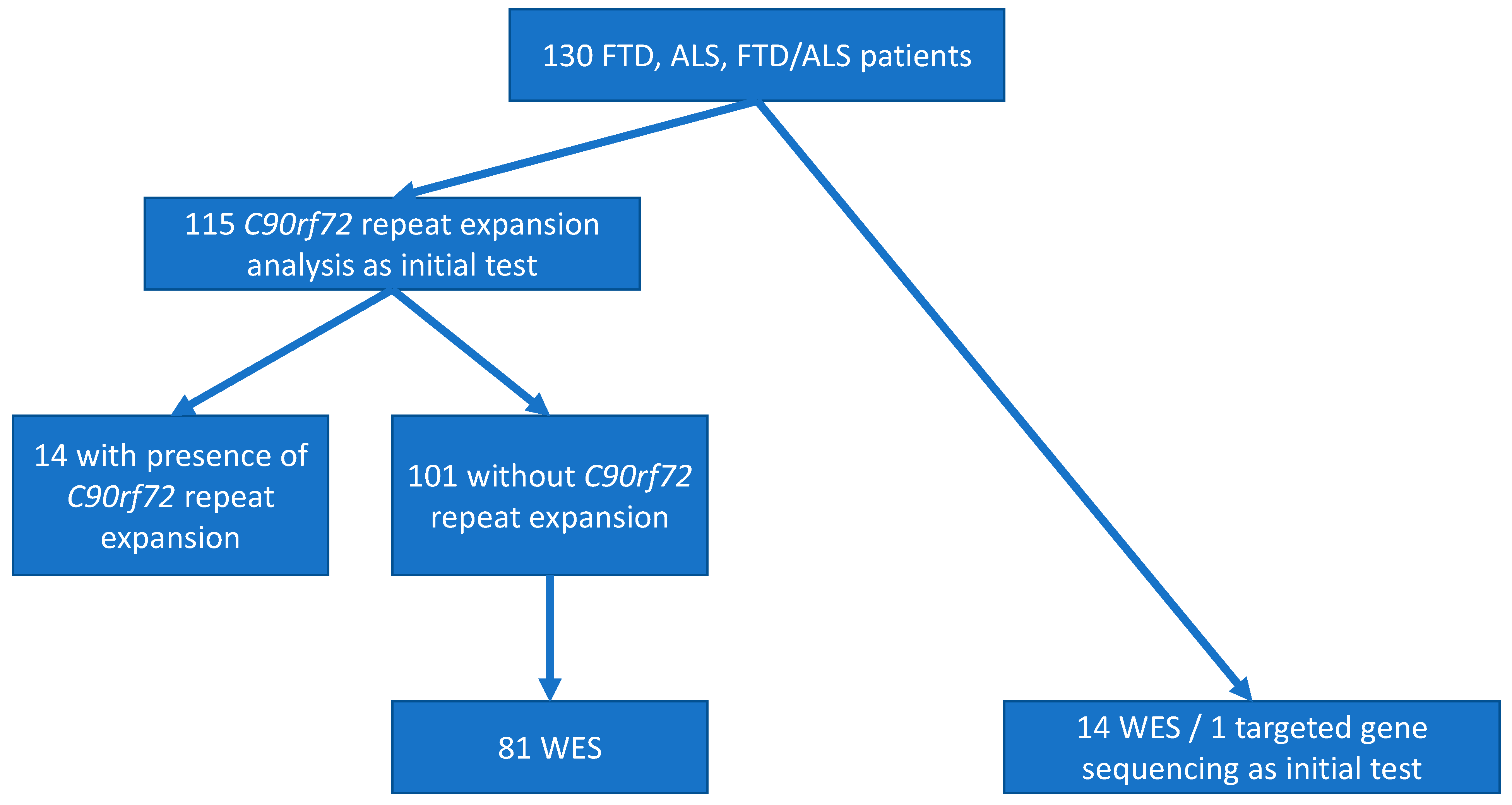
2.1. Participants
2.2. Blood Collection and DNA Extraction
2.3. C9orf72 Repeat Expansions
2.4. Whole Exome Sequencing
- (1)
- At Minotech Genomics Facility, Institute of Molecular Biology and Biotechnology (IMBB-FORTH, Crete) with the use of the Illumina NextSeq500 platform (n = 60). In detail, sequencing of 2 × 75 bp DNA fragments with at least 50× coverage, targeting regions of 45.3 Mb size, was performed. Libraries were prepared with the TruSeq® Rapid Exome Library prep kit (Illumina, San Diego, CA, USA). Bioinformatics processing of the data derived from mapping on the hg19 reference genome and quality control of the results (e.g., number of readings and coverage quality) were performed by the BaseSpace® software (Illumina, San Diego, CA, USA). Finally, genetic variation was identified with the VariantStudio® software after comparison with the hg19 reference genome and drawing information from genetic databases, e.g., Human Genome Mutation Database (HGMD), ClinVar® (National Center for Biotechnology Information, Bethesda MD, USA) (Annotation Excel file).
- (2)
- At Macrogen (Seoul, Korea), using the Illumina HiSeq4000 platform (n = 24). In specific, 2 × 100 bp DNA fragments were sequenced with an aim of at least 50x coverage. For the construction of genomic libraries, the Agilent Sure-Select Human All Exon V5 (not including UTRs) Target Enrichment System was used.
- (3)
- At Otogenetics (GA, USA), using the Illumina HiSeq2500 platform (n = 11). In detail, sequencing of 2 × 100 bp DNA fragments was performed aiming at coverage of at least 50× and targeting a region of 45.3 Mb, that represents >98% of the human coding sequence according to the Consensus Coding Sequences (CCDS) and Ensembl. Exon-enriched library preparation was performed with the use of the Agilent V5 (51Mb) Sure-Select Target Enrichment System.
2.5. Gene Variant Identification and Verification
2.6. Measurements of CSF and Plasma Biomarkers
2.7. Statistical Analysis
3. Results
3.1. Demographic Data
3.2. Family History
3.3. Pathogenic and Likely Pathogenic Variants in FTD-ALS Genes
3.3.1. C9orf72 Repeat Expansion
3.3.2. Other Causative Variants
3.4. Association of Variants in FTD -ALS Genes with CSF Biomarkers
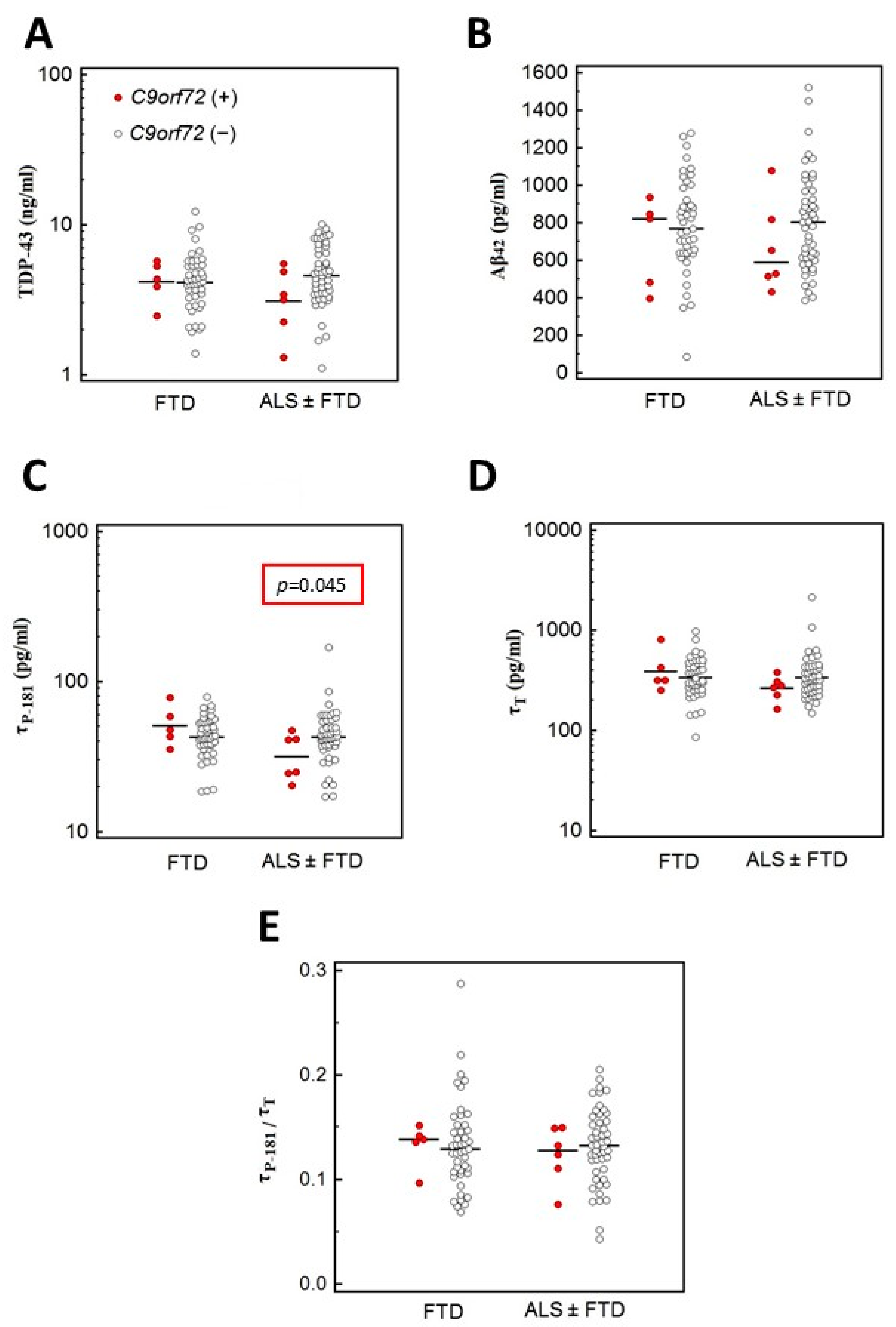
3.4.1. Association of C9orf72 Variants with CSF Biomarkers
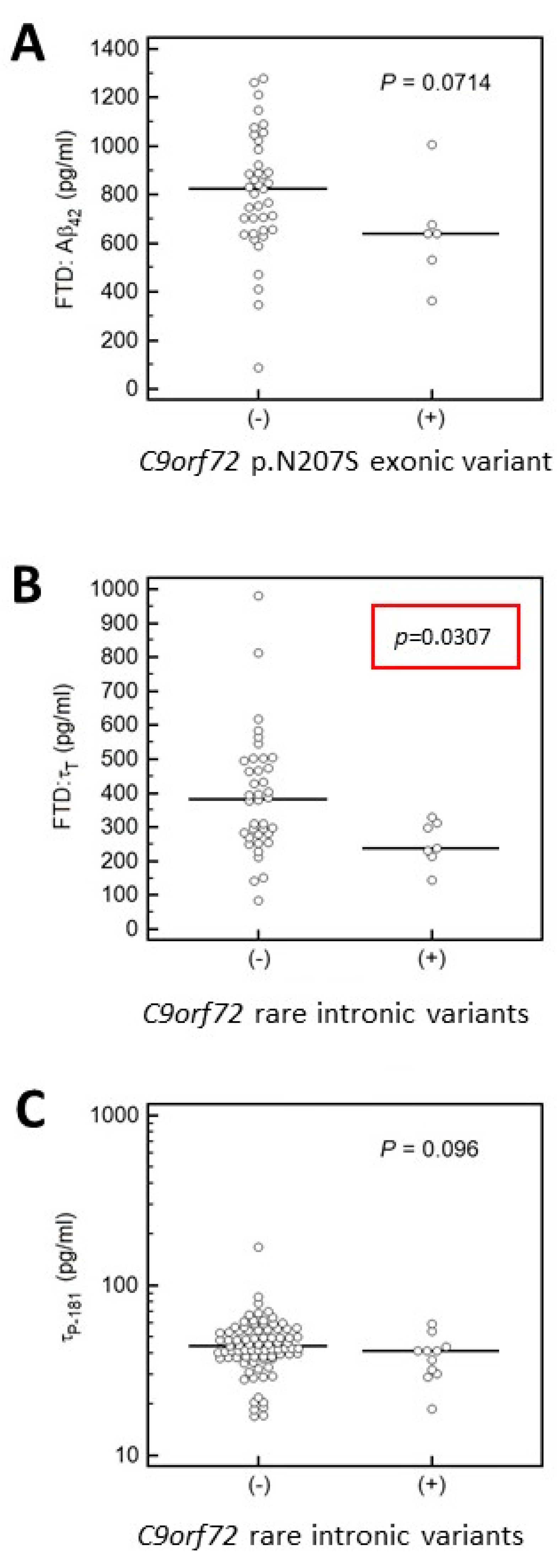
3.4.2. Association of MAPT and APP Gene Variants with CSF Biomarkers
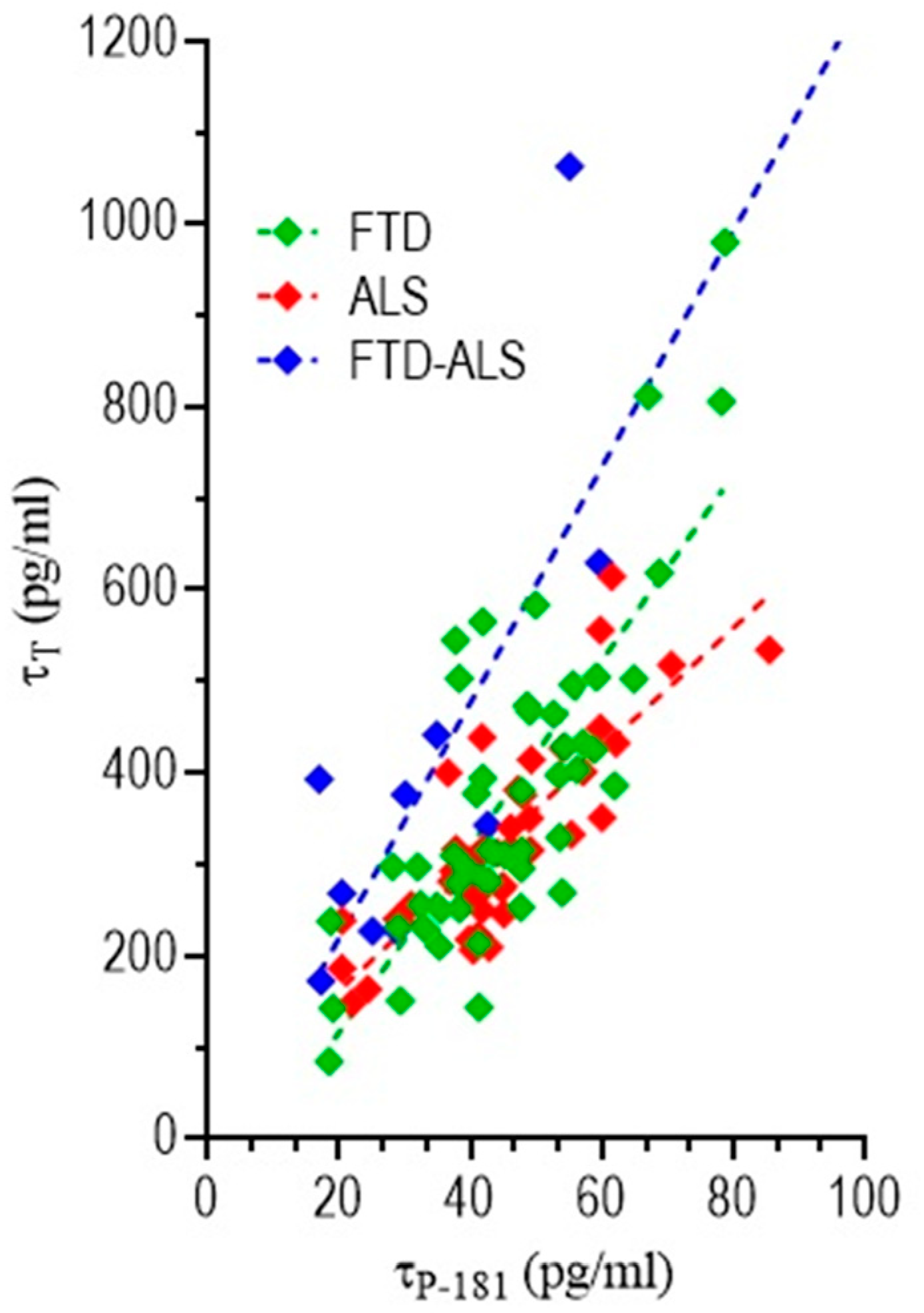
3.4.3. Correlation of CSF Biomarkers Values
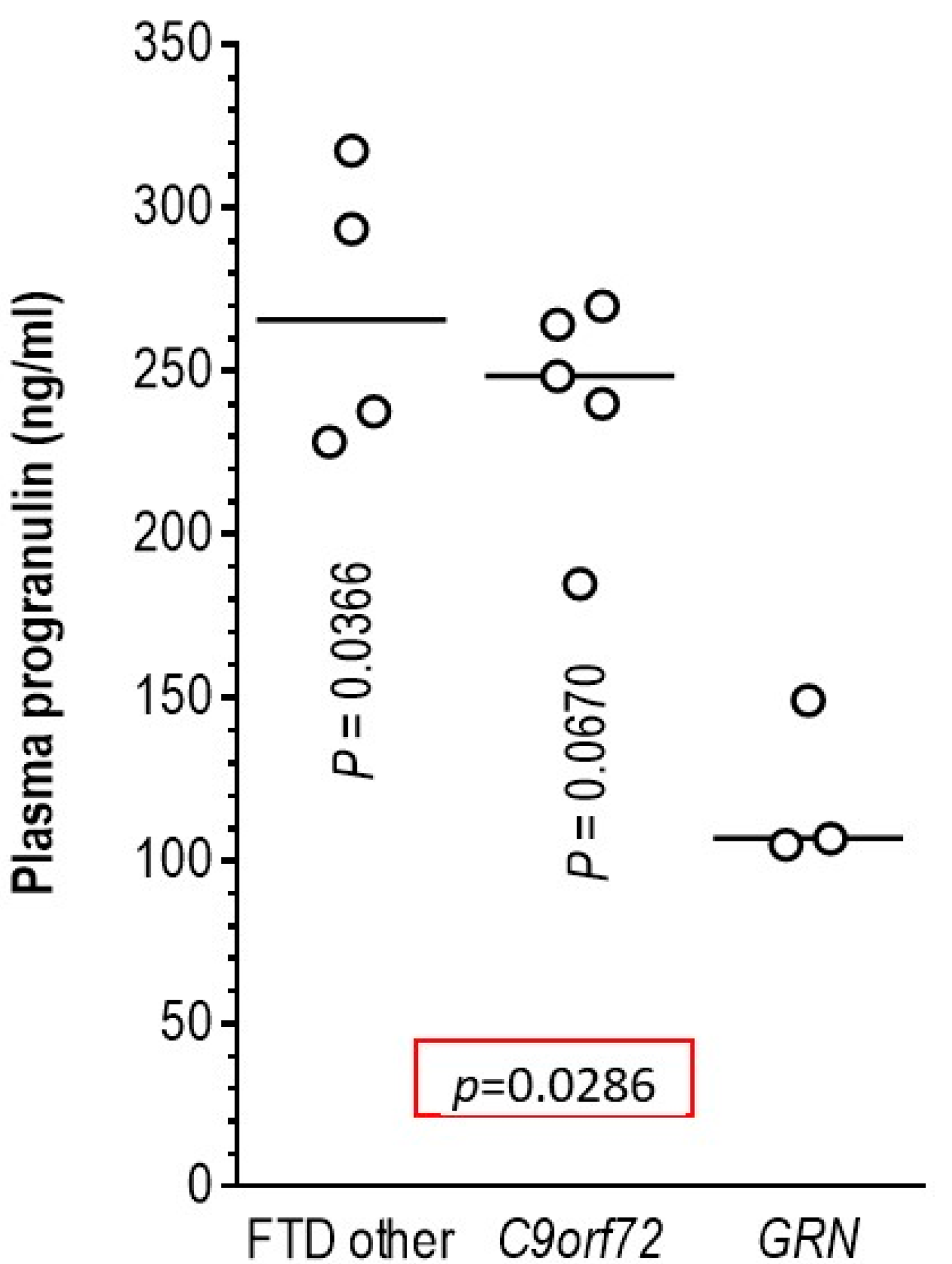
3.5. Association of Pathogenic Variants in the GRN Gene with Plasma Progranulin Levels
4. Discussion
Strengths and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weishaupt, J.H.; Hyman, T.; Dikic, I. Common Molecular Pathways in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Trends Mol. Med. 2016, 22, 769–783. [Google Scholar] [CrossRef]
- Hinz, F.I.; Geschwind, D.H. Molecular Genetics of Neurodegenerative Dementias. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauwendraat, C.; Wilke, C.; Simón-Sánchez, J.; Jansen, I.E.; Reifschneider, A.; Capell, A.; Haass, C.; Castillo-Lizardo, M.; Biskup, S.; Maetzler, W.; et al. The wide genetic landscape of clinical frontotemporal dementia: Systematic combined sequencing of 121 consecutive subjects. Genet. Med. 2018, 20, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, J.S.; Van Deerlin, V.M. Alzheimer’s Disease and Frontotemporal Dementia: The Current State of Genetics and Genetic Testing Since the Advent of Next-Generation Sequencing. Mol. Diagn. Ther. 2018, 22, 505–513. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Pastor, P.; Cooper, B.; Cervantes, S.; Benitez, B.A.; Razquin, C.; Goate, A.; Cruchaga, C.; Ibero-American Alzheimer Disease Genetics Group, R. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res. Ther. 2012, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guven, G.; Lohmann, E.; Bras, J.; Gibbs, J.R.; Gurvit, H.; Bilgic, B.; Hanagasi, H.; Rizzu, P.; Heutink, P.; Emre, M.; et al. Mutation Frequency of the Major Frontotemporal Dementia Genes, MAPT, GRN and C9ORF72 in a Turkish Cohort of Dementia Patients. PLoS ONE 2016, 11, e0162592. [Google Scholar] [CrossRef] [PubMed]
- Pastor, P.; Moreno, F.; Clarimón, J.; Ruiz, A.; Combarros, O.; Calero, M.; de Munain, A.L.; Bullido, M.J.; de Pancorbo, M.M.; Carro, E.; et al. MAPT H1 Haplotype is Associated with Late-Onset Alzheimer’s Disease Risk in APOE ε4 Noncarriers: Results from the Dementia Genetics Spanish Consortium. J. Alzheimers Dis. 2016, 49, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhou, Y.; Tu, L.; Ba, Z.; Huang, J.; Huang, N.; Luo, Y. TDP-43: From Alzheimer’s Disease to Limbic-Predominant Age-Related TDP-43 Encephalopathy. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, B.; Feldman, H.H.; Jacova, C.; DeKosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS–ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal Fluid TAR DNA-Binding Protein 43 Combined with Tau Proteins as a Candidate Biomarker for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Spectrum Disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Kasselimis, D.; Kourtidou, E.; Constantinides, V.; Bougea, A.; Potagas, C.; Evdokimidis, I.; Kapaki, E. Cerebrospinal Fluid Biomarkers as a Diagnostic Tool of the Underlying Pathology of Primary Progressive Aphasia. J. Alzheimers Dis. 2017, 55, 1453–1461. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; McLaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobáGyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimas, G.K.; Paraskevas, G.P.; Zambelis, T.; Karagiaouris, C.; Bourbouli, M.; Bougea, A.; Walter, M.C.; Schumacher, N.U.; Krause, S.; Kapaki, E. The multifaceted clinical presentation of VCP-proteinopathy in a Greek family. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2017, 36, 203–206. [Google Scholar]
- Kapaki, E.; Paraskevas, G.P.; Papageorgiou, S.G.; Bonakis, A.; Kalfakis, N.; Zalonis, I.; Vassilopoulos, D. Diagnostic value of CSF biomarker profile in frontotemporal lobar degeneration. Alzheimers Dis. Assoc. Disord. 2008, 22, 47–53. [Google Scholar] [CrossRef]
- Thijssen, E.H.; La Joie, R.; Strom, A.; Fonseca, C.; Iaccarino, L.; Wolf, A.; Spina, S.; Allen, I.E.; Cobigo, Y.; Heuer, H.; et al. Plasma phosphorylated tau 217 and phosphorylated tau 181 as biomarkers in Alzheimer’s disease and frontotemporal lobar degeneration: A retrospective diagnostic performance study. Lancet Neurol. 2021, 20, 739–752. [Google Scholar] [CrossRef]
- Neddens, J.; Daurer, M.; Loeffler, T.; Alzola Aldamizetxebarria, S.; Flunkert, S.; Hutter-Paier, B. Constant Levels of Tau Phosphorylation in the Brain of htau Mice. Front. Mol. Neurosci. 2020, 13, 136. [Google Scholar] [CrossRef]
- Ghidoni, R.; Benussi, L.; Glionna, M.; Franzoni, M.; Binetti, G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 2008, 71, 1235–1239. [Google Scholar] [CrossRef]
- Sokratous, M.; Lucia, S.; Bourinaris, T.; Marogianni, C.; Arnaoutoglou, M.; Patrikiou, E.; Ralli, S.; Markou, A.; Dardiotis, E.; Houlden, H.; et al. Prevalence of C9orf72 hexanucleotide repeat expansion in Greek patients with sporadic ALS. Amyotroph. Lateral Scler. Front. Degener. 2020, 1–3. [Google Scholar] [CrossRef]
- Kartanou, C.; Karadima, G.; Koutsis, G.; Breza, M.; Papageorgiou, S.G.; Paraskevas, G.P.; Kapaki, E.; Panas, M. Screening for the C9ORF72 repeat expansion in a greek frontotemporal dementia cohort. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 152–154. [Google Scholar] [CrossRef]
- Ramos, E.M.; Koros, C.; Dokuru, D.R.; Van Berlo, V.; Kroupis, C.; Wojta, K.; Wang, Q.; Andronas, N.; Matsi, S.; Beratis, I.N.; et al. Frontotemporal dementia spectrum: First genetic screen in a Greek cohort. Neurobiol. Aging 2019, 75, 224.e221–224.e228. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.P.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef]
- Mok, K.Y.; Koutsis, G.; Schottlaender, L.V.; Polke, J.; Panas, M.; Houlden, H. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol. Aging 2012, 33, 1851.e1851–1851.e1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, M.O.; van Rooij, J.G.J.; Wong, T.H.; Melhem, S.; Verkerk, A.J.M.H.; Kievit, A.J.A.; van Minkelen, R.; Rademakers, R.; Pottier, C.; Kaat, L.D.; et al. Underlying genetic variation in familial frontotemporal dementia: Sequencing of 198 patients. Neurobiol. Aging 2020. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Tosat-Bitrian, C.; Nozal, V.; Nagaraj, S.; Martin-Requero, A.; Martinez, A. TDP-43: A Key Therapeutic Target beyond Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2019, 10, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Valdmanis, P.; Millecamps, S.; Lionnet, C.; Blasco, H.; Mouzat, K.; Daoud, H.; Belzil, V.; Morales, R.; Pageot, N.; et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology 2012, 78, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Caroppo, P.; Camuzat, A.; Guillot-Noel, L.; Thomas-Antérion, C.; Couratier, P.; Wong, T.H.; Teichmann, M.; Golfier, V.; Auriacombe, S.; Belliard, S.; et al. Defining the spectrum of frontotemporal dementias associated with TARDBPmutations. Neurol. Genet. 2016, 2, e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P.; et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Ticozzi, N.; LeClerc, A.L.; van Blitterswijk, M.; Keagle, P.; McKenna-Yasek, D.M.; Sapp, P.C.; Silani, V.; Wills, A.-M.; Brown, R.H.; Landers, J.E. Mutational analysis of TARDBP in neurodegenerative diseases. Neurobiol. Aging 2011, 32, 2096–2099. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.-W.; Lee, M.-J.; Chen, T.-F.; Cheng, T.-W.; Lai, Y.-M.; Hua, M.-S.; Chiu, M.-J. A single nucleotide TDP-43 mutation within a Taiwanese family: A multifaceted demon. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 292–294. [Google Scholar] [CrossRef]
- Ramos, E.M.; Dokuru, D.R.; Van Berlo, V.; Wojta, K.; Wang, Q.; Huang, A.Y.; Deverasetty, S.; Qin, Y.; van Blitterswijk, M.; Jackson, J.; et al. Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimer’s Dement. 2020, 16, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-r.; Hu, W.; Zhan, L.-L.; Wang, C.; Xu, L.-Q.; Lin, M.-T.; Chen, W.-J.; Wang, N.; Zhang, Q.-J. High frequency of the TARDBP p.M337 V mutation among south-eastern Chinese patients with familial amyotrophic lateral sclerosis. BMC Neurol. 2018, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.-Y.; Hsu, J.S.; Teo, K.-C.; Li, Y.; Kung, M.H.W.; Cheah, K.S.E.; Chan, D.; Cheung, K.M.C.; Li, M.; Sham, P.-C.; et al. Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol. Aging 2017, 58, 238.e9. [Google Scholar] [CrossRef]
- Narain, P.; Pandey, A.; Gupta, S.; Gomes, J.; Bhatia, R.; Vivekanandan, P. Targeted next-generation sequencing reveals novel and rare variants in Indian patients with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 71, 265.e9. [Google Scholar] [CrossRef]
- Tamaoka, A.; Arai, M.; Itokawa, M.; Arai, T.; Hasegawa, M.; Tsuchiya, K.; Takuma, H.; Tsuji, H.; Ishii, A.; Watanabe, M.; et al. TDP-43 M337V Mutation in Familial Amyotrophic Lateral Sclerosis in Japan. Intern. Med. 2010, 49, 331–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühnlein, P.; Sperfeld, A.-D.; Vanmassenhove, B.; Van Deerlin, V.; Lee, V.M.-Y.; Trojanowski, J.Q.; Kretzschmar, H.A.; Ludolph, A.C.; Neumann, M. Two German Kindreds With Familial Amyotrophic Lateral Sclerosis Due to TARDBP Mutations. Arch. Neurol. 2008, 65, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Czell, D.; Andersen, P.M.; Morita, M.; Neuwirth, C.; Perren, F.; Weber, M. Phenotypes in Swiss Patients with Familial ALS Carrying TARDBP Mutations. Neurodegener. Dis. 2013, 12, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Nagaoka, U.; Kawata, A.; Mochizuki, Y.; Kawakami, H.; Maruyama, H.; Matsubara, S.; Komori, T. Neuropathological features of Japanese familial amyotrophic lateral sclerosis with p.N352S mutation in TARDBP. Neuropathol. Appl. Neurobiol. 2014, 40, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated Disease Onset with Stabilized Familial Amyotrophic Lateral Sclerosis (ALS)-linked Mutant TDP-43 Proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef] [Green Version]
- Budini, M.; Romano, V.; Avendaño-Vázquez, S.E.; Bembich, S.; Buratti, E.; Baralle, F.E. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res. 2012, 1462, 139–150. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mossevelde, S.; Van der Zee, J.; Cruts, M.; Van Broeckhoven, C. Modifiers of GRN-Associated Frontotemporal Lobar Degeneration. Trends Mol. Med. 2017, 23, 962–979. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.-J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.-M. Primary Progressive Aphasia: A 25-year Retrospective. Alzheimer Dis. Assoc. Disord. 2007, 21, S8–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, J.D.; Ridgway, G.R.; Crutch, S.J.; Hailstone, J.; Goll, J.C.; Clarkson, M.J.; Mead, S.; Beck, J.; Mummery, C.; Ourselin, S.; et al. Progressive logopenic/phonological aphasia: Erosion of the language network. NeuroImage 2010, 49, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliebus, G.; Bigio, E.H.; Gasho, K.; Mishra, M.; Caplan, D.; Mesulam, M.-M.; Geula, C. Asymmetric TDP-43 distribution in primary progressive aphasia with progranulin mutation. Neurology 2010, 74, 1607–1610. [Google Scholar] [CrossRef] [Green Version]
- Sassi, C.; Capozzo, R.; Gibbs, R.; Crews, C.; Zecca, C.; Arcuti, S.; Copetti, M.; Barulli, M.R.; Brescia, V.; Singleton, A.B.; et al. A Novel Splice-Acceptor Site Mutation in GRN (c.709-2 A>T) Causes Frontotemporal Dementia Spectrum in a Large Family from Southern Italy. J. Alzheimers Dis. 2016, 53, 475–485. [Google Scholar] [CrossRef]
- van der Zee, J.; Pirici, D.; Van Langenhove, T.; Engelborghs, S.; Vandenberghe, R.; Hoffmann, M.; Pusswald, G.; Van den Broeck, M.; Peeters, K.; Mattheijssens, M.; et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 2009, 73, 626–632. [Google Scholar] [CrossRef]
- Evangelista, T.; Weihl, C.C.; Kimonis, V.; Lochmüller, H.; Clemen, C.; Deshaies, R.; Evangelista, T.; Eymard, B.; Greensmith, L.; Hilton-Jones, D.; et al. 215th ENMC International Workshop VCP-related multi-system proteinopathy (IBMPFD) 13–15 November 2015, Heemskerk, The Netherlands. Neuromuscul. Disord. 2016, 26, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Al-Obeidi, E.; Al-Tahan, S.; Surampalli, A.; Goyal, N.; Wang, A.K.; Hermann, A.; Omizo, M.; Smith, C.; Mozaffar, T.; Kimonis, V. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet. 2018, 93, 119–125. [Google Scholar] [CrossRef]
- Gitcho, M.A.; Strider, J.; Carter, D.; Taylor-Reinwald, L.; Forman, M.S.; Goate, A.M.; Cairns, N.J. VCP Mutations Causing Frontotemporal Lobar Degeneration Disrupt Localization of TDP-43 and Induce Cell Death. J. Biol. Chem. 2009, 284, 12384–12398. [Google Scholar] [CrossRef] [Green Version]
- Manno, A.; Noguchi, M.; Fukushi, J.; Motohashi, Y.; Kakizuka, A. Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 2010, 15, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Wu, H.C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.J.; Fox, N.C.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP Mutations Induce Mitochondrial Uncoupling and Reduced ATP Levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gui, L.; Zhang, X.; Bulfer, S.L.; Sanghez, V.; Wong, D.E.; Lee, Y.; Lehmann, L.; Lee, J.S.; Shih, P.-Y.; et al. Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E1705–E1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, C.; Kirby, J.; Highley, J.R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; McDermott, C.J.; Shaw, P.J. Novel FUS/TLS Mutations and Pathology in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2010, 67, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Sorarù, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2010, 47, 190–194. [Google Scholar] [CrossRef]
- Lai, S.-L.; Abramzon, Y.; Schymick, J.C.; Stephan, D.A.; Dunckley, T.; Dillman, A.; Cookson, M.; Calvo, A.; Battistini, S.; Giannini, F.; et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2011, 32, 550.e551–550.e554. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Ma, Y.; Liu, X.L.; Chen, L.; Fan, D.S. Better survival in female SOD1-mutant patients with ALS: A study of SOD1-related natural history. Transl. Neurodegener. 2019, 8, 2. [Google Scholar] [CrossRef]
- Andersen, P.M.; Sims, K.B.; Xin, W.W.; Kiely, R.; O’Neill, G.; Ravits, J.; Pioro, E.; Harati, Y.; Brower, R.D.; Levine, J.S.; et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2003, 4, 62–73. [Google Scholar] [CrossRef]
- Lysogorskaia, E.V.; Rossokhin, A.V.; Abramycheva, N.; Zakharova, M.N.; Illarioshkin, S.N. SOD1 gene mutations in patients with amyotrophic lateral sclerosis: Potential for the method of molecular. Mol. Biol. 2013, 47, 861–867. [Google Scholar] [CrossRef]
- Goossens, J.; Bjerke, M.; Van Mossevelde, S.; Van den Bossche, T.; Goeman, J.; De Vil, B.; Sieben, A.; Martin, J.J.; Cras, P.; De Deyn, P.P.; et al. Diagnostic value of cerebrospinal fluid tau, neurofilament, and progranulin in definite frontotemporal lobar degeneration. Alzheimers Res. Ther. 2018, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kämäläinen, A.; Herukka, S.K.; Hartikainen, P.; Helisalmi, S.; Moilanen, V.; Knuuttila, A.; Jansson, L.; Tienari, P.J.; Remes, A.M. Cerebrospinal fluid biomarkers for Alzheimer’s disease in patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis with the C9ORF72 repeat expansion. Dement Geriatr. Cogn. Disord. 2015, 39, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Junttila, A.; Kuvaja, M.; Hartikainen, P.; Siloaho, M.; Helisalmi, S.; Moilanen, V.; Kiviharju, A.; Jansson, L.; Tienari, P.J.; Remes, A.M.; et al. Cerebrospinal Fluid TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Patients with and without the C9ORF72 Hexanucleotide Expansion. Dement Geriatr. Cogn Dis. Extra 2016, 6, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Kauwe, J.S.K.; Cruchaga, C.; Mayo, K.; Fenoglio, C.; Bertelsen, S.; Nowotny, P.; Galimberti, D.; Scarpini, E.; Morris, J.C.; Fagan, A.M.; et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc. Natl. Acad. Sci. USA 2008, 105, 8050–8054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekris, L.M.; Tsuang, D.W.; Peskind, E.R.; Yu, C.E.; Montine, T.J.; Zhang, J.; Zabetian, C.P.; Leverenz, J.B. Cerebrospinal fluid Aβ42 levels and APP processing pathway genes in Parkinson’s disease. Mov Disord 2015, 30, 936–944. [Google Scholar] [CrossRef] [Green Version]
- van Etten, E.S.; Verbeek, M.M.; van der Grond, J.; Zielman, R.; van Rooden, S.; van Zwet, E.W.; van Opstal, A.M.; Haan, J.; Greenberg, S.M.; van Buchem, M.A.; et al. β-Amyloid in CSF: Biomarker for preclinical cerebral amyloid angiopathy. Neurology 2017, 88, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Thordardottir, S.; Kinhult Ståhlbom, A.; Almkvist, O.; Thonberg, H.; Eriksdotter, M.; Zetterberg, H.; Blennow, K.; Graff, C. The effects of different familial Alzheimer’s disease mutations on APP processing in vivo. Alzheimers Res. Ther. 2017, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Cognat, E.; De Schaepdryver, M.; Hugon, J.; Poesen, K.; Paquet, C. Elevated ALS Biomarker Levels in CSF of a FTD Patient at the Presymptomatic Stage of ALS. Alzheimers Dis. Assoc. Disord. 2018, 32, 156–157. [Google Scholar] [CrossRef]
- Ashton, N.J.; Janelidze, S.; Al Khleifat, A.; Leuzy, A.; van der Ende, E.L.; Karikari, T.K.; Benedet, A.L.; Pascoal, T.A.; Lleó, A.; Parnetti, L.; et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat. Commun. 2021, 12, 3400. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.C.; Wang, P.; Staffaroni, A.M.; Heller, C.; Cobigo, Y.; Wolf, A.; Goh, S.-Y.M.; Ljubenkov, P.A.; Heuer, H.W.; Fong, J.C.; et al. Plasma Neurofilament Light for Prediction of Disease Progression in Familial Frontotemporal Lobar Degeneration. Neurology 2021, 96, e2296–e2312. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FTD | ALS | FTD-ALS | p-Value | |
|---|---|---|---|---|
| n (m/f) | 56 (32/24) | 58 (26/32) | 16 (7/9) | NS † |
| Age (y) | 60.2 ± 10.8 | 61.2 ± 11.8 | 60.7 ± 10.7 | NS ‡ |
| Disease Duration (y) | 3.0 (1.3–6.0) a | 1.0 (0.7–2.0) | 3.0 (1.0–4.0) | <0.001 § |
| Family History, 1st degree relative (%) | 16 (28.6) | 9 (15.5) | 4 (25.0) | NS † |
| Patient ID | Sex | Phenotype | Age at Onset | Age at Diagnosis | Family History | MRI | HMPAO-SPECT |
|---|---|---|---|---|---|---|---|
| 1 | F | FTD | 61 | 63 | Sister and 3 cousins ALS | Mild frontal atrophy and left sylvius and temporal pole. | NA |
| 2 | F | bvFTD | 69 | 70 | Mother with dementia | Mild frontal, temporal (L > R) and parietal atrophy | Frontal hypoperfusion (R > L) and right parietal |
| 3 | F | FTD-psychiatric symptoms | 45 | 54 | Mother FTD-ALS Maternal uncle ALS and aunt dementia | Mildfrontal atrophy and white matter lesions | Frontal hypoperfusion (L > R) |
| 4 | M | bvFTD | 51 | 58 | Mother ALS | Bilateral frontal strokes and frontal atrophy | NA |
| 5 | M | bvFTD with psychiatric symptoms | 38 | 41 | No | Frontal, temporal and parietal atrophy | NA |
| 6 | M | ALS | 71 | 72 | No | Mild global atrophy | NA |
| 7 | F | ALS | 59 | 61 | No | Frontal and parietal atrophy | NA |
| 8 | F | ALS | 63 | 64 | Sister ALS | NA | NA |
| 9 | F | ALS | 42 | 43 | Mother and 2 maternal aunts with ALS | NA | NA |
| 10 | M | FTD-ALS | 48 | 50 | Father with FTD | Diffuse atrophy, temporal > frontal | NA |
| 11 | F | FTD-ALS | 43 | 44 | Grandmother and 7/9 uncles with ALS; mother with dementia | Frontal, perisylvian atrophy (L > R), mild increase in signal intensity along the corticospinal tract | Diffuse frontal, temporal and parietal hypoperfusion |
| 12 | F | ALS | 56 | 58 | Mother with ALS; maternal cousin with ALS and C9orf72 (+) | Mild ischemic microangiopathy | NA |
| 13 | M | FTD-ALS | 44 | 45 | Mother and 2 maternal uncles with motor disorder | Frontotemporal atrophy | NA (DATSCAN+) |
| 14 | F | ALS | 41 | 43 | Father with dementia; paternal aunt with ALS | Midline cerebellar dysplasia | NA |
| Patient ID | Sex | Phenotype | Age at Onset | Age at Diagnosis | Family History | Brain MRI | Gene | Transcript | Variant | gnomAD Frequency (%) | rs | CADD Score | MaxEnt-Scan (Splice Site Loss) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | ALS (bulbar onset) | 67 | 69 | 2 brothers; sister; father with ALS | Unremarkable | TARDBP | NM_007375.4 | p.Met337Val (c.1009A>G) | ≤0.001 | 80356730 | 22.4 | - |
| 2 | F | ALS | 63 | 64 | No | Unremarkable | p.Asn352Ser (c.1055A>G) | 0.000 | 80356734 | 18.6 | - | ||
| 3 | M | FTD-ALS | 57 | 60 | No | Frontal, temporal atrophy | p.Ile383Val (c.1147A>G) | 0.002 | 80356740 | 17.2 | - | ||
| 4 | F | PPA | 60.3 | 61 | No | Frontal, temporal atrophy (L > R) | GRN | NM_002087.4 | c.463-2A>G | 0.000 | - | 33.0 | From 3.77 to −4.19 |
| 5 | M | PPA | 50 | 60 | Yes (grandmother) | Frontal, temporal, parietal atrophy (L > R) | c.934-1G>A | 0.000 | - | 34.0 | From 9.63 to 0.88 | ||
| 6 | F | PPA | 61 | 62 | No | Perisylvian atrophy (L > R) | p.Cys482Tyr (c.1445G>A) | 0.000 | - | 30.0 | - | ||
| 7 | M | IBM/FTD | 47 | 63 | Yes (brother ALS; brother bvFTD) | Frontal lobe atrophy | VCP | NM_007126.5 | p.Arg159His (c.476G>A) | ≤0.001 | 121909335 | 23.2 | - |
| 8 | M | IBM/FTD | 58 | 68 | Yes (mother ALS) | - | |||||||
| 9 | M | IBM/FTD/PaD | 43 | 53 | No | - | p.Arg155His (c.464G>A) | 0.000 | 121909329 | 24.6 | - | ||
| 10 | M | ALS | 79 | 81 | No | Unremarkable | FUS | NM_001170634.1 | p.Gly506Val (c.1517G>T) | 0.000 | - | 23.7 | - |
| 11 | M | ALS | 36 | 37 | Yes (brother ALS) | Unremarkable | SOD1 | NM_000454.5 | p.Ser106Leu (c.317C>T) | ~0.000 | 1378590183 | 22.4 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourbouli, M.; Paraskevas, G.P.; Rentzos, M.; Mathioudakis, L.; Zouvelou, V.; Bougea, A.; Tychalas, A.; Kimiskidis, V.K.; Constantinides, V.; Zafeiris, S.; et al. Genotyping and Plasma/Cerebrospinal Fluid Profiling of a Cohort of Frontotemporal Dementia–Amyotrophic Lateral Sclerosis Patients. Brain Sci. 2021, 11, 1239. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091239
Bourbouli M, Paraskevas GP, Rentzos M, Mathioudakis L, Zouvelou V, Bougea A, Tychalas A, Kimiskidis VK, Constantinides V, Zafeiris S, et al. Genotyping and Plasma/Cerebrospinal Fluid Profiling of a Cohort of Frontotemporal Dementia–Amyotrophic Lateral Sclerosis Patients. Brain Sciences. 2021; 11(9):1239. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091239
Chicago/Turabian StyleBourbouli, Mara, George P. Paraskevas, Mihail Rentzos, Lambros Mathioudakis, Vasiliki Zouvelou, Anastasia Bougea, Athanasios Tychalas, Vasilios K. Kimiskidis, Vasilios Constantinides, Spiros Zafeiris, and et al. 2021. "Genotyping and Plasma/Cerebrospinal Fluid Profiling of a Cohort of Frontotemporal Dementia–Amyotrophic Lateral Sclerosis Patients" Brain Sciences 11, no. 9: 1239. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091239