Neural Hyperexcitability in Autism Spectrum Disorders

1
Department of Psychiatry, University of Texas Southwestern, Dallas, TX 75390, USA
2
Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH 45220, USA
*
Author to whom correspondence should be addressed.
Brain Sci. 2017, 7(10), 129; https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci7100129
Submission received: 5 September 2017
/
Revised: 29 September 2017
/
Accepted: 5 October 2017
/
Published: 13 October 2017
(This article belongs to the Special Issue Autism Spectrum Disorder: From Etio-Pathology to Treatment)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Despite the progress that has been made in research on autism spectrum disorders (ASD), the understanding of the biological basis of ASD to identify targets for novel, effective treatment remains limited. One of the leading biological theories of autism is a model of cortical hyperexcitability. While numerous genetic and epigenetic studies support this model, how this particular biological alteration relates to known phenotypes in ASD is not well established. Using examples of sensory processing alterations, this review illustrates how cortical excitability may affect neural processes to result eventually in some core clinical phenotypes in ASD. Applications of the cortical excitability model for translational research and drug development are also discussed.
1. Introduction
Frequent seizures and sensory hyperreactivity in autism spectrum disorders (ASD) suggest cortical hyperexcitability in this population. Accordingly, an elevation in cortical excitability has been proposed to be a fundamental neurobiological characteristic for many patients with ASD [1,2]. While the model of cortical excitability has gained widespread support from genetic and epigenetic studies, its specific effects on observed behavioral phenotypes in ASD are not well established. This review will use examples of sensory processing abnormalities to illustrate how cortical excitability can influence neuronal processes eventually to affect behavioral phenotypes. Changes in cortical excitability, as seen in many animal models and clinical disorders, produce profound effects on sensory functions. This is because sensory systems rely on a dynamic, yet precise, balance of excitatory and inhibitory drives to discriminate and code features of incoming signals. In turn, sensory assessment can be a powerful tool for identifying changes in cortical excitability in clinical research and practice. We will first review evidence that supports elevation in cortical excitability in ASD, then discuss how increased cortical excitability can influence sensory processes via various neural processes. Finally, we will discuss therapeutic and translational applications of the cortical excitability model.
2. Evidence Supporting the Cortical Excitability Model in ASD
Several lines of genetic and epigenetic evidence suggest an elevation in cortical excitability in ASD by documenting gamma-aminobutyric acid (GABA) [3,4,5,6,7,8,9] and glutamate [10,11,12,13] system alterations. First, one of the most commonly observed copy number variations (CNV) in ASD is located on the chromosome 15q11 to q13 region, which contains several genes coding for subunit variants for GABA receptors [4]. Deletions involving these regions have been associated with Angelman or Prader–Willi syndromes, depending on the parental origin of the gene, and these syndromes are known for high incidence of ASD [8]. Duplications in the 15q11–13 region have also been observed by several studies with ASD [14,15,16]. The duplication of genes in this region leads to decreases, not increases, in GABA signaling, and are associated with epilepsy [8,17,18].
Second, the reduced expression of GABA-related genes, such as glutamic acid decarboxylase (GAD) 65 and GAD67 proteins has been observed in the cerebellum and parietal cortex in histological studies of ASD [5]. In addition, lower counts of GABAergic neurons, including Purkinje [19,20] and parvalbumin (PV) positive cells [21] have been reported in ASD. Further, reduced binding at GABA receptors is observed in the hippocampus [3] and anterior and posterior cingulate cortex [6,22]. These differences in gene expression and microstructures indicate a reduced GABA signaling in ASD. Third, increased glutamate receptors and glutamate transfer proteins have been additionally reported in ASD to further support the bias toward excitation. The mRNA levels of glutamate-system-related genes, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) 1 and amino acid transporter 1, are elevated in ASD [23]. Histone modification changes have also been associated with a glutamate receptor gene, glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A), to indicate alterations in the glutamate pathways in ASD [13].
Fourth, macrolevel analyses using magnetic resonance spectroscopy (MRS) are also consistent with elevated excitability in ASD. Lower GABA concentrations have been reported in the motor [24], somatosensory [25], visual [26], and auditory [24,27] cortices. Increases in glutamate concentrations have also been reported in the auditory cortex [28] and occipital [26] areas. Reduced inhibitory effects or paradoxical facilitation following transcranial magnetic stimulation (TMS) have been observed in ASD participants [29,30], which corroborate the findings of neurotransmitter imbalance and the resulting bias toward excitation.
3. Effects of Elevated Excitability on Neurophysiological Function in ASD
3.1. Neural Synchronization
While there is increasingly robust evidence for an elevation in cortical excitability in ASD, cortical excitability has many mechanisms to affect cortical networks, via changes in synaptic formation, neural plasticity, and neural communication [21,31,32]. Thus, the influence of cortical excitability on cognition and perception is multifaceted, and its impact may vary across brain regions, in different behavioral conditions, and across different individuals.
One of the best-documented impacts of increased cortical excitability is on neural communication within brain networks. Control of excitability affects neural activity in both phasic and tonic fashions [32,33]. The fast phasic form of control results from synaptically mediated effects at fast-spiking GABAergic neurons, and resulting inhibitory postsynaptic currents have a very rapid onset that allows for immediate effects on postsynaptic activation [32,34,35]. This rapid phasic influence enables precise control for neural activity timing, and thus is essential in coordinating neural signals to produce rhythmic, synchronized oscillations [36,37]. Synchronized oscillation is critical in coordinating the timing of interactions between neurons, which is essential to optimal neural communication [38,39].
Past electroencephalogram (EEG) studies have shown reduced neural oscillation in ASD [40,41,42], which suggests reduced phasic control. In particular, reduction in gamma oscillation during auditory and linguistic stimulus processing has been replicated in ASD by several groups [43,44,45]. Abnormal gamma oscillation is associated with impairments in language functioning in ASD [46] and is also seen in first-degree relatives [47,48]. These findings indicate that reduction in gamma oscillation is likely a shared neurophysiological feature for the population with hereditary components.
Reduced temporal control over neural activity also predicts more variable neural response timing across trials. Many neurons need to fire in synchrony to produce large enough physiological signal changes to be detected by techniques such as EEG and magnetic resonance imaging (MRI). Reduced neural synchrony results in more variable responses when the system is repeatedly stimulated. ASD participants are known to have greater intertrial variability in Blood-oxygen-level dependent (BOLD) responses during MRI sensory studies, and this is not accounted for by general signal-to-noise ratio differences [49,50]. Similar increases in the intertrial variability in ASD have also been documented in visually evoked responses using EEG techniques [51].
Reduced temporal control over the timing of neural activity is also likely to have effects on behavioral phenotypes in ASD. Because conscious perception of stimuli depends on the duration of cortical sensory responses [52,53], precise control for the onset and offset of the neural response is essential for an accurate perception of stimulus durations. Consistent with this idea, studies of typically developing (TD) adults have shown that the ability to differentiate temporal characteristics of stimuli decreases as levels of cortical excitability increase [54,55]. Hence, lower discrimination ability for temporal information is expected in populations with elevated cortical excitability, such as ASD. This is consistent with impairments in the detection and discrimination of subsecond or fast oscillating stimuli that have been reported in ASD [25,56,57]. These impairments in processing temporal aspects of stimuli appear to be more severe in individuals with ASD with clinically evident sensory hypersensitivity [56] or lower GABA levels in the relevant sensory cortex [25].
3.2. Neural Response Scaling
In addition to the fast phasic influence, cortical excitability can affect gain control of pyramidal cell activity in a more tonic fashion. This occurs through the generation of inhibitory currents via drive from extrasynaptic GABA receptors, which are located on soma, dendrites, and axons that are distant from synaptic neurotransmitter release sites [31,32]. These inhibitory currents affect levels of resting neuronal depolarization, and thus modulate neuronal readiness to produce action potentials [32,33]. Hence, tonic excitability control affects the likelihood of local neural communication and the magnitude of response, rather than the precise timing of neural communication. This effect is typically observed as a change in scaling of response magnitude per stimulation (i.e., response gain), which results in an enhanced response at middle- to high-intensity stimulation and reduced response saturation [33]. Drugs that affect neuronal excitability are known to produce similar disproportionate changes at middle- to high-intensity stimulation [58,59,60]. Consistently, some populations with known cortical hyperexcitability, such as epilepsy, show abnormal regulation of neural system output that is characterized by disproportionate scaling and reduced response saturation [61,62].
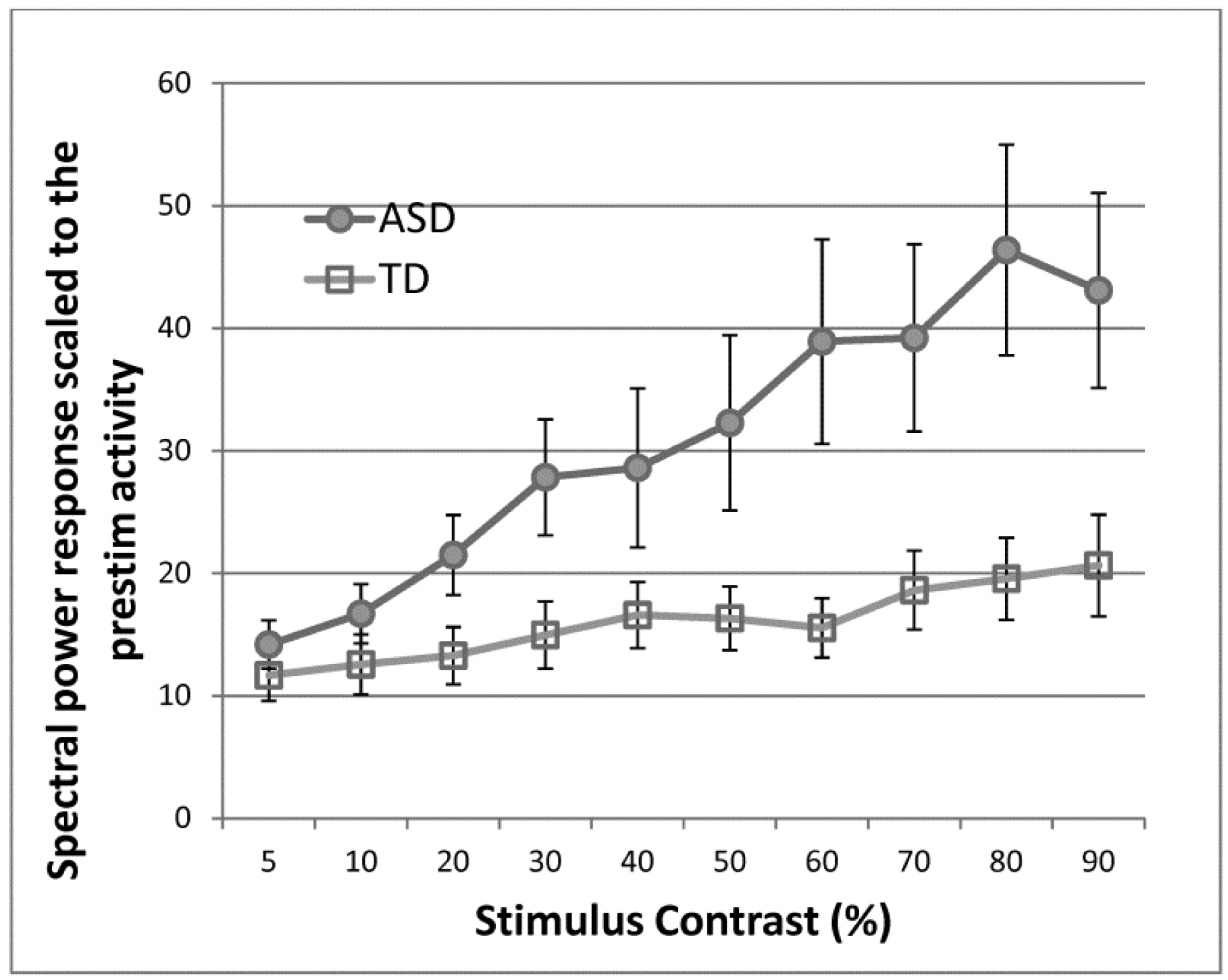
We have shown that abnormal response scaling under visual stimulus contrast manipulation is present in ASD participants who are seizure-free [63]. We measured steady-state visually evoked responses with a wide range of stimulus contrasts in participants with ASD and age- and IQ-matched TD control participants (Figure 1). The spectral response at the stimulus frequency was greater in ASD than in control participants. More importantly, ASD participants showed a greater rate of response increase as the stimulus contrast increased, which is similar to the pattern observed in epilepsy patients.
Additional evidence indicating the difficulty in regulating neural output includes enhanced activation in the visual cortex in functional MRI (fMRI) studies, where suprathreshold, high-contrast stimuli were used [64,65]. Enhanced visual responses are considered as one of the most common fMRI findings in ASD [65]. These reports of heightened neural responses to sensory events may relate to clinical reports of sensory hypersensitivity or overresponsivity in everyday situations.
Psychophysical studies in ASD are consistent with a disproportionately elevated neural scaling when a wide range of suprathreshold stimuli are used. Greater sensitivity to middle- to high-intensity stimuli has been documented by lower thresholds for blink startle responses to loud sounds [66,67] and compressed auditory dynamic range, where stimuli were judged as being uncomfortable at a lower intensity [68]. Visual motion perception is also enhanced under high-contrast conditions in ASD [69].
3.3. Neural Habituation and Adaptation
Problems with the dysregulation of neural response amplitude may be exacerbated when the sensory system is repeatedly stimulated. Repeated stimulation typically results in habituation. Habituation, or repetition suppression, refers to a reduction of neural activity when stimuli are repeated. This is an important neural mechanism to differentiate “old” and “new” stimuli to code history with the particular stimulus. ASD individuals are known to show less neural habituation [70], or even increased responses [71], to repeated stimuli. This trait for the reduced ability for habituation may be familial [72] and is also seen in a related genetic condition, Fragile X [73].
Although habituation is a complex phenomenon that is probably related to both perceptual and postperceptual processes, reduced habituation could contribute to both hyper- and hyposensitivity to sensory events that are observed in ASD [72]. Reduced habituation results in smaller neural signal differences between new and old stimuli. Thus, old repeated stimuli do not earn an “old” status and continue to evoke relatively high responses like “new” stimuli, which may lead to more exaggerated behavioral responses. At the same time, this could result in the failure to assign a “new” status to new stimuli, which could lead to failure in executing appropriate orienting or other exploratory behaviors that are typically performed with new stimuli. This lack of orienting and exploratory behaviors to new stimuli may be regarded as sensory hyposensitivity.
ASD individuals also show reduced adaptation [64,74,75,76]. Habituation and adaptation are related concepts, and both are a part of activity-dependent neural plasticity to adjust neural responses dynamically for the optimal processing of sensory information [77,78,79]. While habituation paradigms typically examine response suppression to a single class of repeating stimuli, adaptation paradigms typically investigate changes in response selectivity of the system after extensive exposure to stimuli. The response property changes because extensive exposure could change the balance between sets of neurons with opposing response selectivity. Sensory neurons with different response selectivity are known to inhibit each other and also to receive inhibitory input from each other; inhibitory influence over other neurons is a function of the activity level of the neuron. This process of mutual inhibition, called opponency, instantiates inhibitory interactions within local circuitries. Opponency is especially critical in determining how much influence single neurons have in the population response because each neuron’s signal is weighted by both the relevant activation and inhibition processes [80,81]. Thus, the disruption of opponency by sensory adaptation could change how the sensory system responds to incoming signals.
Sensory adaptation has been well studied in the context of visual motion perception, and we have shown that sensory adaptation to visual motion is reduced in ASD [64]. In a typical experiment to induce visual motion adaptation, participants view directional movement for a prolonged period. After prolonged viewing, visual neurons that are sensitive to the movement direction experience response saturation. As a result, both their activity and their ability to maintain inhibitory influence over other neurons with opposite direction tuning decrease [82,83]. This temporary disruption in inhibitory interaction results in a relative increase in activation, via disinhibition, in the neurons with opposing direction tuning. This relative disinhibition yields the illusory perception of movement in the opposite direction (motion aftereffect, also called the waterfall illusion) [82,84]. Time course of activity in V5, extrastriate areas known to be sensitive to visual movement, is critically related to the motion aftereffect [85,86]. Decay of the V5 BOLD signal after motion adaptation is related to dissipation of the disinhibition resulting from the temporal disruption of inhibitory interactions (thus resumption of mutual inhibition) [85,86]. This V5 BOLD response after motion adaptation decays faster in ASD individuals [64]. Because the magnitude of disinhibition depends on the strength of the existing inhibitory interactions, a faster recovery suggests reduced inhibitory interactions. Consistent with these concepts, lower GABA levels obtained from MRS are associated with an increased magnitude and a shorter duration of BOLD responses in TD adults after viewing visual stimuli [87,88].
Similar principles based on disruption in mutual inhibition apply to adaptation to other stimulus features, such as orientation [89,90,91], and even higher-level stimuli, such as faces [92]. Laboratory studies with tactile stimulation have reported reduced neural adaptation in both adults [74] and children [76] with ASD. ASD individuals are also known to have less adaptation to faces [75]. Adaptation decreases neural responses to the adapted stimulus, while increasing responses to one that is orthogonal to the adapted stimuli. Thus, adaptation distorts the response property of the system and yields to temporary hyper- and hyposensitivity along the relevant stimulus dimension.
4. Individual Differences
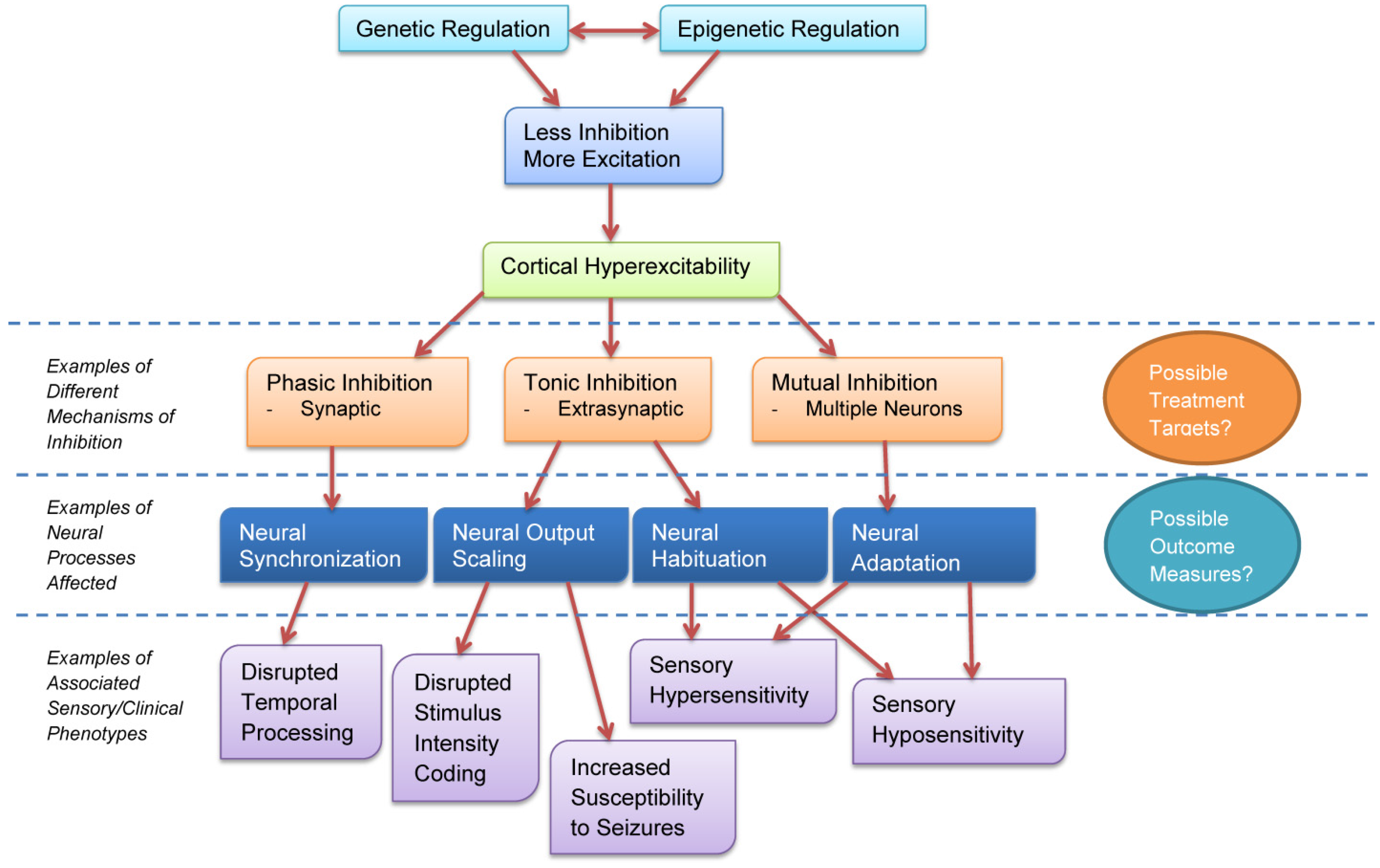
We have discussed how cortical excitability influences neural synchronization, output scaling, habituation, and adaptation, and how these neural processes are altered in ASD. These neural changes are consistent with documented perceptual changes in ASD and are consistent with sensory abnormalities observed clinically. Figure 2 shows a schematic diagram of how these different neural changes affect behavioral phenotypes in ASD.
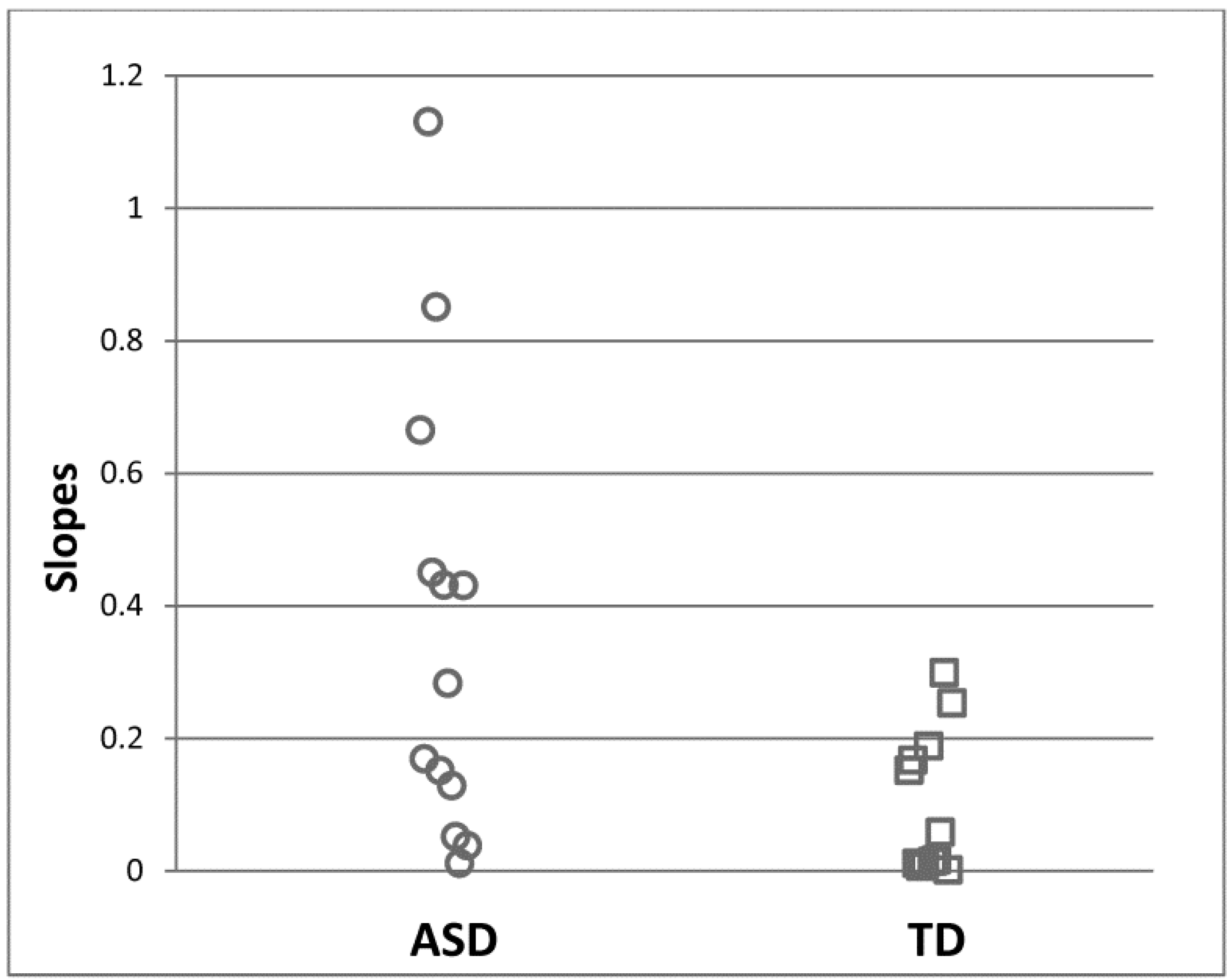
It is worth noting that alterations in cortical excitability seem to exist to varying degrees in individuals with ASD, and only a subset of affected individuals may have neuronal hyperreactivity [29,30,63]. In the previously discussed steady-state study of visually evoked potentials [63], individual slopes for response increases to the visual contrast manipulation were quantified by fitting linear functions. The linear slope was greater in the ASD group than in the control group, indicating greater neural responsivity to visual stimuli at the group level. Here, 44% of ASD participants had slopes larger than any control participants (Figure 3), but the remaining participants had slopes that overlapped with the control group. This observation suggests that only a subgroup of individuals with ASD have neuronal hypersensitivity. Other studies also found that changes in cortical excitability were not universally observed in the participants. Post-TMS inhibitory effects were reduced in 37% of participants [30] and were more common in individuals with a history of early language delay [29]. Thus, there appears to be a fairly large range of cortical excitability in ASD, and perhaps only a specific subgroup with distinct clinical characteristics shows elevated cortical excitability. Thus, the ability to capture individual differences in cortical excitability may have a significant impact when cortical excitability is considered as a treatment target, as patients might be stratified before a treatment based on their pretreatment neurophysiological testing.
In addition to individual differences in overall levels of cortical excitability, further examining specific aspects of cortical excitability may clarify which potential neural mechanisms contribute to cortical hyperexcitability in ASD and possibly inform us about biological heterogeneity in the disorder. Different aspects of excitability control can result from distinct genetic and epigenetic factors, as well as atypical developmental trajectories [31,32]. For example, phasic inhibition is critical for neural synchronization and is primarily associated with GAD65, which is predominantly expressed in axons [93]. Tonic inhibition is critical in regulating neural output and is associated with GAD67, which is primarily expressed in soma [93]. Further, GAD65 and GAD67 have different regional distributions and developmental trajectories, suggesting that they might have distinctive effects on brain systems [94,95]. Thus, differentiating between specific effects of cortical excitability could provide new insights into the pathophysiology and possible pharmacological targets for individuals with ASD.
5. Cross-Species Comparisons
It has been shown that many preclinical models of ASD have abnormal sensory processing and altered balances for excitation and inhibition. The similar elevation of cortical excitability to the human data warrants well-controlled cross-species studies using identical tasks to seek parallel neurophysiological characteristics, such as changes in neural synchronization and neural output scaling. In particular, EEG and event related potentials (ERP) provide excellent tools for the investigation because homologues of human ERPs and oscillatory activity have been identified in rodents. This provides a particularly strong framework to link findings from rodent models to studies of human diseases and to develop novel translational research programs. ERP and oscillatory activity have a high reliability, which is suited for such investigations [96,97,98,99]. The framework may also be useful for investigating the molecular basis for specific symptoms in syndromic forms of ASD. For example, both human and rodent forms of Fragile X show sensory hypersensitivity [100,101]. Increased amplitude of ERP components, such as N1 and P2, are observed in both human participants and the fmr1 knock out mouse model of Fragile X [101]. The abnormally large ERP response becomes normalized by GABA agonists [102]. The enhanced neural and sensory responses are also consistent with the findings from slice physiology that show decreases in PV-positive inhibitory neuron activity in Fragile X [103]. The cross-species homologue at multiple levels of analysis brings opportunities to address cellular and circuit level abnormalities in the disorder to facilitate greatly the development of pharmacological targets.
6. Conclusions
Elevation in cortical excitability is observed in ASD at genetic, epigenetic, neural, and behavioral levels. While changes in cortical excitability affect general cognitive function, cortical excitability has especially profound effects on sensory phenotypes in ASD. The effect on sensory phenotypes is complex, affecting sensory processes through multiple different neural processes, which may contribute to the heterogeneous expression of symptoms. Despite the complex effect, close homologues in human and animal models offer great promise for developing models of cortical excitability and their neural mechanisms and creating novel therapeutic strategies. When combined with the ability to capture individual differences, these measures of cortical excitability have high potential for predicting and evaluating therapeutic outcomes to such therapies.
Acknowledgments
This work was supported by NIH grants (K01MH087720 and U54HD082008).
Author Contributions
Y.T. was involved in the initial conceptualization and drafting of the manuscript. J.A.S. was involved in revising the manuscript.
Conflicts of Interest
J.A.S. serves on an advisory board to Takeda Pharmaceuticals for work that is unrelated to this manuscript. The authors report no actual or potential conflict of interest in relation to this manuscript.
References
- Rubenstein, J.L. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr. Opin. Neurol. 2010, 23, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Blatt, G.J.; Fitzgerald, C.M.; Guptill, J.T.; Booker, A.B.; Kemper, T.L.; Bauman, M.L. Density and distribution of hippocampal neurotransmitter receptors in autism: An autoradiographic study. J. Autism Dev. Disord. 2001, 31, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, S.; Horder, J.; Inkster, B.; Mendez, M.A.; Murphy, D.G.; Nutt, D.J. GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 2012, 36, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.C.; Realmuto, G.R. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Oblak, A.; Gibbs, T.T.; Blatt, G.J. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2009, 2, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.; Soghomonian, J.J.; Blatt, G.J. Increased GAD67 mRNA expression in cerebellar interneurons in autism: Implications for Purkinje cell dysfunction. J. Neurosci. Res. 2008, 86, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; LaSalle, J.M. Epigenetic dysregulation of 15q11–13 GABAA receptor genes in autism. In The Neurochemical Basis of Autism: From Molecules to Minicolumns, 1st ed.; Blatt, G.J., Ed.; Springer: Boston, MA, USA, 2010; pp. 113–127. [Google Scholar]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Burnashev, N.; Szepetowski, P. NMDA receptor subunit mutations in neurodevelopmental disorders. Curr. Opin. Pharmacol. 2014, 20C, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Rojas, D.C. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural Transm. 2014, 121, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, M.J.; Ey, E.; Wegener, S.; Bockmann, J.; Stempel, A.V.; Kuebler, A.; Janssen, A.L.; Udvardi, P.T.; Shiban, E.; Spilker, C.; et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Poschmann, J.; Cruz-Herrera del Rosario, R.; Parikshak, N.N.; Hajan, H.S.; Kumar, V.; Ramasamy, R.; Belgard, T.G.; Elanggovan, B.; Wong, C.C.Y.; et al. Histone acetylome-wide association study of autism spectrum disorder. Cell 2016, 167, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Silverman, J.M.; Smith, C.J.; Greenberg, D.A.; Kilifarski, M.; Reichert, J.; Cook, E.H., Jr.; Fang, Y.; Song, C.Y.; Vitale, R. Association between a GABRB3 polymorphism and autism. Mol. Psychiatry 2002, 7, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H., Jr.; Courchesne, R.Y.; Cox, N.J.; Lord, C.; Gonen, D.; Guter, S.J.; Lincoln, A.; Nix, K.; Haas, R.; Leventhal, B.L.; et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11–13 markers. Am. J. Hum. Genet. 1998, 62, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- McCauley, J.L.; Olson, L.M.; Delahanty, R.; Amin, T.; Nurmi, E.L.; Organ, E.L.; Jacobs, M.M.; Folstein, S.E.; Haines, J.L.; Sutcliffe, J.S. A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2004, 131B, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.W.; Haas-Givler, B.; Finucane, B. A longitudinal follow-up study of autistic symptoms in children and adults with duplications of 15q11–13. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, Y.; Provenzano, G.; Casarosa, S. Neurobiological bases of autism-epilepsy comorbidity: A focus on excitation/inhibition imbalance. Eur. J. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Palmen, S.J.; van Engeland, H.; Hof, P.R.; Schmitz, C. Neuropathological findings in autism. Brain 2004, 127 Pt 12, 2572–2583. [Google Scholar] [CrossRef] [PubMed]
- Kemper, T.L.; Bauman, M. Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol. 1998, 57, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Zikopoulos, B.; Barbas, H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 2013, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L.; Gibbs, T.T.; Blatt, G.J. Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J. Neurochem. 2010, 114, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Gaetz, W.; Bloy, L.; Wang, D.J.; Port, R.G.; Blaskey, L.; Levy, S.E.; Roberts, T.P. GABA estimation in the brains of children on the autism spectrum: Measurement precision and regional cortical variation. NeuroImage 2014, 86, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Puts, N.A.J.; Wodka, E.L.; Harris, A.D.; Crocetti, D.; Tommerdahl, M.; Mostofsky, S.H.; Edden, R.A.E. Reduced GABA and altered somatosensory function in children with autism spectrum disorder. Autism Res. 2017, 10, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Drenthen, G.S.; Barendse, E.M.; Aldenkamp, A.P.; Van Veenendaal, T.M.; Puts, N.A.; Edden, R.A.; Zinger, S.; Thoonen, G.; Hendriks, M.P.; Kessels, R.P.; et al. Altered neurotransmitter metabolism in adolescents with high-functioning autism. Psychiatry Res. 2016, 256, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Rojas, D.C.; Singel, D.; Steinmetz, S.; Hepburn, S.; Brown, M.S. Decreased left perisylvian GABA concentration in children with autism and unaffected siblings. NeuroImage 2014, 86, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Singel, D.; Hepburn, S.; Rojas, D.C. Increased glutamate concentration in the auditory cortex of persons with autism and first-degree relatives: A (1)H-MRS study. Autism Res. 2013, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Enticott, P.G.; Kennedy, H.A.; Rinehart, N.J.; Tonge, B.J.; Bradshaw, J.L.; Fitzgerald, P.B. GABAergic activity in autism spectrum disorders: An investigation of cortical inhibition via transcranial magnetic stimulation. Neuropharmacology 2013, 68, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Oberman, L.M.; Pascual-Leone, A.; Rotenberg, A. Modulation of corticospinal excitability by transcranial magnetic stimulation in children and adolescents with autism spectrum disorder. Front. Hum. Neurosci. 2014, 8, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deidda, G.; Bozarth, I.F.; Cancedda, L. Modulation of GABAergic transmission in development and neurodevelopmental disorders: Investigating physiology and pathology to gain therapeutic perspectives. Front. Cell. Neurosci. 2014, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Walker, M.C.; Kullmann, D.M.; Silver, R.A. Tonically active GABA A receptors: Modulating gain and maintaining the tone. Trends Neurosci. 2004, 27, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Nusser, Z.; Cull-Candy, S.; Farrant, M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 1997, 19, 697–709. [Google Scholar] [CrossRef]
- Brickley, S.G.; Cull-Candy, S.G.; Farrant, M. Single-channel properties of synaptic and extrasynaptic GABAA receptors suggest differential targeting of receptor subtypes. J. Neurosci. 1999, 19, 2960–2973. [Google Scholar] [PubMed]
- Klausberger, T.; Magill, P.J.; Marton, L.F.; Roberts, J.D.; Cobden, P.M.; Buzsaki, G.; Somogyi, P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 2003, 421, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Tukker, J.J.; Fuentealba, P.; Hartwich, K.; Somogyi, P.; Klausberger, T. Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J. Neurosci. 2007, 27, 8184–8189. [Google Scholar] [CrossRef] [PubMed]
- Fries, P. A mechanism for cognitive dynamics: Neuronal communication through neuronal coherence. Trends Cogn. Sci. 2005, 9, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Klimesch, W.; Sauseng, P.; Hanslmayr, S. EEG alpha oscillations: The inhibition-timing hypothesis. Brain Res. Rev. 2007, 53, 63–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barstein, J.; Ethridge, L.E.; Mosconi, M.W.; Takarae, Y.; Sweeney, J.A. Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Grutzner, C.; Bolte, S.; Wibral, M.; Tozman, T.; Schlitt, S.; Poustka, F.; Singer, W.; Freitag, C.M.; Uhlhaas, P.J. Impaired gamma-band activity during perceptual organization in adults with autism spectrum disorders: Evidence for dysfunctional network activity in frontal-posterior cortices. J. Neurosci. 2012, 32, 9563–9573. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.R.; Simpson, A.; Till, A.; Simmons, K.; Puzzo, I. Beta event-related desynchronization as an index of individual differences in processing human facial expression: Further investigations of autistic traits in typically developing adults. Front. Hum. Neurosci. 2013, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Port, R.G.; Edgar, J.C.; Ku, M.; Bloy, L.; Murray, R.; Blaskey, L.; Levy, S.E.; Roberts, T.P. Maturation of auditory neural processes in autism spectrum disorder—A longitudinal MEG study. NeuroImage Clin. 2016, 11, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Rojas, D.C.; Maharajh, K.; Teale, P.; Rogers, S.J. Reduced neural synchronization of gamma-band MEG oscillations in first-degree relatives of children with autism. BMC Psychiatry 2008, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Jochaut, D.; Lehongre, K.; Saitovitch, A.; Devauchelle, A.D.; Olasagasti, I.; Chabane, N.; Zilbovicius, M.; Giraud, A.L. Atypical coordination of cortical oscillations in response to speech in autism. Front. Hum. Neurosci. 2015, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.C.; Khan, S.Y.; Blaskey, L.; Chow, V.Y.; Rey, M.; Gaetz, W.; Cannon, K.M.; Monroe, J.F.; Cornew, L.; Qasmieh, S.; et al. Neuromagnetic oscillations predict evoked-response latency delays and core language deficits in autism spectrum disorders. J. Autism Dev. Disord. 2015, 45, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Rojas, D.C.; Teale, P.D.; Maharajh, K.; Kronberg, E.; Youngpeter, K.; Wilson, L.B.; Wallace, A.; Hepburn, S. Transient and steady-state auditory gamma-band responses in first-degree relatives of people with autism spectrum disorder. Mol. Autism 2011, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.L.; Hepburn, S.; Winterrowd, E.; Schmidt, G.L.; Rojas, D.C. Abnormalities in gamma-band responses to language stimuli in first-degree relatives of children with autism spectrum disorder: An MEG study. BMC Psychiatry 2012, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Dinstein, I.; Heeger, D.J.; Lorenzi, L.; Minshew, N.J.; Malach, R.; Behrmann, M. Unreliable evoked responses in autism. Neuron 2012, 75, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Haigh, S.M.; Heeger, D.J.; Dinstein, I.; Minshew, N.; Behrmann, M. Cortical Variability in the Sensory-Evoked Response in Autism. J. Autism Dev. Disord. 2014. [Google Scholar] [CrossRef] [PubMed]
- Milne, E. Increased intra-participant variability in children with autistic spectrum disorders: Evidence from single-trial analysis of evoked EEG. Front. Psychol. 2011, 2, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagleman, D.M.; Pariyadath, V. Is subjective duration a signature of coding efficiency? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2009, 364, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.P.; Sommer, M.A. Neuronal correlates of visual time perception at brief timescales. Proc. Natl. Acad. Sci. USA 2013, 110, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Giersch, A.; Herzog, M.H. Lorazepam strongly prolongs visual information processing. Neuropsychopharmacology 2004, 29, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Terhune, D.B.; Russo, S.; Near, J.; Stagg, C.J.; Cohen Kadosh, R. GABA predicts time perception. J. Neurosci. 2014, 34, 4364–4370. [Google Scholar] [CrossRef] [PubMed]
- Bhatara, A.; Babikian, T.; Laugeson, E.; Tachdjian, R.; Sininger, Y.S. Impaired timing and frequency discrimination in high-functioning autism spectrum disorders. J. Autism Dev. Disord. 2013, 43, 2312–2328. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, D.A.; Gordon Green, C.; Flores, H.; Burack, J.A. Time estimation among low-functioning individuals with autism spectrum disorders: Evidence of poor sensitivity to variability of short durations. Autism Res. 2014, 7, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Berardi, N.; Morrone, M.C. The role of gamma-aminobutyric acid mediated inhibition in the response properties of cat lateral geniculate nucleus neurones. J. Physiol. 1984, 357, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.; Sato, H.; Daw, N. The effect of varying stimulus intensity on NMDA-receptor activity in cat visual cortex. J. Neurophysiol. 1990, 64, 1413–1428. [Google Scholar] [PubMed]
- Katzner, S.; Busse, L.; Carandini, M. GABAA inhibition controls response gain in visual cortex. J. Neurosci. 2011, 31, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Porciatti, V.; Bonanni, P.; Fiorentini, A.; Guerrini, R. Lack of cortical contrast gain control in human photosensitive epilepsy. Nat. Neurosci. 2000, 3, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.J.; Norcia, A.M.; Ales, J.M.; Wade, A.R. Contrast gain control abnormalities in idiopathic generalized epilepsy. Ann. Neurol. 2011, 70, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Takarae, Y.; Sablich, S.R.; White, S.P.; Sweeney, J.A. Neurophysiological hyperresponsivity to sensory input in autism spectrum disorders. J. Neurodev. Disord. 2016, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Takarae, Y.; Luna, B.; Minshew, N.J.; Sweeney, J.A. Visual motion processing and visual sensorimotor control in autism. J. Int. Neuropsychol. Soc. 2014, 20, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Samson, F.; Mottron, L.; Soulieres, I.; Zeffiro, T.A. Enhanced visual functioning in autism: An ALE meta-analysis. Hum. Brain Mapp. 2012, 33, 1553–1581. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Nakahachi, T.; Komatsu, S.; Ogino, K.; Iida, Y.; Kamio, Y. Hyperreactivity to weak acoustic stimuli and prolonged acoustic startle latency in children with autism spectrum disorders. Mol. Autism 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Komatsu, S.; Nakahachi, T.; Ogino, K.; Kamio, Y. Relationship of the acoustic startle response and its modulation to emotional and behavioral problems in typical development children and those with autism spectrum disorders. J. Autism Dev. Disord. 2016, 46, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Khalfa, S.; Bruneau, N.; Roge, B.; Georgieff, N.; Veuillet, E.; Adrien, J.L.; Barthelemy, C.; Collet, L. Increased perception of loudness in autism. Hear. Res. 2004, 198, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Foss-Feig, J.H.; Tadin, D.; Schauder, K.B.; Cascio, C.J. A substantial and unexpected enhancement of motion perception in autism. J. Neurosci. 2013, 33, 8243–8249. [Google Scholar] [CrossRef] [PubMed]
- Kemner, C.; Oranje, B.; Verbaten, M.N.; van Engeland, H. Normal P50 gating in children with autism. J. Clin. Psychiatry 2002, 63, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Kagitani-Shimono, K.; Sugata, H.; Hirata, M.; Hanaie, R.; Nagatani, F.; Tachibana, M.; Tominaga, K.; Mohri, I.; Taniike, M. Progressively increased M50 responses to repeated sounds in autism spectrum disorder with auditory hypersensitivity: A magnetoencephalographic study. PLoS ONE 2014, 9, e102599. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, J.A.; Kushnerenko, E.; Tomalski, P.; Davies, K.; Ribeiro, H.; Johnson, M.H. Differential habituation to repeated sounds in infants at high risk for autism. Neuroreport 2011, 22, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Byerly, M.J.; Sweeney, J.A. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry 2016, 6, e787. [Google Scholar] [CrossRef] [PubMed]
- Tommerdahl, M.; Tannan, V.; Cascio, C.J.; Baranek, G.T.; Whitsel, B.L. Vibrotactile adaptation fails to enhance spatial localization in adults with autism. Brain Res. 2007, 1154, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, E.; Jeffery, L.; Burr, D.; Rhodes, G. Abnormal adaptive face-coding mechanisms in children with autism spectrum disorder. Curr. Biol. 2007, 17, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Puts, N.A.; Wodka, E.L.; Tommerdahl, M.; Mostofsky, S.H.; Edden, R.A. Impaired tactile processing in children with autism spectrum disorder. J. Neurophysiol. 2014, 111, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.L.; Sun, P.; Waggoner, R.A.; Ueno, K.; Tanaka, K.; Cheng, K. Contrast adaptation and representation in human early visual cortex. Neuron 2005, 47, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Menzel, C.; Hayn-Leichsenring, G.U.; Redies, C.; Nemeth, K.; Kovacs, G. When noise is beneficial for sensory encoding: Noise adaptation can improve face processing. Brain Cogn. 2017, 117, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Jamann, N.; Jordan, M.; Engelhardt, M. Activity-dependent axonal plasticity in sensory systems. Neuroscience 2017. [Google Scholar] [CrossRef] [PubMed]
- Bair, W.; Cavanaugh, J.R.; Movshon, J.A. Time course and time-distance relationships for surround suppression in macaque V1 neurons. J. Neurosci. 2003, 23, 7690–7701. [Google Scholar] [PubMed]
- Heeger, D.J.; Boynton, G.M.; Demb, J.B.; Seidemann, E.; Newsome, W.T. Motion opponency in visual cortex. J. Neurosci. 1999, 19, 7162–7174. [Google Scholar] [PubMed]
- Krekelberg, B.; Boynton, G.M.; van Wezel, R.J. Adaptation: From single cells to BOLD signals. Trends Neurosci. 2006, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Van Wezel, R.J.; Britten, K.H. Motion adaptation in area MT. J. Neurophysiol. 2002, 88, 3469–3476. [Google Scholar] [CrossRef] [PubMed]
- Anstis, S.; Verstraten, F.A.; Mather, G. The motion aftereffect. Trends Cogn. Sci. 1998, 2, 111–117. [Google Scholar] [CrossRef]
- Tootell, R.B.; Reppas, J.B.; Dale, A.M.; Look, R.B.; Sereno, M.I.; Malach, R.; Brady, T.J.; Rosen, B.R. Visual motion aftereffect in human cortical area MT revealed by functional magnetic resonance imaging. Nature 1995, 375, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, M.; Kozak, L.R.; Formisano, E.; Teixeira, J.; Xavier, J.; Goebel, R. Type of featural attention differentially modulates hMT+ responses to illusory motion aftereffects. J. Neurophysiol. 2009, 102, 3016–3025. [Google Scholar] [CrossRef] [PubMed]
- Donahue, M.J.; Near, J.; Blicher, J.U.; Jezzard, P. Baseline GABA concentration and fMRI response. NeuroImage 2010, 53, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Muthukumaraswamy, S.D.; Evans, C.J.; Edden, R.A.; Wise, R.G.; Singh, K.D. Individual variability in the shape and amplitude of the BOLD-HRF correlates with endogenous GABAergic inhibition. Hum. Brain Mapp. 2012, 33, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Z.; Dragoi, V.; Sur, M.; Seung, H.S. Tilt aftereffect and adaptation-induced changes in orientation tuning in visual cortex. J. Neurophysiol. 2005, 94, 4038–4050. [Google Scholar] [CrossRef] [PubMed]
- Mathot, S.; Theeuwes, J. A reinvestigation of the reference frame of the tilt-adaptation aftereffect. Sci. Rep. 2013, 3, 1152. [Google Scholar] [CrossRef] [PubMed]
- Clifford, C.W.; Wenderoth, P.; Spehar, B. A functional angle on some after-effects in cortical vision. Proc. Biol. Sci. R. Soc. 2000, 267, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.A.; MacLeod, D.I. Visual adaptation and face perception. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2011, 366, 1702–1725. [Google Scholar] [CrossRef] [PubMed]
- Esclapez, M.; Tillakaratne, N.J.; Kaufman, D.L.; Tobin, A.J.; Houser, C.R. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. J. Neurosci. 1994, 14 3 Pt 2, 1834–1855. [Google Scholar] [PubMed]
- Feldblum, S.; Erlander, M.G.; Tobin, A.J. Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J. Neurosci. Res. 1993, 34, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.G.; Hornby, K.R.; Jones, D.G.; Murphy, K.M. Developmental changes in GABAergic mechanisms in human visual cortex across the lifespan. Front. Cell. Neurosci. 2010, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.L.; Steinmetz, S.E.; Carroll, A.M.; Simon, S.T.; Wallace, A.; Rojas, D.C. Test-retest reliability of the 40 Hz EEG auditory steady-state response. PLoS ONE 2014, 9, e85748. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.M.; Robertson, I.H.; O’Connell, R.G. Retest reliability of event-related potentials: Evidence from a variety of paradigms. Psychophysiology 2012, 49, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, J.; Jockers-Scherubl, M.C.; Boutros, N.N.; Gallinat, J. Test-retest reliability of P50, N100 and P200 auditory sensory gating in healthy subjects. Int. J. Psychophysiol. 2008, 67, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Sarnthein, J.; Andersson, M.; Zimmermann, M.B.; Zumsteg, D. High test-retest reliability of checkerboard reversal visual evoked potentials (VEP) over 8 months. Clin. Neurophysiol. 2009, 120, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Pedapati, E.V.; Erickson, C.A.; Byerly, M.J.; Sweeney, J.A. Neural synchronization deficits linked to cortical hyper-excitability and auditory hypersensitivity in fragile X syndrome. Mol. Autism 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, D.; Oranje, B.; Razak, K.A.; Siegel, S.J.; Schmid, S. Sensory processing in autism spectrum disorders and Fragile X syndrome-From the clinic to animal models. Neurosci. Biobehav. Rev. 2017, 76 Pt B, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Leigh, M.J.; Adams, P.; Nanakul, R.; Chechi, T.; Olichney, J.; Hagerman, R.; Hessl, D. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J. Psychopharmacol. 2013, 27, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Hays, S.A.; Huber, K.M.; Gibson, J.R. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 2011, 31, 14223–14234. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A greater increase in electroencephalogram (EEG) spectral power at the frequency of stimulus oscillation (monochromatic sinewave gratings) over varying levels of visual stimulus contrast in autism spectrum disorders (ASD) and typically developing (TD) groups, indicating greater cortical reactivity in ASD. From Takarae et al. 2016 [63], with permission.
Figure 1.
A greater increase in electroencephalogram (EEG) spectral power at the frequency of stimulus oscillation (monochromatic sinewave gratings) over varying levels of visual stimulus contrast in autism spectrum disorders (ASD) and typically developing (TD) groups, indicating greater cortical reactivity in ASD. From Takarae et al. 2016 [63], with permission.

Figure 2.
Cortical excitability affects neural inhibition through different mechanisms, each of which has cascading effects on sensory and clinical phenotypes in ASD.
Figure 2.
Cortical excitability affects neural inhibition through different mechanisms, each of which has cascading effects on sensory and clinical phenotypes in ASD.

Figure 3.
44% of ASD participants showed greater spectral power increases over contrast manipulation of the visual stimuli than any TD participant. Linear slopes for individual EEG responses across increasing visual contrast were estimated. From Takarae et al. 2016 [63], with permission.
Figure 3.
44% of ASD participants showed greater spectral power increases over contrast manipulation of the visual stimuli than any TD participant. Linear slopes for individual EEG responses across increasing visual contrast were estimated. From Takarae et al. 2016 [63], with permission.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Takarae, Y.; Sweeney, J. Neural Hyperexcitability in Autism Spectrum Disorders. Brain Sci. 2017, 7, 129. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci7100129
AMA Style
Takarae Y, Sweeney J. Neural Hyperexcitability in Autism Spectrum Disorders. Brain Sciences. 2017; 7(10):129. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci7100129
Chicago/Turabian StyleTakarae, Yukari, and John Sweeney. 2017. "Neural Hyperexcitability in Autism Spectrum Disorders" Brain Sciences 7, no. 10: 129. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci7100129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.