Neuroinflammation in Post-Traumatic Epilepsy: Pathophysiology and Tractable Therapeutic Targets

 ,
,
Abstract
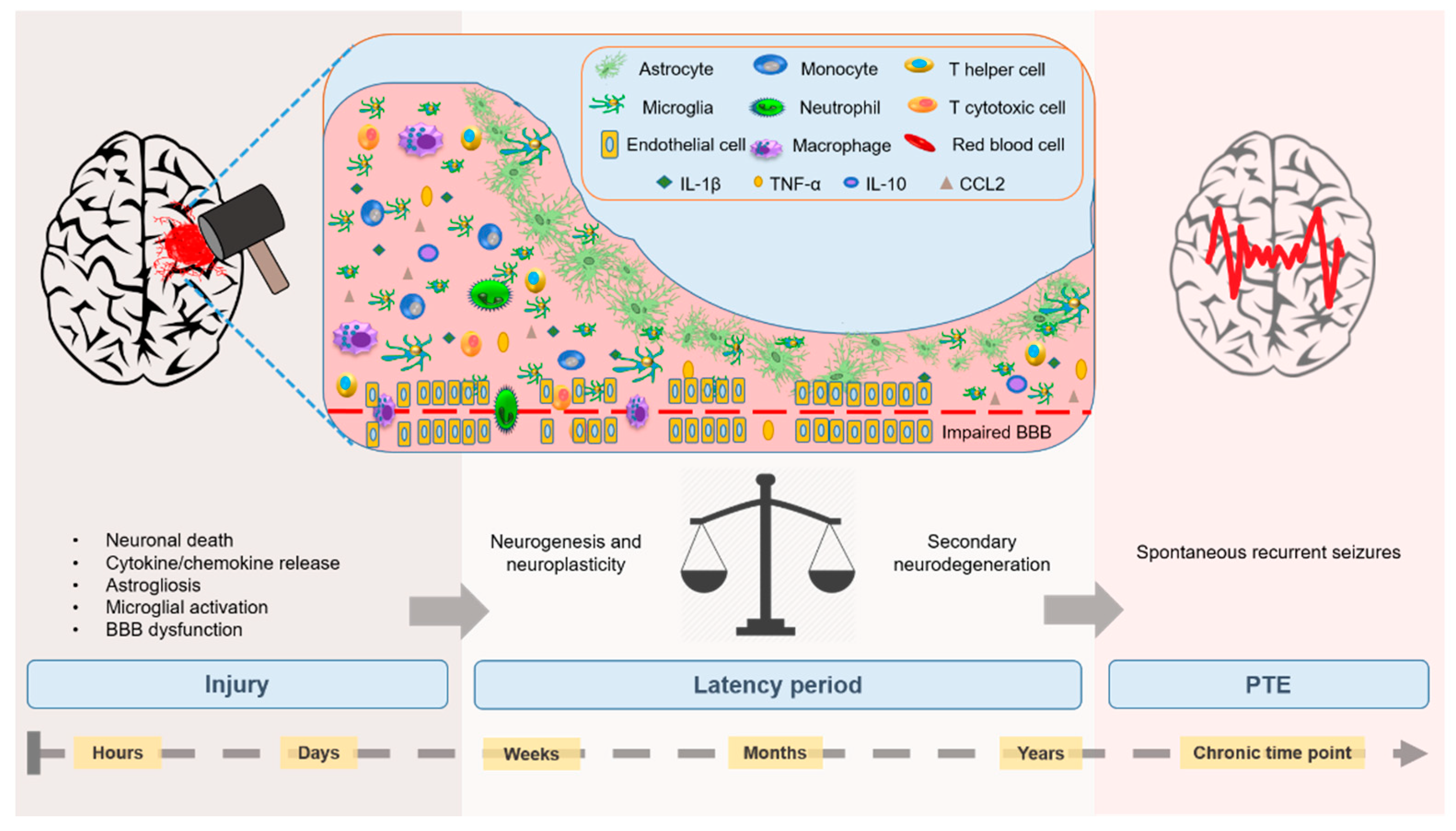
:1. Introduction
2. Neuroinflammatory Mechanisms Driving PTE
2.1. Cytokines
2.1.1. Interleukin-1 (IL-1)
2.1.2. Tumor Necrosis Factor (TNFα)
2.1.3. Interleukin-6 (IL-6)
2.1.4. High Mobility Group Box Protein-1 (HMGB1)
2.1.5. Interleukin-10 (IL-10)
2.2. Chemokines
2.3. Microglia
2.4. Astrocytes
2.5. Endothelial Cells and Blood-Brain Barrier Dysfunction
2.6. Blood-Derived Leukocytes
2.6.1. Neutrophils
2.6.2. T-Cells
2.6.3. Monocytes and Macrophages
3. Inflammatory Mediators as Biomarkers of PTE
4. Inflammation as an Anti-Epileptic Therapeutic Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lowenstein, D.H. Epilepsy after head injury: An overview. Epilepsia 2009, 50 (Suppl. 2), 4–9. [Google Scholar] [CrossRef]
- Bhalla, D.; Godet, B.; Druet-Cabanac, M.; Preux, P.M. Etiologies of epilepsy: A comprehensive review. Expert Rev. Neurother. 2011, 11, 861–876. [Google Scholar] [CrossRef]
- Agrawal, A.; Timothy, J.; Pandit, L.; Manju, M. Post-traumatic epilepsy: An overview. Clin. Neurol. Neurosurg. 2006, 108, 433–439. [Google Scholar] [CrossRef]
- Ferguson, P.L.; Smith, G.M.; Wannamaker, B.B.; Thurman, D.J.; Pickelsimer, E.E.; Selassie, A.W. A population-based study of risk of epilepsy after hospitalization for traumatic brain injury. Epilepsia 2010, 51, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Salazar, A.M.; Grafman, J. Post-traumatic epilepsy: Clinical clues to pathogenesis and paths to prevention. Handb. Clin. Neurol. 2015, 128, 525–538. [Google Scholar] [PubMed]
- Appleton, R.E.; Demellweek, C. Post-traumatic epilepsy in children requiring inpatient rehabilitation following head injury. J. Neurol. Neurosurg. Psychiatry 2002, 72, 669. [Google Scholar] [CrossRef] [PubMed]
- Statler, K.D.; Scheerlinck, P.; Pouliot, W.; Hamilton, M.; White, H.S.; Dudek, F.E. A potential model of pediatric posttraumatic epilepsy. Epilepsy Res. 2009, 86, 221–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ILAE. The 2014 Definition of Epilepsy: A Perspective for Patients and Caregivers. 2014. Available online: https://www.ilae.org/guidelines/definition-and-classification/the-2014-definition-of-epilepsy-a-perspective-for-patients-and-caregivers (accessed on 12 August 2019).
- Caplan, R.; Sagun, J.; Siddarth, P.; Gurbani, S.; Koh, S.; Gowrinathan, R.; Sankar, R. Social competence in pediatric epilepsy: Insights into underlying mechanisms. Epilepsy Behav. 2005, 6, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A. Difficulties in Treatment and Management of Epilepsy and Challenges in New Drug Development. Pharmaceuticals 2010, 3, 2090–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, M.M. Overview of drugs used for epilepsy and seizures: Etiology, diagnosis, and treatment. Pharm. Ther. 2010, 35, 392–415. [Google Scholar]
- Schmidt, D.; Loscher, W. Drug resistance in epilepsy: Putative neurobiologic and clinical mechanisms. Epilepsia 2005, 46, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.; Meyer, R.M.; Szuflita, N.S.; Severson, M.A.; Levine, Z.T. Post-Traumatic, Drug-Resistant Epilepsy and Review of Seizure Control Outcomes from Blinded, Randomized Controlled Trials of Brain Stimulation Treatments for Drug-Resistant Epilepsy. Cureus 2016, 8, e744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, A.I.R.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Webster, K.M.; Sun, M.; Crack, P.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Inflammation in epileptogenesis after traumatic brain injury. J. Neuroinflamm. 2017, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, E.J.; Gupta, A.; Subramanian, D.; Korgaonkar, A.A.; Santhakumar, V. Converging early responses to brain injury pave the road to epileptogenesis. J. Neurosc. Res. 2017, 97, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Shultz, S.R.; Robinson, M.J.; Belli, A.; Hibbs, M.L.; O’Brien, T.J.; Semple, B.D. Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav. Immun. 2019, 79, 63–74. [Google Scholar] [CrossRef] [PubMed]
- DeGrauw, X.; Thurman, D.; Xu, L.; Kancherla, V.; DeGrauw, T. Epidemiology of traumatic brain injury-associated epilepsy and early use of anti-epilepsy drugs: An analysis of insurance claims data, 2004–2014. Epilepsy Res. 2018, 146, 41–49. [Google Scholar] [CrossRef]
- Ritter, A.C.; Wagner, A.K.; Fabio, A.; Pugh, M.J.; Walker, W.C.; Szaflarski, J.P.; Zafonte, R.D.; Brown, A.W.; Hammond, F.M.; Bushnik, T.; et al. Incidence and risk factors of posttraumatic seizures following traumatic brain injury: A Traumatic Brain Injury Model Systems Study. Epilepsia 2016, 57, 1968–1977. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, D.H.; Thomas, M.J.; Smith, D.H.; McIntosh, T.K. Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef]
- Kelly, K.M.; Miller, E.R.; Lepsveridze, E.; Kharlamov, E.A.; McHedlishvili, Z. Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res. 2015, 117, 104–116. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Agostini, M.A.; Frol, A.B.; Mickey, B.; Fleckenstein, J.; Bigio, E.; Van Ness, P.C. Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch Neurol. 2000, 57, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Hudak, A.M.; Trivedi, K.; Harper, C.R.; Booker, K.; Caesar, R.R.; Agostini, M.; Van Ness, P.C.; Diaz-Arrastia, R. Evaluation of seizure-like episodes in survivors of moderate and severe traumatic brain injury. J. Head Trauma Rehabil. 2004, 19, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Afroz, S.; Michelson, H.B.; Goodman, J.H.; Valsamis, H.A.; Ling, D.S. Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J. Neurotrauma 2010, 27, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Walker, K.; Andresen, L.; Taylor-Weiner, A.; Hampton, D.; Tesco, G.; Dulla, C.G. Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb. Cortex 2015, 25, 2306–2320. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Murphy, K.; Travers, P.; Walport, M.; Janeway, C. Janeway’s Immunobiology, 8th ed.; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Lucas, S.-M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S232–S240. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil is in the Details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef]
- Semple, B.D.; Morganti-Kossmann, M.C. Cerebral inflammation after traumatic injury: Regulation of secondary damage, repair or both. In Traumatic Brain and Spinal Cord Injury: Challenges and Developments; Morganti-Kossmann, M.C., Raghupathi, R., Maas, A., Eds.; Cambridge University Press: New York, NY, USA, 2012; pp. 155–168. [Google Scholar]
- de Vries, E.E.; van den Munckhof, B.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Dhote, F.; Peinnequin, A.; Carpentier, P.; Baille, V.; Delacour, C.; Foquin, A.; Lallement, G.; Dorandeu, F. Prolonged inflammatory gene response following soman-induced seizures in mice. Toxicology 2007, 238, 166–176. [Google Scholar] [CrossRef]
- Fan, L.; Young, P.R.; Barone, F.C.; Feuerstein, G.Z.; Smith, D.H.; McIntosh, T.K. Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res. Mol. Brain Res. 1995, 30, 125–130. [Google Scholar] [CrossRef]
- Yatsiv, I.; Morganti-Kossmann, M.C.; Perez, D.; Dinarello, C.A.; Novick, D.; Rubinstein, M.; Otto, V.I.; Rancan, M.; Kossmann, T.; Redaelli, C.A.; et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J. Cereb. Blood Flow Metab. 2002, 22, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.; Tyrrell, P.J.; Allan, S.M. Regulation of interleukin-1 in acute brain injury. Trends Pharmacol. Sci. 2011, 32, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Holmin, S.; Schalling, M.; Hojeberg, B.; Nordqvist, A.C.; Skeftruna, A.K.; Mathiesen, T. Delayed cytokine expression in rat brain following experimental contusion. J. Neurosurg. 1997, 86, 493–504. [Google Scholar] [CrossRef]
- Csuka, E.; Hans, V.H.; Ammann, E.; Trentz, O.; Kossmann, T.; Morganti-Kossmann, M.C. Cell activation and inflammatory response following traumatic axonal injury in the rat. Neuroreport 2000, 11, 2587–2590. [Google Scholar] [CrossRef]
- Pinteaux, E.; Parker, L.C.; Rothwell, N.J.; Luheshi, G.N. Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J. Neurochem. 2002, 83, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.T.; Wang, Y.W.; Wo, Y.Y.; Yang, Y.L. Extracellular signal-regulated kinase-mediated IL-1-induced cortical neuron damage during traumatic brain injury. Neurosci. Lett. 2005, 386, 40–45. [Google Scholar] [CrossRef]
- Frugier, T.; Morganti-Kossmann, M.C.; O’Reilly, D.; McLean, C.A. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J. Neurotrauma 2010, 27, 497–507. [Google Scholar] [CrossRef]
- Kinoshita, K.; Chatzipanteli i, K.; Vitarbo, E.; Truettner, J.S.; Alonso, O.F.; Dietrich, W.D. Interleukin-1beta messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: Importance of injury severity and brain temperature. Neurosurgery 2002, 51, 195–203. [Google Scholar] [CrossRef]
- Lu, K.T.; Wang, Y.W.; Yang, J.T.; Yang, Y.L.; Chen, H.I. Effect of interleukin-1 on traumatic brain injury-induced damage to hippocampal neurons. J. Neurotrauma 2005, 22, 885–895. [Google Scholar] [CrossRef]
- Kamm, K.; Vanderkolk, W.; Lawrence, C.; Jonker, M.; Davis, A.T. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J. Trauma 2006, 60, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Relton, J.K.; Rothwell, N.J. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res. Bull. 1992, 29, 243–246. [Google Scholar] [CrossRef]
- Toulmond, S.; Rothwell, N.J. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995, 671, 261–266. [Google Scholar] [CrossRef]
- Yang, G.Y.; Liu, X.H.; Kadoya, C.; Zhao, Y.J.; Mao, Y.; Davidson, B.L.; Betz, A.L. Attenuation of ischemic inflammatory response in mouse brain using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist. J. Cereb. Blood Flow Metab. 1998, 18, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.; Morganti-Kossmann, M.C. The role of markers of inflammation in traumatic brain injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.Y.; Benveniste, E.N. Tumor necrosis factor-alpha production by astrocytes. Induction by lipopolysaccharide, IFN-gamma, and IL-1 beta. J. Immunol. 1990, 144, 2999–3007. [Google Scholar]
- Aloisi, F.; Care, A.; Borsellino, G.; Gallo, P.; Rosa, S.; Bassani, A.; Cabibbo, A.; Testa, U.; Levi, G.; Peschle, C. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J. Immunol. 1992, 149, 2358–2366. [Google Scholar]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A., Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J. Clin. Investig. 1985, 76, 2003–2011. [Google Scholar] [CrossRef]
- Tehranian, R.; Andell-Jonsson, S.; Beni, S.M.; Yatsiv, I.; Shohami, E.; Bartfai, T.; Lundkvist, J.; Iverfeldt, K. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J. Neurotrauma 2002, 19, 939–951. [Google Scholar] [CrossRef]
- Quagliarello, V.J.; Wispelwey, B.; Long, W.J., Jr.; Scheld, W.M. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J. Clin. Investig. 1991, 87, 1360–1366. [Google Scholar] [CrossRef]
- Holmin, S.; Mathiesen, T. Intracerebral administration of interleukin-1beta and induction of inflammation, apoptosis, and vasogenic edema. J. Neurosurg. 2000, 92, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, Q.; Malik, S.; Yang, J. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J. Pharmacol. Exp. Ther. 2000, 292, 497–504. [Google Scholar] [PubMed]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.; Cumiskey, D.; O’Connor, J.J. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005, 90, 663–670. [Google Scholar] [CrossRef]
- Balosso, S.; Ravizza, T.; Pierucci, M.; Calcagno, E.; Invernizzi, R.; Di Giovanni, G.; Esposito, E.; Vezzani, A. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: Implications for seizure susceptibility. Neuroscience 2009, 161, 293–300. [Google Scholar] [CrossRef]
- Dube, C.; Vezzani, A.; Behrens, M.; Bartfai, T.; Baram, T.Z. Interleukin-1beta contributes to the generation of experimental febrile seizures. Ann. Neurol. 2005, 57, 152–155. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Iori, V.; Wright, C.I.; French, J.; Vezzani, A. Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 2011, 8, 304–315. [Google Scholar] [CrossRef]
- Vezzani, A.; Moneta, D.; Conti, M.; Richichi, C.; Ravizza, T.; De Luigi, A.; De Simoni, M.G.; Sperk, G.; Andell-Jonsson, S.; Lundkvist, J.; et al. Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11534–11539. [Google Scholar] [CrossRef] [Green Version]
- Semple, B.D.; O’Brien, T.J.; Gimlin, K.; Wright, D.K.; Kim, S.E.; Casillas-Espinosa, P.M.; Webster, K.M.; Petrou, S.; Noble-Haeusslein, L.J. Interleukin-1 Receptor in Seizure Susceptibility after Traumatic Injury to the Pediatric Brain. J. Neurosci. 2017, 37, 7864–7877. [Google Scholar] [CrossRef]
- Ramilo, O.; Saez-Llorens, X.; Mertsola, J.; Jafari, H.; Olsen, K.D.; Hansen, E.J.; Yoshinaga, M.; Ohkawara, S.; Nariuchi, H.; McCracken, G.H., Jr. Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J. Exp. Med. 1990, 172, 497–507. [Google Scholar] [CrossRef]
- Kim, K.S.; Wass, C.A.; Cross, A.S.; Opal, S.M. Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 1992, 11, 293–298. [Google Scholar] [PubMed]
- Shohami, E.; Bass, R.; Wallach, D.; Yamin, A.; Gallily, R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J. Cereb. Blood Flow Metab. 1996, 16, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Knoblach, S.M.; Fan, L.; Faden, A.I. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J. Neuroimmunol. 1999, 95, 115–125. [Google Scholar] [CrossRef]
- Trembovler, V.; Beit-Yannai, E.; Younis, F.; Gallily, R.; Horowitz, M.; Shohami, E. Antioxidants attenuate acute toxicity of tumor necrosis factor-alpha induced by brain injury in rat. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 1999, 19, 791–795. [Google Scholar] [CrossRef]
- Carlson, N.G.; Wieggel, W.A.; Chen, J.; Bacchi, A.; Rogers, S.W.; Gahring, L.C. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J. Immunol. 1999, 163, 3963–3968. [Google Scholar]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [Green Version]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef]
- Nikolic, L.; Shen, W.; Nobili, P.; Virenque, A.; Ulmann, L.; Audinat, E. Blocking TNFalpha-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia 2018, 66, 2673–2683. [Google Scholar] [CrossRef]
- Balosso, S.; Ravizza, T.; Perego, C.; Peschon, J.; Campbell, I.L.; De Simoni, M.G.; Vezzani, A. Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann. Neurol. 2005, 57, 804–812. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef]
- Aderka, D.; Le, J.M.; Vilcek, J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J. Immunol. 1989, 143, 3517–3523. [Google Scholar] [PubMed]
- Wang, X.Q.; Peng, Y.P.; Lu, J.H.; Cao, B.B.; Qiu, Y.H. Neuroprotection of interleukin-6 against NMDA attack and its signal transduction by JAK and MAPK. Neurosci. Lett. 2009, 450, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Kossmann, T.; Hans, V.; Imhof, H.G.; Trentz, O.; Morganti-Kossmann, M.C. Interleukin-6 released in human cerebrospinal fluid following traumatic brain injury may trigger nerve growth factor production in astrocytes. Brain Res. 1996, 713, 143–152. [Google Scholar] [CrossRef]
- Lehtimaki, K.A.; Peltola, J.; Koskikallio, E.; Keranen, T.; Honkaniemi, J. Expression of cytokines and cytokine receptors in the rat brain after kainic acid-induced seizures. Brain Res. Mol. Brain Res. 2003, 110, 253–260. [Google Scholar] [CrossRef]
- Lehtimaki, K.A.; Keranen, T.; Huhtala, H.; Hurme, M.; Ollikainen, J.; Honkaniemi, J.; Palmio, J.; Peltola, J. Regulation of IL-6 system in cerebrospinal fluid and serum compartments by seizures: The effect of seizure type and duration. J. Neuroimmunol. 2004, 152, 121–125. [Google Scholar] [CrossRef]
- Peltola, J.; Hurme, M.; Miettinen, A.; Keranen, T. Elevated levels of interleukin-6 may occur in cerebrospinal fluid from patients with recent epileptic seizures. Epilepsy Res. 1998, 31, 129–133. [Google Scholar] [CrossRef]
- Peltola, J.; Palmio, J.; Korhonen, L.; Suhonen, J.; Miettinen, A.; Hurme, M.; Lindholm, D.; Keranen, T. Interleukin-6 and interleukin-1 receptor antagonist in cerebrospinal fluid from patients with recent tonic-clonic seizures. Epilepsy Res. 2000, 41, 205–211. [Google Scholar] [CrossRef]
- Peltola, J.; Laaksonen, J.; Haapala, A.M.; Hurme, M.; Rainesalo, S.; Keranen, T. Indicators of inflammation after recent tonic-clonic epileptic seizures correlate with plasma interleukin-6 levels. Seizure 2002, 11, 44–46. [Google Scholar] [CrossRef]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Samland, H.; Huitron-Resendiz, S.; Masliah, E.; Criado, J.; Henriksen, S.J.; Campbell, I.L. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 2003, 73, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, S.C.; Campbell, I.L.; Henriksen, S.J. Site-specific hippocampal pathophysiology due to cerebral overexpression of interleukin-6 in transgenic mice. Brain Res. 1994, 652, 149–153. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [PubMed]
- de Waal Malefyt, R.; Abrams, J.; Bennett, B.; Figdor, C.G.; de Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.; Aste-Amezaga, M.; Valiante, N.M.; Ma, X.; Kubin, M.; Trinchieri, G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 1993, 178, 1041–1048. [Google Scholar] [CrossRef]
- de Waal Malefyt, R.; Figdor, C.G.; Huijbens, R.; Mohan-Peterson, S.; Bennett, B.; Culpepper, J.; Dang, W.; Zurawski, G.; de Vries, J.E. Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J. Immunol. 1993, 151, 6370–6381. [Google Scholar]
- Gruber, M.F.; Williams, C.C.; Gerrard, T.L. Macrophage-colony-stimulating factor expression by anti-CD45 stimulated human monocytes is transcriptionally up-regulated by IL-1 beta and inhibited by IL-4 and IL-10. J. Immunol. 1994, 152, 1354–1361. [Google Scholar]
- Knoblach, S.M.; Faden, A.I. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp. Neurol. 1998, 153, 143–151. [Google Scholar] [CrossRef]
- Levin, S.G.; Godukhin, O.V. Protective effects of interleukin-10 on the development of epileptiform activity evoked by transient episodes of hypoxia in rat hippocampal slices. Neurosci. Behav. Physiol. 2007, 37, 467–470. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Kira, R.; Fukuda, M.; Torisu, H.; Sakai, Y.; Sanefuji, M.; Yukaya, N.; Hara, T. Interleukin-10 is associated with resistance to febrile seizures: Genetic association and experimental animal studies. Epilepsia 2009, 50, 761–767. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Manfredi, A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 2007, 220, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.M.; Sun, M.; Crack, P.J.; O’Brien, T.J.; Shultz, S.R.; Semple, B.D. Age-dependent release of high-mobility group box protein-1 and cellular neuroinflammation after traumatic brain injury in mice. J. Comp. Neurol. 2019, 527, 1102–1117. [Google Scholar] [CrossRef] [PubMed]
- Balosso, S.; Liu, J.; Bianchi, M.E.; Vezzani, A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid. Redox Signal. 2014, 21, 1726–1740. [Google Scholar] [CrossRef] [PubMed]
- Iori, V.; Frigerio, F.; Vezzani, A. Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 2016, 26, 118–123. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Semple, B.D.; Jones, N.C.; Othman, I.; Shaikh, M.F. High mobility group box 1 (HMGB1) as a novel frontier in epileptogenesis: From pathogenesis to therapeutic approaches. J Neurochem. 2019. [Google Scholar] [CrossRef]
- Semple, B.D.; Bye, N.; Rancan, M.; Ziebell, J.M.; Morganti-Kossmann, M.C. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): Evidence from severe TBI patients and CCL2−/−mice. J. Cereb. Blood Flow Metab. 2010, 30, 769–782. [Google Scholar] [CrossRef]
- Semple, B.D.; Frugier, T.; Morganti-Kossmann, M.C. CCL2 modulates cytokine production in cultured mouse astrocytes. J. Neuroinflamm. 2010, 7, 67. [Google Scholar] [CrossRef]
- van Gassen, K.L.; Netzeband, J.G.; de Graan, P.N.; Gruol, D.L. The chemokine CCL2 modulates Ca2+ dynamics and electrophysiological properties of cultured cerebellar Purkinje neurons. Eur. J. Neurosci. 2005, 21, 2949–2957. [Google Scholar] [CrossRef]
- Gosselin, R.D.; Varela, C.; Banisadr, G.; Mechighel, P.; Rostene, W.; Kitabgi, P.; Melik-Parsadaniantz, S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J. Neurochem. 2005, 95, 1023–1034. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, H.; Liu, J.; Dong, J.; Xiong, H. Chemokine CCL2 modulation of neuronal excitability and synaptic transmission in rat hippocampal slices. J. Neurochem. 2011, 116, 406–414. [Google Scholar] [CrossRef]
- Cerri, C.; Genovesi, S.; Allegra, M.; Pistillo, F.; Puntener, U.; Guglielmotti, A.; Perry, V.H.; Bozzi, Y.; Caleo, M. The Chemokine CCL2 Mediates the Seizure-enhancing Effects of Systemic Inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 3777–3788. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Shuaib, A. Involvement of inflammatory cytokines in central nervous system injury. Prog. Neurobiol. 2002, 67, 161–172. [Google Scholar] [CrossRef]
- Stahel, P.F.; Shohami, E.; Younis, F.M.; Kariya, K.; Otto, V.I.; Lenzlinger, P.M.; Grosjean, M.B.; Eugster, H.P.; Trentz, O.; Kossmann, T.; et al. Experimental closed head injury: Analysis of neurological outcome, blood-brain barrier dysfunction, intracranial neutrophil infiltration, and neuronal cell death in mice deficient in genes for pro-inflammatory cytokines. J. Cereb. Blood Flow Metab. 2000, 20, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Scherbel, U.; Raghupathi, R.; Nakamura, M.; Saatman, K.E.; Trojanowski, J.Q.; Neugebauer, E.; Marino, M.W.; McIntosh, T.K. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. USA 1999, 96, 8721–8726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.G.; Bruce-Keller, A.J.; Rabchevsky, A.G.; Christakos, S.; Clair, D.K.; Mattson, M.P.; Scheff, S.W. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 6248–6256. [Google Scholar] [CrossRef]
- Woodroofe, M.N.; Sarna, G.S.; Wadhwa, M.; Hayes, G.M.; Loughlin, A.J.; Tinker, A.; Cuzner, M.L. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: Evidence of a role for microglia in cytokine production. J. Neuroimmunol. 1991, 33, 227–236. [Google Scholar] [CrossRef]
- Sebire, G.; Emilie, D.; Wallon, C.; Hery, C.; Devergne, O.; Delfraissy, J.F.; Galanaud, P.; Tardieu, M. In vitro production of IL-6, IL-1 beta, and tumor necrosis factor-alpha by human embryonic microglial and neural cells. J. Immunol. 1993, 150, 1517–1523. [Google Scholar]
- Benveniste, E.N.; Sparacio, S.M.; Norris, J.G.; Grenett, H.E.; Fuller, G.M. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J. Neuroimmunol. 1990, 30, 201–212. [Google Scholar] [CrossRef]
- Van Wagoner, N.J.; Benveniste, E.N. Interleukin-6 expression and regulation in astrocytes. J. Neuroimmunol. 1999, 100, 124–139. [Google Scholar] [CrossRef]
- Schobitz, B.; de Kloet, E.R.; Sutanto, W.; Holsboer, F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur. J. Neurosci. 1993, 5, 1426–1435. [Google Scholar] [CrossRef]
- Gadient, R.A.; Otten, U. Identification of interleukin-6 (IL-6)-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci. Lett. 1994, 182, 243–246. [Google Scholar] [CrossRef]
- Ringheim, G.E.; Burgher, K.L.; Heroux, J.A. Interleukin-6 mRNA expression by cortical neurons in culture: Evidence for neuronal sources of interleukin-6 production in the brain. J. Neuroimmunol. 1995, 63, 113–123. [Google Scholar] [CrossRef]
- Sallmann, S.; Juttler, E.; Prinz, S.; Petersen, N.; Knopf, U.; Weiser, T.; Schwaninger, M. Induction of interleukin-6 by depolarization of neurons. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 8637–8642. [Google Scholar] [CrossRef]
- Islam, O.; Gong, X.; Rose-John, S.; Heese, K. Interleukin-6 and neural stem cells: More than gliogenesis. Mol. Biol. Cell 2009, 20, 188–199. [Google Scholar] [CrossRef]
- Bell, M.J.; Kochanek, P.M.; Doughty, L.A.; Carcillo, J.A.; Adelson, P.D.; Clark, R.S.; Wisniewski, S.R.; Whalen, M.J.; DeKosky, S.T. Interleukin-6 and interleukin-10 in cerebrospinal fluid after severe traumatic brain injury in children. J. Neurotrauma 1997, 14, 451–457. [Google Scholar] [CrossRef]
- Crespel, A.; Coubes, P.; Rousset, M.C.; Brana, C.; Rougier, A.; Rondouin, G.; Bockaert, J.; Baldy-Moulinier, M.; Lerner-Natoli, M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002, 952, 159–169. [Google Scholar] [CrossRef]
- Iori, V.; Iyer, A.M.; Ravizza, T.; Beltrame, L.; Paracchini, L.; Marchini, S.; Cerovic, M.; Hill, C.; Ferrari, M.; Zucchetti, M.; et al. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol. Dis. 2017, 99, 12–23. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Aronica, E.; Iyer, A.M.; Rossetti, C.; Molteni, M.; Casalgrandi, M.; Manfredi, A.A.; et al. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 2010, 16, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Wang, L.; Zhang, B.; Gao, F.; Yang, C.M. Glycyrrhizin, an HMGB1 inhibitor, exhibits neuroprotective effects in rats after lithium-pilocarpine-induced status epilepticus. J. Pharm. Pharmacol. 2019, 71, 390–399. [Google Scholar] [CrossRef]
- Luo, L.; Jin, Y.; Kim, I.D.; Lee, J.K. Glycyrrhizin suppresses HMGB1 inductions in the hippocampus and subsequent accumulation in serum of a kainic acid-induced seizure mouse model. Cell Mol. Neurobiol. 2014, 34, 987–997. [Google Scholar] [CrossRef]
- Keystone, E.; Wherry, J.; Grint, P. IL-10 as a therapeutic strategy in the treatment of rheumatoid arthritis. Rheum. Dis. Clin. N. Am. 1998, 24, 629–639. [Google Scholar] [CrossRef]
- O’Garra, A.; Barrat, F.J.; Castro, A.G.; Vicari, A.; Hawrylowicz, C. Strategies for use of IL-10 or its antagonists in human disease. Immunol. Rev. 2008, 223, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, V.M.; Stanton, E.H.; Nothdurfter, C.; Rupprecht, R.; Wetzel, C.H. The Role of Chemokines in the Pathophysiology of Major Depressive Disorder. Int. J. Mol. Sci. 2019, 20, 2283. [Google Scholar] [CrossRef] [PubMed]
- Cerri, C.; Caleo, M.; Bozzi, Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy. Epilepsy Res. 2017, 136, 77–83. [Google Scholar] [CrossRef]
- Lee, T.S.; Mane, S.; Eid, T.; Zhao, H.; Lin, A.; Guan, Z.; Kim, J.H.; Schweitzer, J.; King-Stevens, D.; Weber, P.; et al. Gene expression in temporal lobe epilepsy is consistent with increased release of glutamate by astrocytes. Mol. Med. 2007, 13, 1–13. [Google Scholar] [CrossRef]
- Arisi, G.M.; Foresti, M.L.; Katki, K.; Shapiro, L.A. Increased CCL2, CCL3, CCL5, and IL-1beta cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine-induced seizures. J. Neuroinflamm. 2015, 12, 129. [Google Scholar] [CrossRef]
- Liu, J.X.; Cao, X.; Tang, Y.C.; Liu, Y.; Tang, F.R. CCR7, CCR8, CCR9 and CCR10 in the mouse hippocampal CA1 area and the dentate gyrus during and after pilocarpine-induced status epilepticus. J. Neurochem. 2007, 100, 1072–1088. [Google Scholar] [CrossRef]
- Fabene, P.F.; Bramanti, P.; Constantin, G. The emerging role for chemokines in epilepsy. J. Neuroimmunol. 2010, 224, 22–27. [Google Scholar] [CrossRef]
- Glabinski, A.R.; Balasingam, V.; Tani, M.; Kunkel, S.L.; Strieter, R.M.; Yong, V.W.; Ransohoff, R.M. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J. Immunol. 1996, 156, 4363–4368. [Google Scholar]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef]
- Di Battista, A.P.; Buonora, J.E.; Rhind, S.G.; Hutchison, M.G.; Baker, A.J.; Rizoli, S.B.; Diaz-Arrastia, R.; Mueller, G.P. Blood Biomarkers in Moderate-To-Severe Traumatic Brain Injury: Potential Utility of a Multi-Marker Approach in Characterizing Outcome. Front. Neurol. 2015, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Dzenko, K.A.; Song, L.; Ge, S.; Kuziel, W.A.; Pachter, J.S. CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc. Res. 2005, 70, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Shakui, P.; Keep, R.F.; Moore, B.B.; Kunkel, S.L.; Van Rooijen, N.; Andjelkovic, A.V. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2005, 25, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, C.M.; McManus, C.M.; Sharron, M.; Gao, Z.Y.; Murphy, D.; Jaffer, S.; Choe, W.; Chen, W.; Hesselgesser, J.; Gaylord, H.; et al. Expression of multiple functional chemokine receptors and monocyte chemoattractant protein-1 in human neurons. Neuroscience 2000, 97, 591–600. [Google Scholar] [CrossRef]
- Gourmala, N.G.; Buttini, M.; Limonta, S.; Sauter, A.; Boddeke, H.W.G.M. Differential and time-dependent expression of monocyte chemoattractant protein-1 mRNA by astrocytes and macrophages in rat brain: Effects of ischemia and peripheral lipopolysaccharide administration. J. Neuroimmunol. 1997, 74, 35–44. [Google Scholar] [CrossRef]
- Wang, C.; Yang, L.; Zhang, J.; Lin, Z.; Qi, J.; Duan, S. Higher expression of monocyte chemoattractant protein 1 and its receptor in brain tissue of intractable epilepsy patients. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2016, 28, 134–140. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, X.; Mo, X.; Xi, Z.; Xiao, F.; Li, J.; Zhu, X.; Luan, G.; Wang, Y.; Li, Y.; et al. Expression of monocyte chemoattractant protein-1 in brain tissue of patients with intractable epilepsy. Clin. Neuropathol. 2008, 27, 55–63. [Google Scholar] [CrossRef]
- Vezzani, A.; Maroso, M.; Balosso, S.; Sanchez, M.A.; Bartfai, T. IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav. Immun. 2011, 25, 1281–1289. [Google Scholar] [CrossRef]
- Tian, D.S.; Peng, J.; Murugan, M.; Feng, L.J.; Liu, J.L.; Eyo, U.B.; Zhou, L.J.; Mogilevsky, R.; Wang, W.; Wu, L.J. Chemokine CCL2-CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1beta Production after Status Epilepticus. J. Neurosci. 2017, 37, 7878–7892. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Raivich, G.; Bohatschek, M.; Kloss, C.U.; Werner, A.; Jones, L.L.; Kreutzberg, G.W. Neuroglial activation repertoire in the injured brain: Graded response, molecular mechanisms and cues to physiological function. Brain Res. Brain Res. Rev. 1999, 30, 77–105. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Braughler, J.M.; Hall, E.D. Central nervous system trauma and stroke. I. Biochemical considerations for oxygen radical formation and lipid peroxidation. Free Radic. Biol. Med. 1989, 6, 289–301. [Google Scholar] [CrossRef]
- Hall, E.D.; Braughler, J.M. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic. Biol. Med. 1989, 6, 303–313. [Google Scholar] [CrossRef]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef]
- Ju, T.C.; Chen, H.M.; Chen, Y.C.; Chang, C.P.; Chang, C.; Chern, Y. AMPK-alpha1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim. Biophys. Acta 2014, 1842, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Perfeito, R.; Lazaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell. Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, A.; Emoto, M.C.; Suzuki, S.; Iwahara, N.; Hisahara, S.; Kawamata, J.; Suzuki, H.; Yamauchi, A.; Sato-Akaba, H.; Fujii, H.G.; et al. Evaluation of oxidative stress in the brain of a transgenic mouse model of Alzheimer disease by in vivo electron paramagnetic resonance imaging. Free Radic. Biol. Med. 2015, 85, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pierozan, P.; Biasibetti, H.; Schmitz, F.; Avila, H.; Parisi, M.M.; Barbe-Tuana, F.; Wyse, A.T.; Pessoa-Pureur, R. Quinolinic acid neurotoxicity: Differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim. Biophys. Acta 2016, 1863, 3001–3014. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Chugh, D.; Ali, I.; Bakochi, A.; Bahonjic, E.; Etholm, L.; Ekdahl, C.T. Alterations in Brain Inflammation, Synaptic Proteins, and Adult Hippocampal Neurogenesis during Epileptogenesis in Mice Lacking Synapsin2. PLoS ONE 2015, 10, e0132366. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Murugan, M.; Wu, L.J. Microglia-Neuron Communication in Epilepsy. GLIA 2017, 65, 5–18. [Google Scholar] [CrossRef]
- Hiragi, T.; Ikegaya, Y.; Koyama, R. Microglia after Seizures and in Epilepsy. Cells 2018, 7, 26. [Google Scholar] [CrossRef]
- Okuneva, O.; Korber, I.; Li, Z.; Tian, L.; Joensuu, T.; Kopra, O.; Lehesjoki, A.E. Abnormal microglial activation in the Cstb(-/-) mouse, a model for progressive myoclonus epilepsy, EPM1. Glia 2015, 63, 400–411. [Google Scholar] [CrossRef]
- Zhao, X.; Liao, Y.; Morgan, S.; Mathur, R.; Feustel, P.; Mazurkiewicz, J.; Qian, J.; Chang, J.; Mathern, G.W.; Adamo, M.A.; et al. Noninflammatory Changes of Microglia Are Sufficient to Cause Epilepsy. Cell Rep. 2018, 22, 2080–2093. [Google Scholar] [CrossRef] [Green Version]
- Tikka, T.; Fiebich, B.L.; Goldsteins, G.; Keinanen, R.; Koistinaho, J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 2580–2588. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Qiao, Y.; Xu, L.; Kacimi, R.; Sun, X.; Giffard, R.G.; Yenari, M.A. Direct protection of cultured neurons from ischemia-like injury by minocycline. Anat. Cell Biol. 2010, 43, 325–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Murugan, M.; Bosco, D.B.; Liu, Y.; Peng, J.; Worrell, G.A.; Wang, H.L.; Ta, L.E.; Richardson, J.R.; Shen, Y.; et al. Microglial proliferation and monocyte infiltration contribute to microgliosis following status epilepticus. Glia 2019, 67, 1434–1448. [Google Scholar] [CrossRef]
- Waltl, I.; Kaufer, C.; Gerhauser, I.; Chhatbar, C.; Ghita, L.; Kalinke, U.; Loscher, W. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun. 2018, 74, 186–204. [Google Scholar] [CrossRef]
- Liberto, C.M.; Albrecht, P.J.; Herx, L.M.; Yong, V.W.; Levison, S.W. Pro-regenerative properties of cytokine-activated astrocytes. J. Neurochem. 2004, 89, 1092–1100. [Google Scholar] [CrossRef]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef]
- Carmen, J.; Magnus, T.; Cassiani-Ingoni, R.; Sherman, L.; Rao, M.S.; Mattson, M.P. Revisiting the astrocyte-oligodendrocyte relationship in the adult CNS. Prog. Neurobiol. 2007, 82, 151–162. [Google Scholar] [CrossRef]
- Eddleston, M.; Mucke, L. Molecular profile of reactive astrocytes—Implications for their role in neurologic disease. Neuroscience 1993, 54, 15–36. [Google Scholar] [CrossRef]
- Rolls, A.; Shechter, R.; Schwartz, M. The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 2009, 10, 235–241. [Google Scholar] [CrossRef]
- Efremova, L.; Chovancova, P.; Adam, M.; Gutbier, S.; Schildknecht, S.; Leist, M. Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Arch. Toxicol. 2017, 91, 231–246. [Google Scholar] [CrossRef] [PubMed]
- McGraw, J.; Hiebert, G.W.; Steeves, J.D. Modulating astrogliosis after neurotrauma. J. Neurosci. Res. 2001, 63, 109–115. [Google Scholar] [CrossRef]
- Shandra, O.; Winemiller, A.R.; Heithoff, B.P.; Munoz-Ballester, C.; George, K.K.; Benko, M.J.; Zuidhoek, I.A.; Besser, M.N.; Curley, D.E.; Edwards, G.F., 3rd; et al. Repetitive Diffuse Mild Traumatic Brain Injury Causes an Atypical Astrocyte Response and Spontaneous Recurrent Seizures. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 1944–1963. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sha, L.; Sun, N.; Shen, Y.; Xu, Q. Deletion of mTOR in Reactive Astrocytes Suppresses Chronic Seizures in a Mouse Model of Temporal Lobe Epilepsy. Mol. Neurobiol. 2017, 54, 175–187. [Google Scholar] [CrossRef]
- Coulter, D.A.; Steinhauser, C. Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022434. [Google Scholar] [CrossRef]
- Ji, Z.G.; Wang, H. Optogenetic control of astrocytes: Is it possible to treat astrocyte-related epilepsy? Brain Res. Bull. 2015, 110, 20–25. [Google Scholar] [CrossRef]
- Robel, S. Astroglial Scarring and Seizures: A Cell Biological Perspective on Epilepsy. Neuroscientist 2017, 23, 152–168. [Google Scholar] [CrossRef]
- Seifert, G.; Schilling, K.; Steinhäuser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194. [Google Scholar] [CrossRef]
- Dadas, A.; Janigro, D. Breakdown of blood brain barrier as a mechanism of post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 20–26. [Google Scholar] [CrossRef]
- Seifert, G.; Carmignoto, G.; Steinhäuser, C. Astrocyte dysfunction in epilepsy. Brain Res. Rev. 2010, 63, 212–221. [Google Scholar] [CrossRef]
- Kivi, A.; Lehmann, T.N.; Kovacs, R.; Eilers, A.; Jauch, R.; Meencke, H.; Von Deimling, A.; Heinemann, U.; Gabriel, S. Effects of barium on stimulus-induced rises of [K+] o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur. J. Neurosci. 2000, 12, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.-S.; McCarthy, K.D. Conditional knock-out of Kir4. 1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef] [PubMed]
- Neusch, C.; Rozengurt, N.; Jacobs, R.E.; Lester, H.A.; Kofuji, P. Kir4. 1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 2001, 21, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Prasad, S. Early down regulation of the glial Kir4. 1 and GLT-1 expression in pericontusional cortex of the old male mice subjected to traumatic brain injury. Biogerontology 2013, 14, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Frassoni, C.; Tassi, L.; Spreafico, R.; Garbelli, R. Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res. 2011, 1367, 330–339. [Google Scholar] [CrossRef]
- Wallraff, A.; Köhling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhäuser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef]
- Tian, G.-F.; Azmi, H.; Takano, T.; Xu, Q.; Peng, W.; Lin, J.; Oberheim, N.; Lou, N.; Wang, X.; Zielke, H.R. An astrocytic basis of epilepsy. Nat. Med. 2005, 11, 973. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Friedman, A.; Kaufer, D.; Heinemann, U. Blood–brain barrier breakdown-inducing astrocytic transformation: Novel targets for the prevention of epilepsy. Epilepsy Res. 2009, 85, 142–149. [Google Scholar] [CrossRef]
- Friedman, A.; Heinemann, U. Role of blood-brain barrier dysfunction in epileptogenesis. In Jasper’s Basic Mechanisms of the Epilepsies [Internet], 4th ed.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- Nag, S.; Kapadia, A.; Stewart, D. Molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol. Appl. Neurobiol. 2011, 37, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cen, J.; Huang, Y.; Shen, H.; Yao, L.; Wang, Y.; Chen, Z. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS ONE 2011, 6, e20599. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Rempe, R.G.; Hartz, A.M.S.; Soldner, E.L.B.; Sokola, B.S.; Alluri, S.R.; Abner, E.L.; Kryscio, R.J.; Pekcec, A.; Schlichtiger, J.; Bauer, B. Matrix Metalloproteinase-Mediated Blood-Brain Barrier Dysfunction in Epilepsy. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 4301–4315. [Google Scholar] [CrossRef] [Green Version]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar] [CrossRef]
- Seiffert, E.; Dreier, J.P.; Ivens, S.; Bechmann, I.; Tomkins, O.; Heinemann, U.; Friedman, A. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef]
- van Vliet, E.A.; da Costa Araujo, S.; Redeker, S.; van Schaik, R.; Aronica, E.; Gorter, J.A. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 2007, 130 Pt 2, 521–534. [Google Scholar] [CrossRef]
- Heinemann, U.; Kaufer, D.; Friedman, A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia 2012, 60, 1251–1257. [Google Scholar] [CrossRef]
- Cornford, E.; Oldendorf, W. Epilepsy and the blood-brain barrier. Adv. Neurol. 1986, 44, 787–812. [Google Scholar]
- Tomkins, O.; Shelef, I.; Kaizerman, I.; Eliushin, A.; Afawi, Z.; Misk, A.; Gidon, M.; Cohen, A.; Zumsteg, D.; Friedman, A. Blood-brain barrier disruption in post-traumatic epilepsy. J. Neurol. Neurosurg. Psychiatry 2008, 79, 774–777. [Google Scholar] [CrossRef] [Green Version]
- Korn, A.; Golan, H.; Melamed, I.; Pascual-Marqui, R.; Friedman, A. Focal Cortical Dysfunction and Blood-Brain Barrier Disruption in Patients with Postconcussion Syndrome. J. Clin. Neurophysiol. 2005, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Guo, Y.; Ha, B.; Zen, K.; Liu, Y. Regulation of the inflammatory response: Enhancing neutrophil infiltration under chronic inflammatory conditions. J. Immunol. 2012, 188, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Bye, N.; Habgood, M.D.; Callaway, J.K.; Malakooti, N.; Potter, A.; Kossmann, T.; Morganti-Kossmann, M.C. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp. Neurol. 2007, 204, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Soares, H.D.; Hicks, R.R.; Smith, D.; McIntosh, T.K. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 8223–8233. [Google Scholar] [CrossRef]
- Scholz, M.; Cinatl, J.; Schadel-Hopfner, M.; Windolf, J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med. Res. Rev. 2007, 27, 401–416. [Google Scholar] [CrossRef]
- Stowe, A.M.; Adair-Kirk, T.L.; Gonzales, E.R.; Perez, R.S.; Shah, A.R.; Park, T.S.; Gidday, J.M. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol. Dis. 2009, 35, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, K.; Dhabhar, F.S.; Sapolsky, R.M. Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc. Natl. Acad.Sci. USA 2004, 101, 331–336. [Google Scholar] [CrossRef]
- Özdemir, H.H.; Akil, E.; Acar, A.; Tamam, Y.; Varol, S.; Cevik, M.U.; Arikanoglu, A. Changes in serum albumin levels and neutrophil-lymphocyte ratio in patients with convulsive status epilepticus. Int. J. Neurosci. 2016, 127, 417–420. [Google Scholar] [CrossRef]
- Kim, J.-E.; Ryu, H.J.; Choi, S.Y.; Kang, T.-C. Tumor necrosis factor-α-mediated threonine 435 phosphorylation of p65 nuclear factor-κB subunit in endothelial cells induces vasogenic edema and neutrophil infiltration in the rat piriform cortex following status epilepticus. J. Neuroinflamm. 2012, 9, 6. [Google Scholar] [CrossRef]
- Pun, P.B.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef]
- Liu, Y.W.; Li, S.; Dai, S.S. Neutrophils in traumatic brain injury (TBI): Friend or foe? J. Neuroinflamm. 2018, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Liao, Y.; Liu, P.; Guo, F.; Zhang, Z.-Y.; Zhang, Z. Oxidative Burst of Circulating Neutrophils Following Traumatic Brain Injury in Human. PLoS ONE 2013, 8, e68963. [Google Scholar]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.C.; Garcia, A.J.; Mochizuki, A.Y.; Chang, J.W.; Reyes, S.D.; Salamon, N.; Prins, R.M.; Mathern, G.W.; Fallah, A. Evidence for Innate and Adaptive Immune Responses in a Cohort of Intractable Pediatric Epilepsy Surgery Patients. Front. Immunol. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Robinson, A.P.; Ishii, T.; Duncan, D.A.S.; Alden, T.D.; Goings, G.E.; Ifergan, I.; Podojil, J.R.; Penaloza-MacMaster, P.; Kearney, J.A.; et al. Peripherally derived T regulatory and γδ T cells have opposing roles in the pathogenesis of intractable pediatric epilepsy. J. Exp. Med. 2018, 215, 1169–1186. [Google Scholar] [CrossRef]
- Yang, J.; Hu, C.; Jiang, X. Activation of peripheral T lymphocytes in children with epilepsy and production of cytokines. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2016, 32, 1234–1237. [Google Scholar]
- Deprez, F.; Zattoni, M.; Mura, M.L.; Frei, K.; Fritschy, J.-M. Adoptive transfer of T lymphocytes in immunodeficient mice influences epileptogenesis and neurodegeneration in a model of temporal lobe epilepsy. Neurobiol. Dis. 2011, 44, 174–184. [Google Scholar] [CrossRef]
- Zattoni, M.; Mura, M.L.; Deprez, F.; Schwendener, R.A.; Engelhardt, B.; Frei, K.; Fritschy, J.M. Brain infiltration of leukocytes contributes to the pathophysiology of temporal lobe epilepsy. J. Neurosci. 2011, 31, 4037–4050. [Google Scholar] [CrossRef]
- Man, S.; Ubogu, E.E.; Ransohoff, R.M. Inflammatory cell migration into the central nervous system: A few new twists on an old tale. Brain Pathol. 2007, 17, 243–250. [Google Scholar] [CrossRef]
- Engelhardt, B. Immune cell entry into the central nervous system: Involvement of adhesion molecules and chemokines. J. Neurol. Sci. 2008, 274, 23–26. [Google Scholar] [CrossRef]
- Makinde, H.M.; Cuda, C.M.; Just, T.B.; Perlman, H.R.; Schwulst, S.J. Nonclassical Monocytes Mediate Secondary Injury, Neurocognitive Outcome, and Neutrophil Infiltration after Traumatic Brain Injury. J. Immunol. 2017, 199, 3583–3591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulian, D.; Chen, J.; Ingeman, J.E.; George, J.K.; Noponen, M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J. Neurosci. Off. J. Soc. Neurosci. 1989, 9, 4416–4429. [Google Scholar] [CrossRef]
- Popovich, P.G.; Guan, Z.; Wei, P.; Huitinga, I.; van Rooijen, N.; Stokes, B.T. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp. Neurol. 1999, 158, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paez, A.C.; Brunschwig, J.P.; Bramlett, H.M. Light and electron microscopic assessment of progressive atrophy following moderate traumatic brain injury in the rat. Acta Neuropathol. 2005, 109, 603–616. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad.Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufer, C.; Chhatbar, C.; Broer, S.; Waltl, I.; Ghita, L.; Gerhauser, I.; Kalinke, U.; Loscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad.Sci. USA 2018, 115, E8929–E8938. [Google Scholar] [CrossRef] [Green Version]
- Agoston, D.V.; Kamnaksh, A. Protein biomarkers of epileptogenicity after traumatic brain injury. Neurobiol. Dis. 2019, 123, 59–68. [Google Scholar] [CrossRef]
- Diamond, M.L.; Ritter, A.C.; Failla, M.D.; Boles, J.A.; Conley, Y.P.; Kochanek, P.M.; Wagner, A.K. IL-1beta associations with posttraumatic epilepsy development: A genetics and biomarker cohort study. Epilepsia 2014, 55, 1109–1119. [Google Scholar] [CrossRef]
- Dedeurwaerdere, S.; Callaghan, P.D.; Pham, T.; Rahardjo, G.L.; Amhaoul, H.; Berghofer, P.; Quinlivan, M.; Mattner, F.; Loc’h, C.; Katsifis, A.; et al. PET imaging of brain inflammation during early epileptogenesis in a rat model of temporal lobe epilepsy. EJNMMI Res. 2012, 2, 60. [Google Scholar] [CrossRef]
- Amhaoul, H.; Hamaide, J.; Bertoglio, D.; Reichel, S.N.; Verhaeghe, J.; Geerts, E.; Van Dam, D.; De Deyn, P.P.; Kumar-Singh, S.; Katsifis, A.; et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol. Dis. 2015, 82, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Brackhan, M.; Bascunana, P.; Postema, J.M.; Ross, T.L.; Bengel, F.M.; Bankstahl, M.; Bankstahl, J.P. Serial Quantitative TSPO-Targeted PET Reveals Peak Microglial Activation up to 2 Weeks after an Epileptogenic Brain Insult. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Mahmud, M.; Owen, D.R.; Johnson, M.R. Microglial positron emission tomography (PET) imaging in epilepsy: Applications, opportunities and pitfalls. Seizure 2017, 44, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravizza, T.; Vezzani, A. Pharmacological targeting of brain inflammation in epilepsy: Therapeutic perspectives from experimental and clinical studies. Epilepsia Open 2018, 3 (Suppl. 2), 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saletti, P.G.; Ali, I.; Casillas-Espinosa, P.M.; Semple, B.D.; Lisgaras, C.P.; Moshe, S.L.; Galanopoulou, A.S. In search of antiepileptogenic treatments for post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.M.; Shultz, S.R.; Ozturk, E.; Dill, L.K.; Sun, M.; Casillas-Espinosa, P.; Jones, N.C.; Crack, P.J.; O’Brien, T.J.; Semple, B.D. Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: Chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp. Neurol. 2019, 320, 112979. [Google Scholar] [CrossRef] [PubMed]
- Vitaliti, G.; Pavone, P.; Mahmood, F.; Nunnari, G.; Falsaperla, R. Targeting inflammation as a therapeutic strategy for drug-resistant epilepsies: An update of new immune-modulating approaches. Hum. Vaccines Immunother. 2014, 10, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Tchekalarova, J.D.; Ivanova, N.M.; Pechlivanova, D.M.; Atanasova, D.; Lazarov, N.; Kortenska, L.; Mitreva, R.; Lozanov, V.; Stoynev, A. Antiepileptogenic and neuroprotective effects of losartan in kainate model of temporal lobe epilepsy. Pharmacol. Biochem. Behav. 2014, 127, 27–36. [Google Scholar] [CrossRef]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138 Pt 11, 3299–3315. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the Eleventh Eilat Conference (EILAT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSena, A.D.; Do, T.; Schulert, G.S. Systemic autoinflammation with intractable epilepsy managed with interleukin-1 blockade. J. Neuroinflamm. 2018, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Kenney-Jung, D.L.; Vezzani, A.; Kahoud, R.J.; LaFrance-Corey, R.G.; Ho, M.L.; Muskardin, T.W.; Wirrell, E.C.; Howe, C.L.; Payne, E.T. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 2016, 80, 939–945. [Google Scholar] [CrossRef]
- Lagarde, S.; Villeneuve, N.; Trebuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.A.; Trommsdorff, V.; Lefranc, J.; et al. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Strzelczyk, A.; Reif, P.S.; Schorlemmer, K.; Bauer, S.; Norwood, B.A.; Oertel, W.H.; Rosenow, F.; Strik, H.; Hamer, H.M. Minocycline as potent anticonvulsant in a patient with astrocytoma and drug resistant epilepsy. Seizure 2012, 21, 227–228. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Mediator | Pro-Inflammatory | Anti-Inflammatory | Role in Seizures/Epilepsy? |
|---|---|---|---|
| IL-1β | Regulates release of cytokines, chemokines, ROS, and proteases [48,49,50,52]. Mediates leukocyte recruitment [51], BBB disruption [53], edema [54], cell apoptosis [54] and glial activation [30] | - | IL-1β: pro-ictogenic [56,57,58,59] IL-1R: anti-seizure and anti-epileptogenic [60,61,62] (experimental and clinical evidence) |
| TNFα | Mediates leukocyte infiltration, BBB disruption, and neuronal degeneration [52,63,64,65,66,67] | Modulates neurotrophin production; protect neutrons against NMDA-mediated calcium influx [68,69] | Pro-ictogenic [70,71] Anti-seizure [72] (receptor-dependent effects) (experimental evidence) |
| IL-6 | Increases secretion of chemokines and adhesion molecules, to enhance leukocyte recruitment [73] | Inhibits TNFα production and reduces NMDA-mediated toxicity [74,75]. Induces synthesis of NGF [76] | Pro-ictogenic [77,78,79,80,81,82,83,84,85] (experimental evidence) |
| IL-10 | - | Inhibits cytokine production [86,87,88,89,90]. Regulates glial activation, inhibits macrophage accumulation, and promotes NGF production [91] | Anti-seizure [92,93] (experimental evidence) |
| HMGB1 | Released passively by necrotic cells, or actively by immune cells; promotes inflammatory cytokine release [94,95] | - | Pro-ictogenic [96,97,98] (experimental and clinical evidence) |
| CCL2 | Release by infiltrated macrophages further promotes peripheral macrophage infiltration [48] | Absence of CCL2 results in delayed secretion of different pro-inflammatory cytokines [99,100] | Pro-ictogenic [101,102,103,104] (experimental evidence) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.; Leung, W.L.; Zamani, A.; O’Brien, T.J.; Casillas Espinosa, P.M.; Semple, B.D. Neuroinflammation in Post-Traumatic Epilepsy: Pathophysiology and Tractable Therapeutic Targets. Brain Sci. 2019, 9, 318. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110318
Sharma R, Leung WL, Zamani A, O’Brien TJ, Casillas Espinosa PM, Semple BD. Neuroinflammation in Post-Traumatic Epilepsy: Pathophysiology and Tractable Therapeutic Targets. Brain Sciences. 2019; 9(11):318. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110318
Chicago/Turabian StyleSharma, Rishabh, Wai Lam Leung, Akram Zamani, Terence J. O’Brien, Pablo M. Casillas Espinosa, and Bridgette D. Semple. 2019. "Neuroinflammation in Post-Traumatic Epilepsy: Pathophysiology and Tractable Therapeutic Targets" Brain Sciences 9, no. 11: 318. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci9110318