
Reverse Phase Protein Arrays—Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use

Abstract
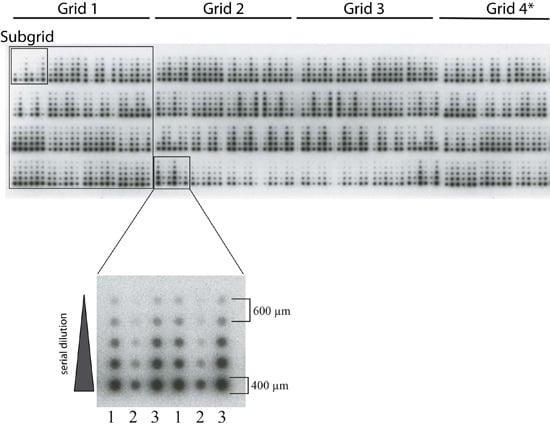
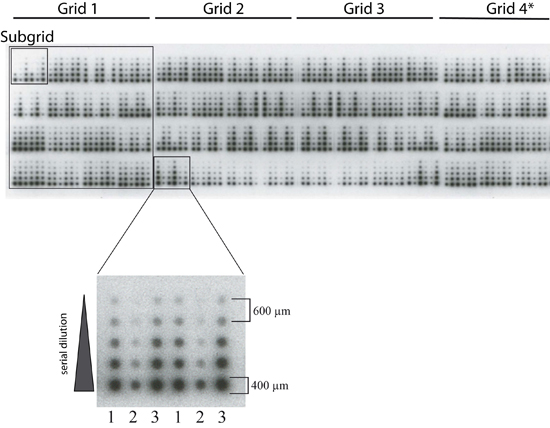
:
1. Introduction

{kind=link}
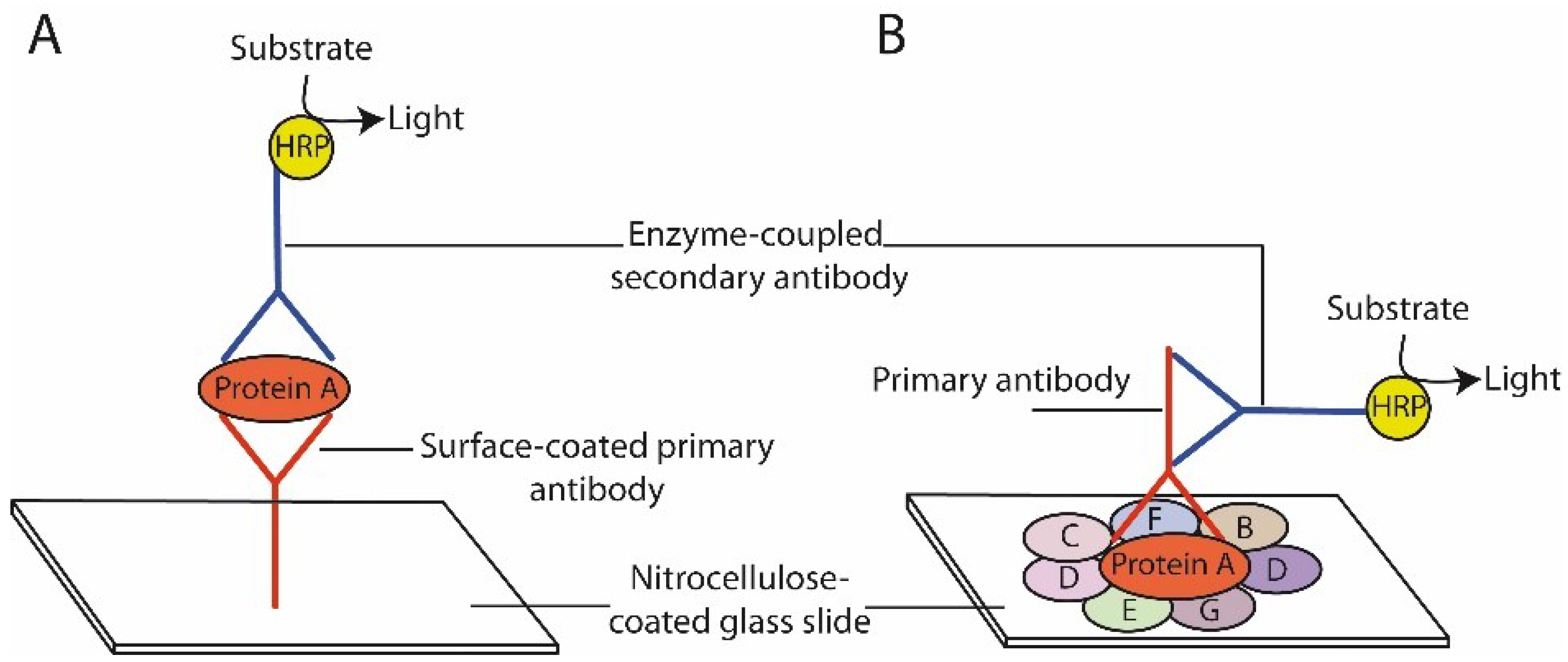{kind=link}
{kind=link}
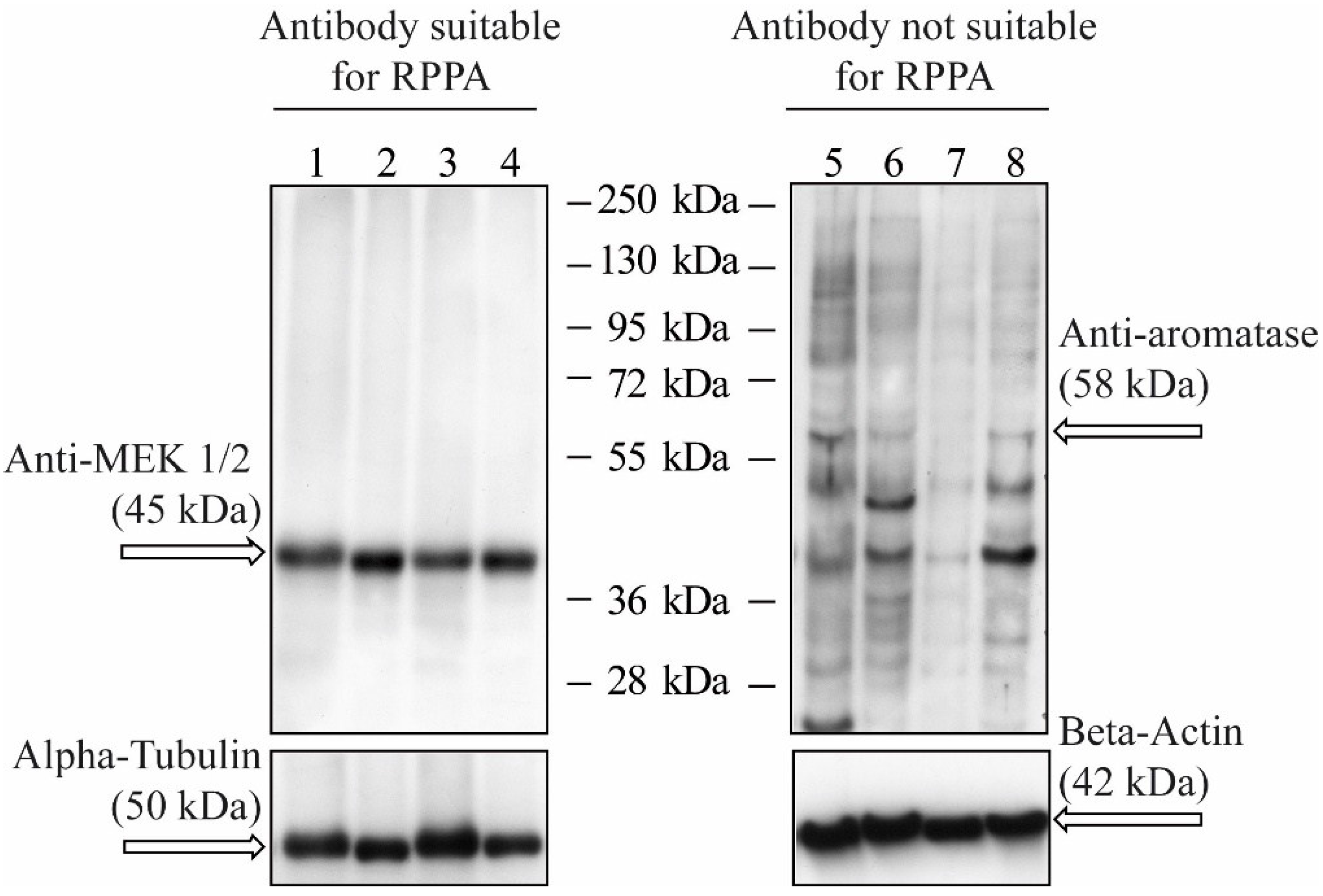{kind=link}
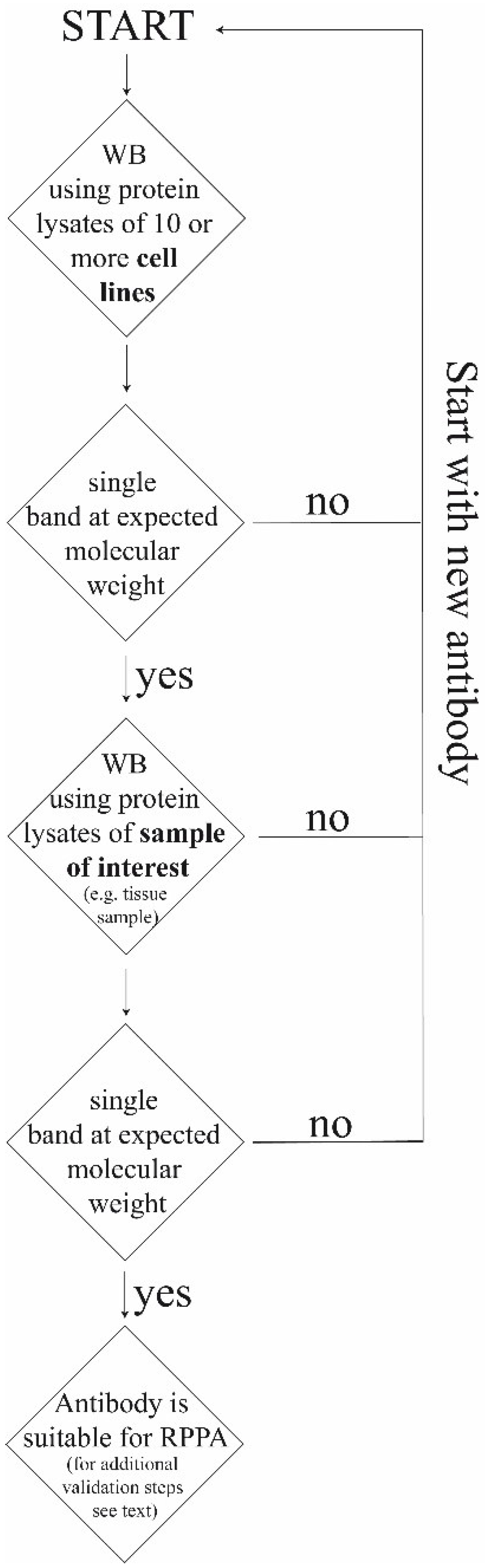{kind=link}
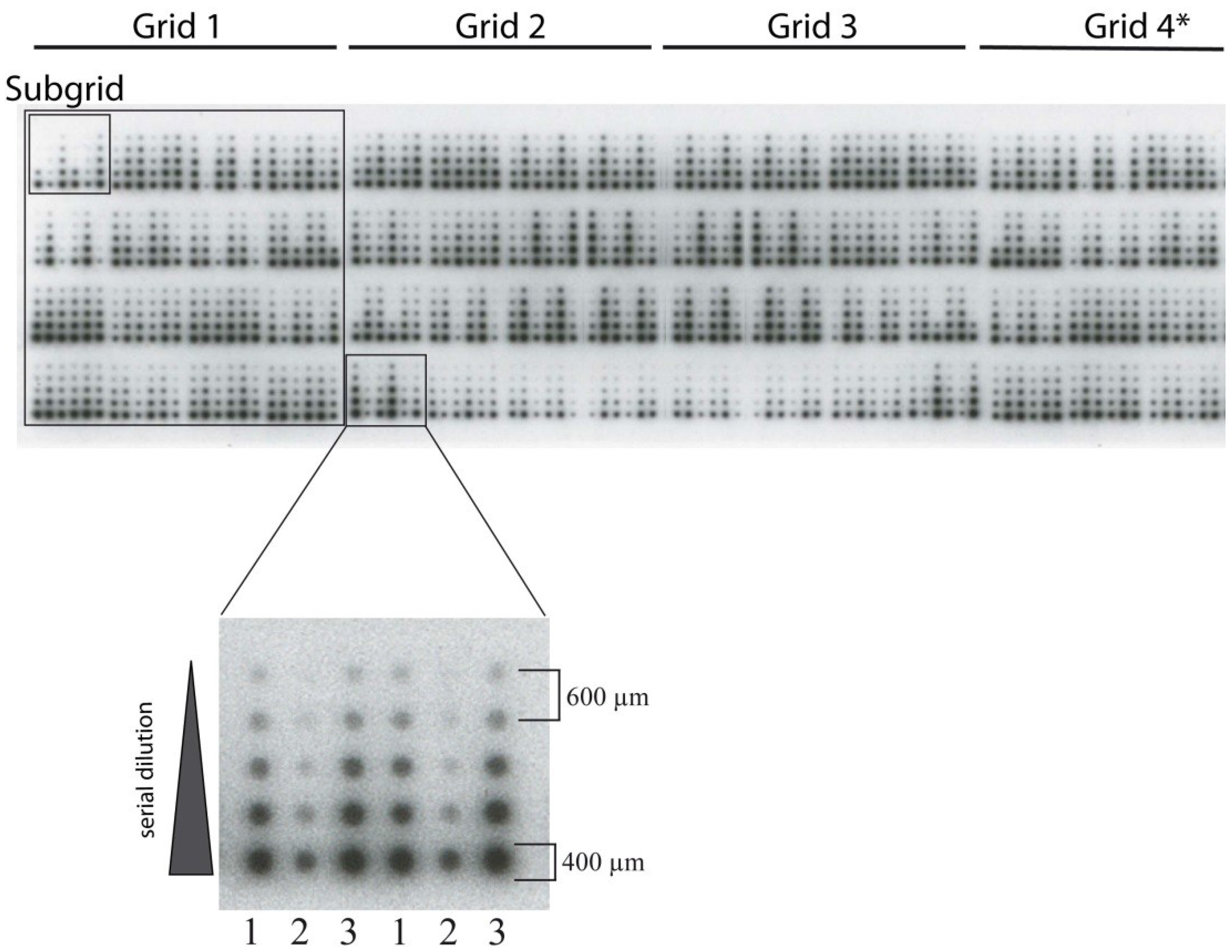{kind=link}
| Protein analysis platform | Advantages | Disadvantages |
|---|---|---|
| Western Blot | Separation of proteins according to molecular weight | Work-intensive, high amounts of protein lysate required, low- or medium-throughput |
| ELISA | Quantitative, very sensitive | High amounts of protein lysate required |
| IHC | Cellular localization of protein of interest | Semi-quantitative, sensitivity often not sufficient to detect phosphorylated proteins |
| Mass spectrometry-based technologies | De novo discovery platform, highly multiplex, protein isoforms can be distinguished, analysis of thousands of proteins, no protein binding reagent required | Complex sample preparation, poor analytical sensitivity compared to immunoassays, low-throughput |
| Forward Phase Protein Arrays | Many analytes can be measured in parallel in a single sample, quantitative | Two highly specific antibodies are needed for every assay, high amounts of protein lysate required |
| Reverse Phase Protein Arrays | Robust quantification, low amount consumption, high-throughput, highly sensitive, detection of phosphoproteins possible | One highly specific antibody is needed for every assay, special devices needed |

2. Clinical Applications of RPPA
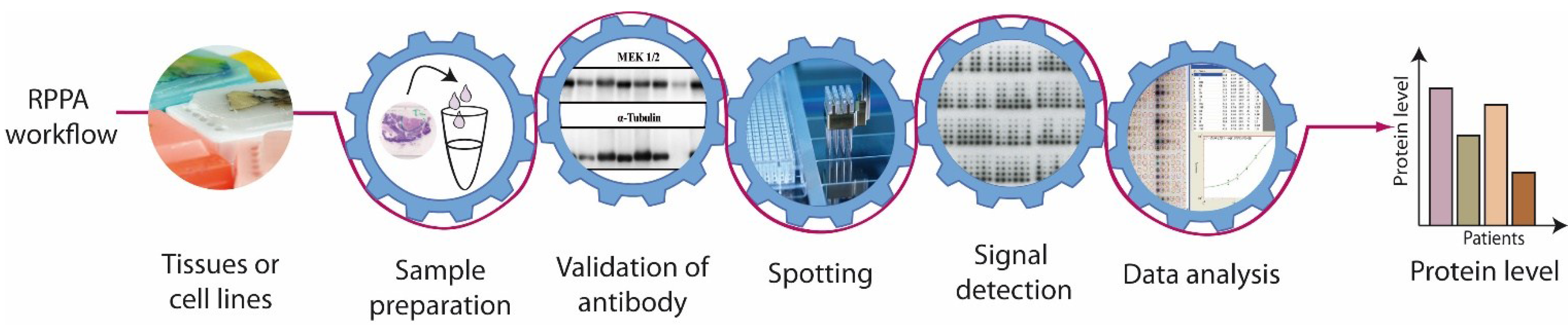
3. Reverse Phase Protein Array-Procedure

3.1. Sample Preparation
3.2. Antibody Validation


3.3. Spotting

3.4. Signal Detection
3.5. Data Analysis
4. Conclusions
5. Outlook
Acknowledgements
Conflicts of Interest
References
- De Gramont, A.; Watson, S.; Ellis, L.M.; Rodon, J.; Tabernero, J.; de Gramont, A.; Hamilton, S.R. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nat. Rev. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Malinowsky, K.; Wolff, C.; Gundisch, S.; Berg, D.; Becker, K. Targeted therapies in cancer—challenges and chances offered by newly developed techniques for protein analysis in clinical tissues. J. Canc. 2010, 2, 26–35. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Pierobon, M.; Wulfkuhle, J.; Liotta, L.; Petricoin, E.F. Application of molecular technologies for phosphoproteomic analysis of clinical samples. Oncogene 2015, 34, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Camp, R.L.; Neumeister, V.; Rimm, D.L. A decade of tissue microarrays: Progress in the discovery and validation of cancer biomarkers. J. Clin. Oncol. 2008, 26, 5630–5637. [Google Scholar] [CrossRef] [PubMed]
- Wulfkuhle, J.D.; Berg, D.; Wolff, C.; Langer, R.; Tran, K.; Illi, J.; Espina, V.; Pierobon, M.; Deng, J.; DeMichele, A.; et al. Molecular analysis of HER2 signaling in human breast cancer by functional protein pathway activation mapping. Clin. Canc. Res. 2012, 18, 6426–6435. [Google Scholar] [CrossRef]
- Murakoshi, Y.; Honda, K.; Sasazuki, S.; Ono, M.; Negishi, A.; Matsubara, J.; Sakuma, T.; Kuwabara, H.; Nakamori, S.; Sata, N.; et al. Plasma biomarker discovery and validation for colorectal cancer by quantitative shotgun mass spectrometry and protein microarray. Canc. Sci. 2011, 102, 630–638. [Google Scholar] [CrossRef]
- Chung, L.; Moore, K.; Phillips, L.; Boyle, F.M.; Marsh, D.J.; Baxter, R.C. Novel serum protein biomarker panel revealed by mass spectrometry and its prognostic value in breast cancer. Breast Canc. Res. 2014, 16, R63. [Google Scholar] [CrossRef]
- Elsner, M.; Rauser, S.; Maier, S.; Schone, C.; Balluff, B.; Meding, S.; Jung, G.; Nipp, M.; Sarioglu, H.; Maccarrone, G.; et al. MALDI imaging mass spectrometry reveals COX7A2, TAGLN2 and S100-A10 as novel prognostic markers in Barrett's adenocarcinoma. J. Proteomics 2012, 75, 4693–4704. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Becker, K.F.; Carragher, N.; Goldstein, T.; de Koning, L.; Korf, U.; Liotta, L.; Mills, G.B.; Nishizuka, S.S.; Pawlak, M.; et al. Realizing the promise of reverse phase protein arrays for clinical, translational, and basic research: A workshop report: the RPPA (Reverse Phase Protein Array) society. Mol. Cell Proteomics 2014, 13, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Espina, V.; Mehta, A.I.; Calvert, V.; Rosenblatt, K.; Geho, D.; Munson, P.J.; Young, L.; Wulfkuhle, J.; Petricoin, E.F. Protein microarrays: Meeting analytical challenges for clinical applications. Canc. Cell 2003, 3, 317–325. [Google Scholar] [CrossRef]
- Paweletz, C.P.; Charboneau, L.; Bichsel, V.E.; Simone, N.L.; Chen, T.; Gillespie, J.W.; Emmert-Buck, M.R.; Roth, M.J.; Petricoin, E.F.; Liotta, L.A. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 2001, 20, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Malinowsky, K.; Wolff, C.; Ergin, B.; Berg, D.; Becker, K.F. Deciphering signaling pathways in clinical tissues for personalized medicine using protein microarrays. J. Cell. Physiol. 2010, 225, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Liotta, L.A.; Petricoin, E.F. Monitoring proteins and protein networks using reverse phase protein arrays. Dis. Markers 2010, 28, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Voshol, H.; Ehrat, M.; Traenkle, J.; Bertrand, E.; van Oostrum, J. Antibody-based proteomics. FEBS J. 2009, 276, 6871–6879. [Google Scholar] [CrossRef] [PubMed]
- Malinowsky, K.; Wolff, C.; Schott, C.; Becker, K. Characterization of Signalling Pathways by Reverse Phase Protein Arrays. Methods Mol. Biol. 2013, 1049, 285–299. [Google Scholar] [PubMed]
- Hong, B.; Lui, V.W.; Hui, E.P.; Lu, Y.; Leung, H.S.; Wong, E.Y.; Cheng, S.H.; Ng, M.H.; Mills, G.B.; Chan, A.T. Reverse phase protein array identifies novel anti-invasion mechanisms of YC-1. Biochem. Pharmacol. 2010, 79, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Gulmann, C.; Sheehan, K.M.; Conroy, R.M.; Wulfkuhle, J.D.; Espina, V.; Mullarkey, M.J.; Kay, E.W.; Liotta, L.A.; Petricoin, E.F., 3rd. Quantitative cell signalling analysis reveals down-regulation of MAPK pathway activation in colorectal cancer. J. Pathol. 2009, 218, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Spurrier, B.; Ramalingam, S.; Nishizuka, S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat. Protocol. 2008, 3, 1796–1808. [Google Scholar] [CrossRef]
- Sonntag, J.; Bender, C.; Soons, Z.; von der Heyde, S.; König, R.; Wiemann, S.; Sinn, H.; Schneeweiss, A.; Beißbarth, T.; Korf, U. Reverse phase protein array based tumor profiling identifies a biomarker signature for risk classification of hormone receptor-positive breast cancer. Trans. Proteomics 2014, 2, 52–59. [Google Scholar] [CrossRef]
- Masuda, M.; Chen, W.Y.; Miyanaga, A.; Nakamura, Y.; Kawasaki, K.; Sakuma, T.; Ono, M.; Chen, C.L.; Honda, K.; Yamada, T. Alternative mammalian target of rapamycin (mTOR) signal activation in sorafenib-resistant hepatocellular carcinoma cells revealed by array-based pathway profiling. Mol. Cell. Proteomics 2014, 13, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Jameson, G.S.; Petricoin, E.F.; Sachdev, J.; Liotta, L.A.; Loesch, D.M.; Anthony, S.P.; Chadha, M.K.; Wulfkuhle, J.D.; Gallagher, R.I.; Reeder, K.A.; et al. A pilot study utilizing multi-omic molecular profiling to find potential targets and select individualized treatments for patients with previously treated metastatic breast cancer. Breast Canc. Res. Treat. 2014, 147, 579–588. [Google Scholar] [CrossRef]
- Honda, K.; Ono, M.; Shitashige, M.; Masuda, M.; Kamita, M.; Miura, N.; Yamada, T. Proteomic approaches to the discovery of cancer biomarkers for early detection and personalized medicine. Jpn. J. Clin. Oncol. 2013, 43, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Liotta, L.A.; Espina, V. Reverse phase protein microarrays advance to use in clinical trials. Mol. Oncol. 2010, 4, 461–481. [Google Scholar] [CrossRef]
- Spruessel, A.; Steimann, G.; Jung, M.; Lee, S.A.; Carr, T.; Fentz, A.K.; Spangenberg, J.; Zornig, C.; Juhl, H.H.; David, K.A. Tissue ischemia time affects gene and protein expression patterns within minutes following surgical tumor excision. BioTechniques 2004, 36, 1030–1037. [Google Scholar] [PubMed]
- Espina, V.; Edmiston, K.H.; Heiby, M.; Pierobon, M.; Sciro, M.; Merritt, B.; Banks, S.; Deng, J.; VanMeter, A.J.; Geho, D.H.; et al. A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol. Cell. Proteomics 2008, 7, 1998–2018. [Google Scholar] [CrossRef] [PubMed]
- Espina, V.; Mueller, C.; Edmiston, K.; Sciro, M.; Petricoin, E.F.; Liotta, L.A. Tissue is alive: New technologies are needed to address the problems of protein biomarker pre-analytical variability. Proteonomics Clin. Appl. 2009, 3, 874–882. [Google Scholar] [CrossRef]
- Gündisch, S.; Hauck, S.; Sarioglu, H.; Schott, C.; Viertler, C.; Kap, M.; Schuster, T.; Reischauer, B.; Rosenberg, R.; Verhoef, C.; et al. Variability of protein and phosphoprotein levels in clinical tissue specimens during the preanalytical phase. J. Proteonomics Res. 2012, 11, 5748–5762. [Google Scholar]
- Espina, V.; Mueller, C.; Liotta, L.A. Phosphoprotein stability in clinical tissue and its relevance for reverse phase protein microarray technology. Methods Mol. Biol. 2011, 785, 23–43. [Google Scholar] [PubMed]
- Silvestri, A.; Calvert, V.; Belluco, C.; Lipsky, M.; De Maria, R.; Deng, J.; Colombatti, A.; De Marchi, F.; Nitti, D.; Mammano, E.; et al. Protein pathway activation mapping of colorectal metastatic progression reveals metastasis-specific network alterations. Clin. Exp. Metastasis 2013, 30, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Amir, E.; Miller, N.; Geddie, W.; Freedman, O.; Kassam, F.; Simmons, C.; Oldfield, M.; Dranitsaris, G.; Tomlinson, G.; Laupacis, A.; et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. 2012, 30, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Niikura, N.; Liu, J.; Hayashi, N.; Mittendorf, E.A.; Gong, Y.; Palla, S.L.; Tokuda, Y.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Ueno, N.T. Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. J. Clin. Oncol. 2012, 30, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Malinowsky, K.; Raychaudhuri, M.; Buchner, T.; Thulke, S.; Wolff, C.; Hofler, H.; Becker, K.F.; Avril, S. Common protein biomarkers assessed by reverse phase protein arrays show considerable intratumoral heterogeneity in breast cancer tissues. PLoS One 2012, 7, e40285. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.F.; Schott, C.; Metzger, V.; Porschewski, P.; Beck, R.; Nährig, J.; Becker, I.; Höfler, H. Quantitative protein analysis from formalin-fixed tissues: implications for translational clinical research and nanoscale molecular diagnosis. J. Pathol. 2007, 211, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R. Immunohistochemistry in surgical pathology: principles and practice. Methods Mol. Biol. 2014, 1180, 81–109. [Google Scholar] [PubMed]
- Silvestri, A.; Colombatti, A.; Calvert, V.S.; Deng, J.; Mammano, E.; Belluco, C.; De Marchi, F.; Nitti, D.; Liotta, L.A.; Petricoin, E.F.; et al. Protein pathway biomarker analysis of human cancer reveals requirement for upfront cellular-enrichment processing. Lab. Investig. 2010, 90, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.F.; Schott, C.; Becker, I.; Hofler, H. Guided protein extraction from formalin-fixed tissues for quantitative multiplex analysis avoids detrimental effects of histological stains. Proteonomics Clin. Appl. 2008, 2, 737–743. [Google Scholar] [CrossRef]
- Addis, M.F.; Tanca, A.; Pagnozzi, D.; Crobu, S.; Fanciulli, G.; Cossu-Rocca, P.; Uzzau, S. Generation of high-quality protein extracts from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 3815–3823. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.S.; Liang, Q.; Liu, J.; Wei, M.Q.; Winters, M.; Liotta, L.; Sandberg, G.; Gong, M. A nondestructive molecule extraction method allowing morphological and molecular analyses using a single tissue section. Lab. Investig. 2005, 85, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Lee, S.J.; Kris, Y.; Braunschweig, T.; Traicoff, J.L.; Hewitt, S.M. A well-based reverse-phase protein array applicable to extracts from formalin-fixed paraffin-embedded tissue. Proteonomics Clin. Appl. 2008, 2, 1539–1547. [Google Scholar] [CrossRef]
- Ikeda, K.; Monden, T.; Kanoh, T.; Tsujie, M.; Izawa, H.; Haba, A.; Ohnishi, T.; Sekimoto, M.; Tomita, N.; Shiozaki, H.; et al. Extraction and analysis of diagnostically useful proteins from formalin-fixed, paraffin-embedded tissue sections. J. Histochem. Cytochem. 1998, 46, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Nirmalan, N.J.; Harnden, P.; Selby, P.J.; Banks, R.E. Development and validation of a novel protein extraction methodology for quantitation of protein expression in formalin-fixed paraffin-embedded tissues using western blotting. J. Pathol. 2009, 217, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.R.; Liu, C.; Balgley, B.M.; Lee, C.; Taylor, C.R. Protein extraction from formalin-fixed, paraffin-embedded tissue sections: quality evaluation by mass spectrometry. J. Histochem. Cytochem. 2006, 54, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Assadi, M.; Lamerz, J.; Jarutat, T.; Farfsing, A.; Paul, H.; Gierke, B.; Breitinger, E.; Templin, M.F.; Essioux, L.; Arbogast, S.; et al. Multiple protein analysis of formalin-fixed and paraffin-embedded tissue samples with reverse phase protein arrays. Mol. Cell Proteomics 2013, 12, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, W.; Ju, Z.; Tamboli, P.; Jonasch, E.; Mills, G.B.; Lu, Y.; Hennessy, B.T.; Tsavachidou, D. An efficient procedure for protein extraction from formalin-fixed, paraffin-embedded tissues for reverse phase protein arrays. Proteome Sci. 2012, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.R.; Taylor, C.R.; Fowler, C.B.; Mason, J.T. Complete solubilization of formalin-fixed, paraffin-embedded tissue may improve proteomic studies. Proteonomics Clin. Appl. 2013, 7, 264–272. [Google Scholar] [CrossRef]
- Becker, K.F. Using tissue samples for proteomic studies—critical considerations. Proteonomics Clin. Appl. 2015. [Google Scholar] [CrossRef]
- Janzi, M.; Odling, J.; Pan-Hammarstrom, Q.; Sundberg, M.; Lundeberg, J.; Uhlen, M.; Hammarstrom, L.; Nilsson, P. Serum microarrays for large scale screening of protein levels. Mol. Cell Proteomics 2005, 4, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Janzi, M.; Sjoberg, R.; Wan, J.; Fischler, B.; von Dobeln, U.; Isaac, L.; Nilsson, P.; Hammarstrom, L. Screening for C3 deficiency in newborns using microarrays. PLoS One 2009, 4, e5321. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Espina, V.; Steinberg, S.M.; Florenes, V.A.; Liotta, L.A.; Kristensen, G.B.; Trope, C.G.; Berner, A.; Kohn, E.C. Proteomic analysis of malignant ovarian cancer effusions as a tool for biologic and prognostic profiling. Clin. Canc. Res. 2006, 12, 791–799. [Google Scholar] [CrossRef]
- Nishizuka, S.; Charboneau, L.; Young, L.; Major, S.; Reinhold, W.C.; Waltham, M.; Kouros-Mehr, H.; Bussey, K.J.; Lee, J.K.; Espina, V.; et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc. Natl. Acad. Sci. USA 2003, 100, 14229–14234. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Zhou, W.; Vanmeter, A.; Heiby, M.; Magaki, S.; Ross, M.M.; Espina, V.; Schrag, M.; Dickson, C.; Liotta, L.A.; et al. The heme degradation pathway is a promising serum biomarker source for the early detection of Alzheimer's disease. J. Alzheim. Dis. 2010, 19, 1081–1091. [Google Scholar]
- MacBeath, G.; Schreiber, S.L. Printing proteins as microarrays for high-throughput function determination. Science 2000, 289, 1760–1763. [Google Scholar] [PubMed]
- Berg, D.; Langer, R.; Tran, K.; Walch, A.; Schuster, T.; Becker, K.F. Protein Microarray-based Comparison of HER2, Estrogen Receptor, and Progesterone Receptor Status in Core Biopsies and Surgial Specimens From FFPE Breast Cancer Tissues. Appl. Immunhistochem. Mol. Morphol. 2011, 19, 300–305. [Google Scholar] [CrossRef]
- Lee, A.H.; Key, H.P.; Bell, J.A.; Hodi, Z.; Ellis, I.O. Concordance of HER2 status assessed on needle core biopsy and surgical specimens of invasive carcinoma of the breast. Histopathology 2012, 60, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.; Malinowsky, K.; Liebmann, S.; Berg, D.; Wolff, C.; Tran, K.; Schott, C.; Reu, S.; Neumann, J.; Faber, C.; et al. Antibody validation by combining immunohistochemistry and protein extraction from formalin-fixed paraffin-embedded tissues. Histopathology 2012, 60, 37–50. [Google Scholar] [CrossRef]
- Berg, D.; Hipp, S.; Malinowsky, K.; Böllner, C.; Becker, K.F. Molecular profiling of signalling pathways in formalin-fixed and paraffin-embedded cancer tissues. Eur. J. Canc. 2010, 46, 47–55. [Google Scholar] [CrossRef]
- Tibes, R.; Qiu, Y.; Lu, Y.; Hennessy, B.; Andreeff, M.; Mills, G.B.; Kornblau, S.M. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Canc. Therapeut. 2006, 5, 2512–2521. [Google Scholar] [CrossRef]
- VanMeter, A.J.; Rodriguez, A.S.; Bowman, E.D.; Jen, J.; Harris, C.C.; Deng, J.; Calvert, V.S.; Silvestri, A.; Fredolini, C.; Chandhoke, V.; et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGPR) phosphorylation events associated with mutated EGFR compared with wild type. Mol. Cell. Proteomics 2008, 7, 1902–1924. [Google Scholar] [CrossRef] [PubMed]
- MD Anderson Cancer Center. Available online: http://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/functional-proteomics-rppa-core/antibody-lists-protocols/functional-proteomics-reverse-phase-protein-array-core-facility-antibody-lists-and-protocols.html (accessed on 19 March 2015).
- Ressine, A.; Corin, I.; Jaras, K.; Guanti, G.; Simone, C.; Marko-Varga, G.; Laurell, T. Porous silicon surfaces: a candidate substrate for reverse protein arrays in cancer biomarker detection. Electrophoresis 2007, 28, 4407–4415. [Google Scholar] [CrossRef] [PubMed]
- Malinowsky, K.; Nitsche, U.; Janssen, K.P.; Bader, F.G.; Spath, C.; Drecoll, E.; Keller, G.; Hofler, H.; Slotta-Huspenina, J.; Becker, K.F. Activation of the PI3K/AKT pathway correlates with prognosis in stage II colon cancer. Brit. J. Canc. 2014, 110, 2081–2089. [Google Scholar] [CrossRef]
- Wang, X.; Dong, Y.; Jiwani, A.J.; Zou, Y.; Pastor, J.; Kuro, O.M.; Habib, A.A.; Ruan, M.; Boothman, D.A.; Yang, C.R. Improved protein arrays for quantitative systems analysis of the dynamics of signaling pathway interactions. Proteome Sci. 2011, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Loebke, C.; Sueltmann, H.; Schmidt, C.; Henjes, F.; Wiemann, S.; Poustka, A.; Korf, U. Infrared-based protein detection arrays for quantitative proteomics. Proteomics 2007, 7, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Espina, V.; Woodhouse, E.C.; Wulfkuhle, J.; Asmussen, H.D.; Petricoin, E.F.; Liotta, L.A. Protein microarray detection strategies: focus on direct detection technologies. J. Immunol. Meth. 2004, 290, 121–133. [Google Scholar] [CrossRef]
- Rapkiewicz, A.; Espina, V.; Zujewski, J.A.; Lebowitz, P.F.; Filie, A.; Wulfkuhle, J.; Camphausen, K.; Petricoin, E.F.; Liotta, L.A.; Abati, A. The needle in the haystack: Application of breast fine-needle aspirate samples to quantitative protein microarray technology. Cancer Cytopathol. 2007, 111, 173–184. [Google Scholar] [CrossRef]
- Kaushik, P.; Molinelli, E.J.; Miller, M.L.; Wang, W.; Korkut, A.; Liu, W.; Ju, Z.; Lu, Y.; Mills, G.; Sander, C. Spatial Normalization of Reverse Phase Protein Array Data. PLoS One 2014, 9, e97213. [Google Scholar] [CrossRef] [PubMed]
- Neeley, E.S.; Kornblau, S.M.; Coombes, K.R.; Baggerly, K.A. Variable slope normalization of reverse phase protein arrays. Bioinformatics 2009, 25, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Mannsperger, H.A.; Gade, S.; Henjes, F.; Beissbarth, T.; Korf, U. RPPanalyzer: Analysis of reverse-phase protein array data. Bioinformatics 2010, 26, 2202–2203. [Google Scholar] [CrossRef] [PubMed]
- Von der Heyde, S.; Sonntag, J.; Kaschek, D.; Bender, C.; Bues, J.; Wachter, A.; Timmer, J.; Korf, U.; Beissbarth, T. RPPanalyzer toolbox: An improved R package for analysis of reverse phase protein array data. BioTechniques 2014, 57, 125–135. [Google Scholar] [PubMed]
- Chiechi, A.; Mueller, C.; Boehm, K.M.; Romano, A.; Benassi, M.S.; Picci, P.; Liotta, L.A.; Espina, V. Improved data normalization methods for reverse phase protein microarray analysis of complex biological samples. BioTechniques 2012. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boellner, S.; Becker, K.-F. Reverse Phase Protein Arrays—Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays 2015, 4, 98-114. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4020098
Boellner S, Becker K-F. Reverse Phase Protein Arrays—Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays. 2015; 4(2):98-114. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4020098
Chicago/Turabian StyleBoellner, Stefanie, and Karl-Friedrich Becker. 2015. "Reverse Phase Protein Arrays—Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use" Microarrays 4, no. 2: 98-114. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4020098