
Data Mining of Gene Arrays for Biomarkers of Survival in Ovarian Cancer

Abstract
:
1. Introduction
2. Experimental Section

2.1. Source Data
2.2. Datasets Used
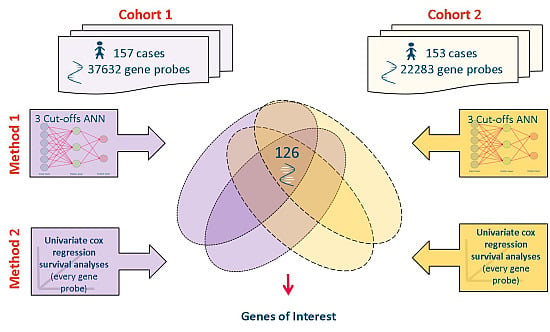
2.3. Meta-Analysis of Microarray Data

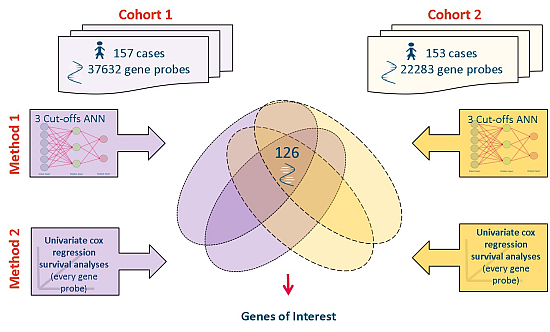{kind=link}
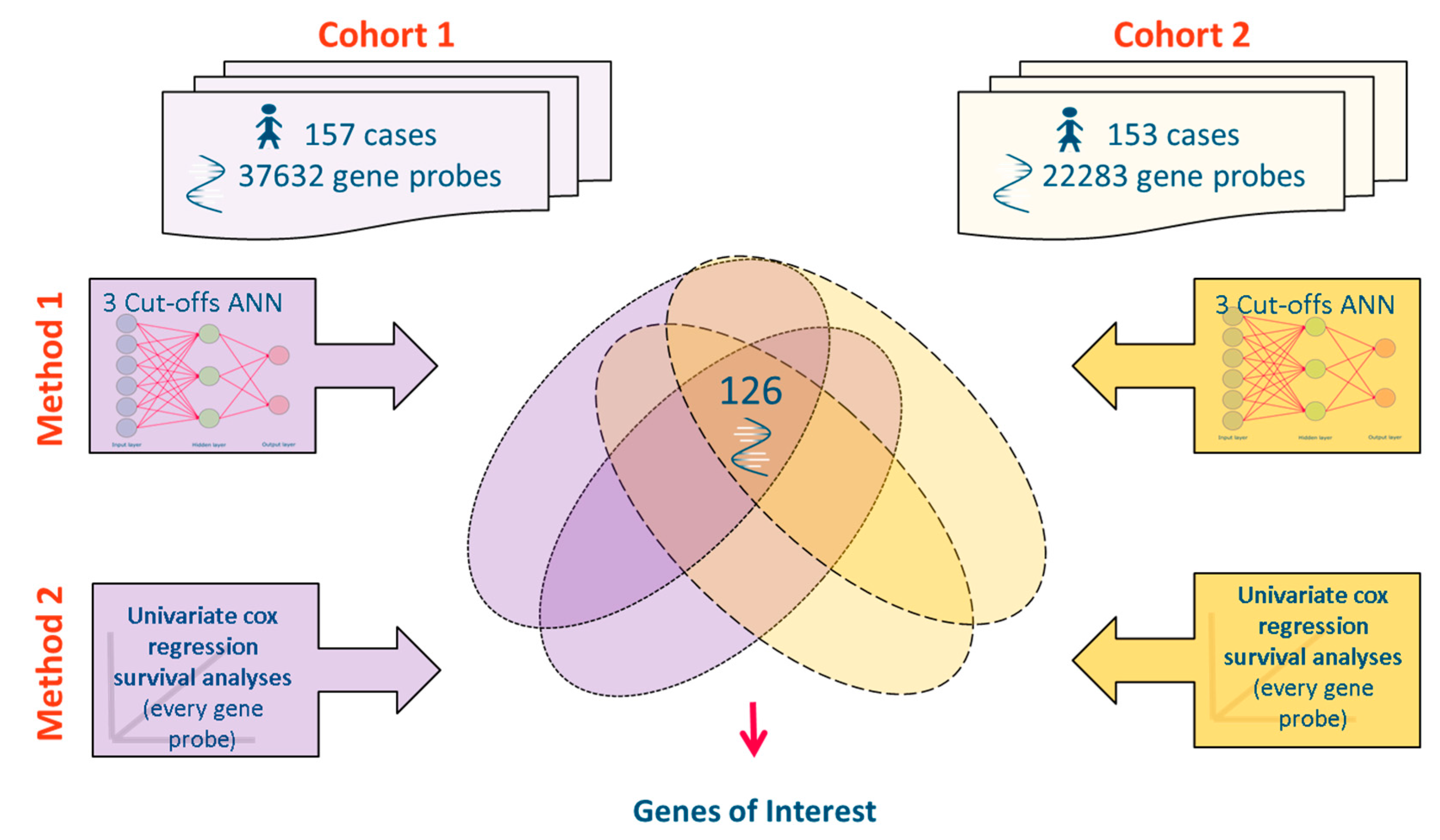{kind=link}
{kind=link}
{kind=link}
| Gene Code | Gene Name | Rank Order of Interest |
|---|---|---|
| DCN | decorin | 1 |
| EDNRA | endothelin receptor type A | 2 |
| GLT8D2 | glycosyltransferase 8 domain containing 2 | 3 |
| IGF2 | insulin-like growth factor 2 (somatomedin A)///INS-IGF2 readthrough | 4 |
| MFAP4 | microfibrillar-associated protein 4 | 5 |
| PDZRN3 | PDZ domain containing ring finger 3 | 6 |
| PKD2 | polycystic kidney disease 2 (autosomal dominant) | 7 |
| SEMA3C | sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3C | 8 |
| IGFBP6 | insulin-like growth factor binding protein 6 | 9 |
| LDB2 | LIM domain binding 2 | 10 |
| NAV3 | neuron navigator 3 | 11 |
| NDN | necdin homolog (mouse) | 12 |
| OLFML3 | olfactomedin-like 3 | 13 |
| PCDH17 | protocadherin 17 | 14 |
| PJA2 | praja ring finger 2, E3 ubiquitin protein ligase | 15 |
| PPFIBP1 | PTPRF interacting protein, binding protein 1 (liprin β 1) | 16 |
| RARRES2 | retinoic acid receptor responder (tazarotene induced) 2 | 17 |
| SFRP4 | secreted frizzled-related protein 4 | 18 |
| BMP4 | bone morphogenetic protein 4 | 19 |
| HNRPDL | heterogeneous nuclear ribonucleoprotein D-like | 20 |
| LRRC17 | leucine rich repeat containing 17 | 21 |
| MAP4K4 | mitogen-activated protein kinase kinase kinase kinase 4 | 22 |
| PPP3CA | protein phosphatase 3, catalytic subunit, α isozyme | 23 |
| COLEC12 | collectin sub-family member 12 | 24 |
| IGFBP3 | insulin-like growth factor binding protein 3 | 25 |
| TNFAIP6 | tumor necrosis factor, α-induced protein 6 | 26 |
| BACH1 | BTB and CNC homology 1, basic leucine zipper transcription factor 1 | 27 |
| INTS5 | integrator complex subunit 5 | 28 |
| TNFRSF14 | tumor necrosis factor receptor superfamily, member 14 | 29 |
| ZFHX4 | zinc finger homeobox 4 | 30 |
| EFNB3 | ephrin-B3 | 31 |
| FYN | FYN oncogene related to SRC, FGR, YES | 32 |
| FZD7 | frizzled family receptor 7 | 33 |
| SCAMP1 | secretory carrier membrane protein 1 | 34 |
| TMEM45A | transmembrane protein 45A | 35 |
| NCOR1 | nuclear receptor corepressor 1 | 36 |
| BACH2 | BTB and CNC homology 1, basic leucine zipper transcription factor 2 | 37 |
| HIST1H3A | histone cluster 1, H3a | 38 |
| CLIP3 | CAP-GLY domain containing linker protein 3 | 39 |
| GULP1 | GULP, engulfment adaptor PTB domain containing 1 | 40 |
| PTPRE | protein tyrosine phosphatase, receptor type, E | 41 |
| SPAG9 | sperm associated antigen 9 | 42 |
| SPCS3 | signal peptidase complex subunit 3 homolog (S. cerevisiae) | 43 |
| CTBP2 | C-terminal binding protein 2 | 44 |
| CDC25B | cell division cycle 25 homolog B (S. pombe) | 45 |
| GJB1 | gap junction protein, β 1, 32 kDa | 46 |
| DCTD | dCMP deaminase | 47 |
| HBD | hemoglobin, delta | 48 |
| SLC11A2 | solute carrier family 11 (proton-coupled divalent metal ion transporters), member 2 | 49 |
| TPM2 | tropomyosin 2 (β) | 50 |
| ZNF45 | zinc finger protein 45 | 51 |
| FHOD3 | formin homology 2 domain containing 3 | 52 |
| H2AFV | H2A histone family, member V | 53 |
| FKBP14 | FK506 binding protein 14, 22 kDa | 54 |
| SMC3 | structural maintenance of chromosomes 3 | 55 |
| WTAP | Wilms tumor 1 associated protein | 56 |
2.4. Verification of Protein Expression
3. Results and Discussion
3.1. Genes of Interest
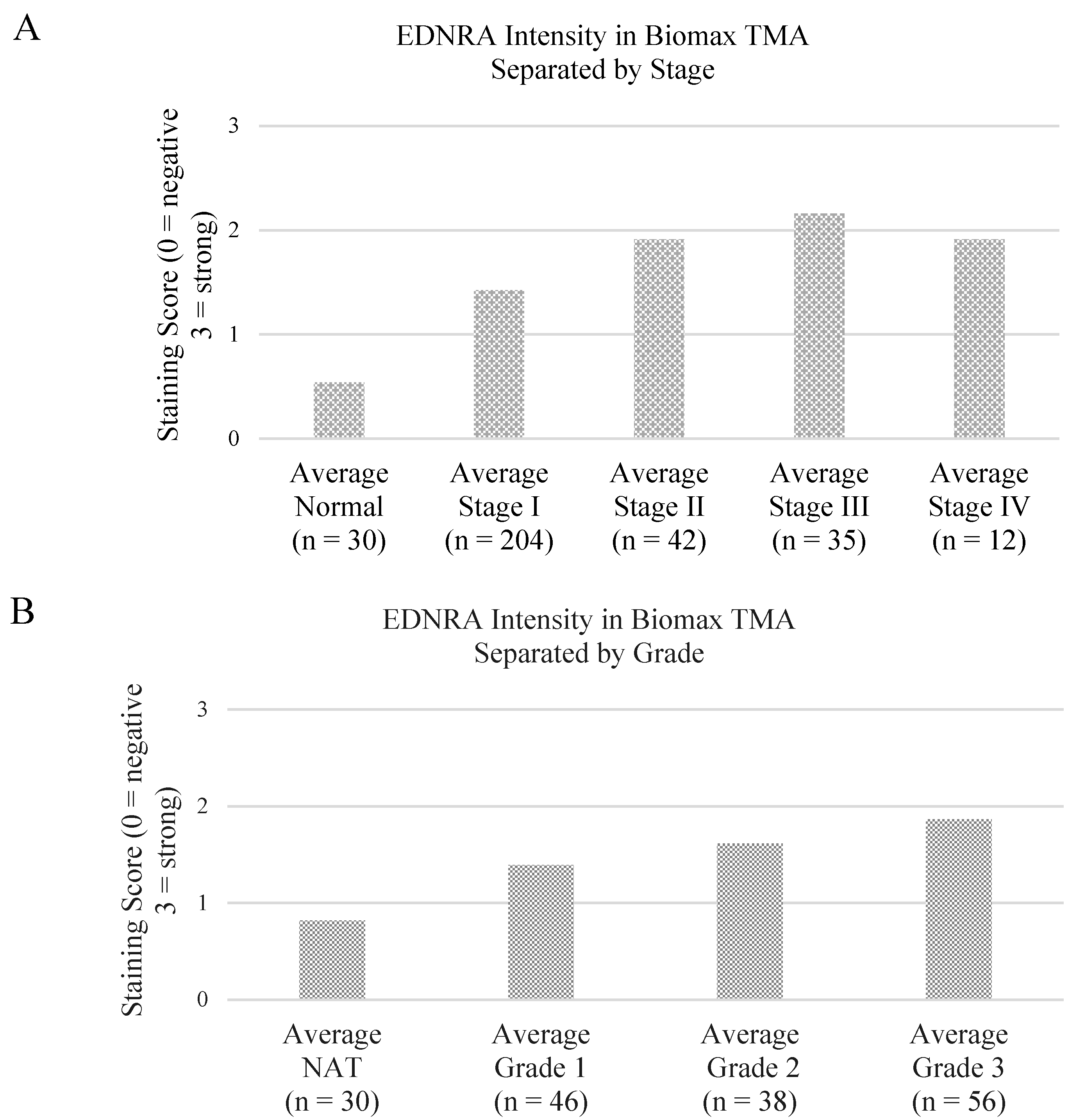
3.2. Preliminary Validation

| p value | Normal | Stage I | Stage II | Stage III | Stage IV |
|---|---|---|---|---|---|
| Normal | - | 2.1974 × 10−5 | 1.00711 × 10−8 | 2.2073 × 10−11 | 9.99574 × 10−7 |
| Stage I | - | - | 0.000137099 | 8.5081 × 10−8 | 0.000137099 |
| Stage II | - | - | - | 0.15060521 | 0.998291248 |
| Stage III | - | - | - | - | 0.316994038 |
| Stage IV | - | - | - | - | - |
| p value | All NAT | All Grade 1 | All Grade 2 | All Grade 3 |
|---|---|---|---|---|
| All NAT | - | 0.005302566 | 4.64816 × 10−6 | 1.36028 × 10−10 |
| All Grade 1 | - | - | 0.244156689 | 0.007596408 |
| All Grade 2 | - | - | - | 0.07998109 |
| All Grade 3 | - | - | - | - |
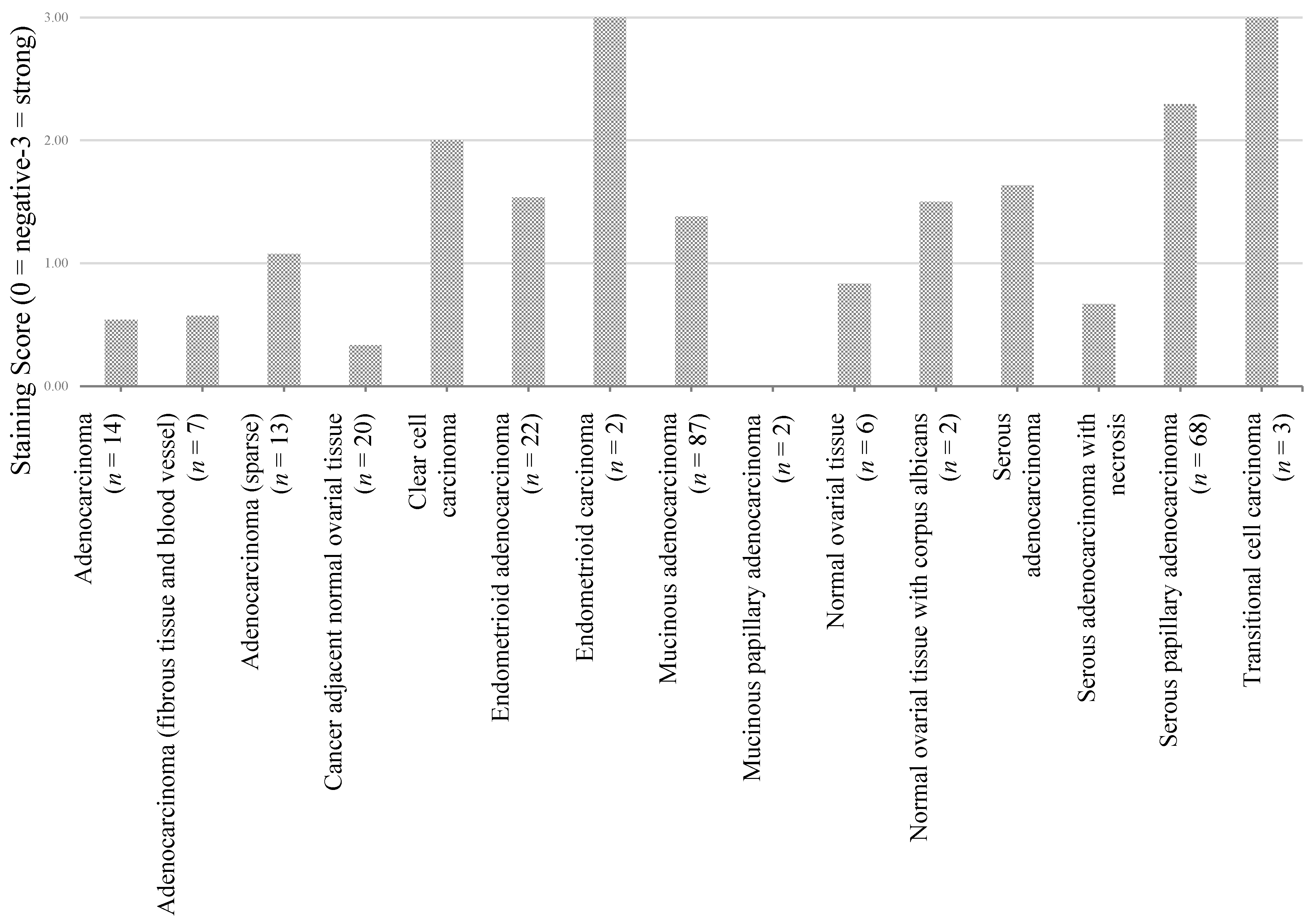
| Adenocarcinoma (n = 14) | Adenocarcinoma (fibrous tissue and blood vessel) (n = 7) | Adenocarcinoma (n = 13) | Cancer adjacent normal ovarial tissue (n = 20) | Clear cell carcinoma (n = 26) | Endometrioid adenocarcinoma (n = 22) | Endometrioid carcinoma (n = 2) | Mucinous adenocarcinoma (n = 87) | Mucinous papillary adenocarcinoma (n = 2) | Normal ovarial tissue (n = 6) | Normal ovarial tissue with corpus albicans (n = 2) | Serous adenocarcinoma (n = 339) | Serous adenocarcinoma ith necrosis (n = 6) | Serous papillary adenocarcinoma (n = 68) | Transitional cell carcinoma (n = 3) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adenocarcinoma (n = 14) | - | 0.91 | 0.18 | 0.37 | 0.01 | 0.06 | 0.00 | 0.07 | 0.19 | 0.35 | 0.08 | 0.01 | 0.81 | 0.00 | 0.00 |
| Adenocarcinoma (fibrous tissue and blood vessel) (n = 7) | - | - | 0.09 | 0.37 | 0.00 | 0.01 | 0.00 | 0.01 | 0.28 | 0.33 | 0.08 | 0.00 | 0.72 | 0.00 | 0.00 |
| Adenocarcinoma (n = 13) | - | - | - | 0.01 | 0.02 | 0.34 | 0.01 | 0.37 | 0.11 | 0.52 | 0.52 | 0.05 | 0.34 | 0.00 | 0.01 |
| Cancer adjacent normal ovarian tissue (n = 20) | - | - | - | - | 0.00 | 0.00 | 0.00 | 0.00 | 0.45 | 0.07 | 0.02 | 0.00 | 0.29 | 0.00 | 0.00 |
| Clear cell carcinoma (n = 26) | - | - | - | - | - | 0.09 | 0.28 | 0.03 | 0.04 | 0.03 | 0.59 | 0.09 | 0.02 | 0.23 | 0.28 |
| Endometrioid adenocarcinoma (n = 22) | - | - | - | - | - | - | 0.05 | 0.92 | 0.08 | 0.21 | 0.59 | 0.32 | 0.12 | 0.00 | 0.05 |
| Endometrioid carcinoma (n = 2) | - | - | - | - | - | - | - | 0.05 | - | 0.00 | 0.10 | 0.06 | 0.01 | 0.26 | - |
| Mucinous adenocarcinoma(n = 87) | - | - | - | - | - | - | - | - | 0.10 | 0.25 | 0.89 | 0.06 | 0.14 | 0.00 | 0.05 |
| Mucinous papillary adenocarcinoma (n = 2) | - | - | - | - | - | - | - | - | - | 0.03 | 0.10 | 0.02 | 0.32 | 0.00 | - |
| Normal ovarian tissue (n = 6) | - | - | - | - | - | - | - | - | - | - | 0.13 | 0.05 | 0.66 | 0.00 | 0.00 |
| Normal ovarian tissue with corpus albicans (n = 2) | - | - | - | - | - | - | - | - | - | - | - | 0.85 | 0.25 | 0.20 | 0.10 |
| Serous adenocarcinoma (n = 339) | - | - | - | - | - | - | - | - | - | - | - | - | 0.02 | 0.00 | 0.06 |
| Serous adenocarcinoma with necrosis (n = 6) | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.00 | 0.01 |
| Serous papillary Adenocarcinoma (n = 68) | - | - | - | - | - | - | - | - | - | - | - | - | - | - | 0.26 |
| Transitional cell Carcinoma (n = 3) | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- CRUK: CancerResearchUK. Available on line: http://info.cancerresearchuk.org (accessed on 20 December 2012).
- Menon, U.; Griffin, M.; Gentry-Maharaj, A. Ovarian cancer screening-Current status, future directions. Gynecol. Oncol. 2014, 132, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.J.; Menon, U. Progress and challenges in screening for early detection of ovarian cancer. Mol. Cell. Proteomics 2004, 3, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.S.; Hacker, N.F. Practical Gynaecologic Oncology. Lippincott Williams & Wilkins (LWW): Philadelphia, PA, USA, 2004. [Google Scholar]
- Chu, C.S.; Rubin, S.C. Screening for ovarian cancer in the general population. Best Pract. Res. Clin. Obstet. Gynaecol. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Nossov, V.; Amneus, M.; Su, F.; Lang, J.; Janco, J.M.T.; Reddy, S.T.; Farias-Eisner, R. The early detection of ovarian cancer: From traditional methods to proteomics. Can we really do better than serum CA-125? Am. J. Obstet. Gynecol. 2008, 199, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Ryan, A.; Kalsi, J.; Gentry-Maharaj, A.; Dawnay, A.; Habib, M.; Apostolidou, S.; Singh, N.; Benjamin, E.; Burnell, M.; et al. Risk algorithm using serial biomarker measurements doubles the number of screen-detected cancers compared with a single-threshold rule in the United Kingdom collaborative trial of ovarian cancer screening. J. Clin. Oncol. 2015, 33, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Braem, M.G.M.; Schouten, L.J.; Peeters, P.H.M.; van den, B.P.A.; Onland-Moret, N.C. Genetic susceptibility to sporadic ovarian cancer: A systematic review. Biochim. Biophys. Acta. 2011, 1816, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Blessing, J.A.; Krasner, C.N.; Mannel, R.S.; Hanjani, P.; Pearl, M.L.; Waggoner, S.E.; Boardman, C.H. Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: A study of the Gynecologic Oncology Group. J. Clin. Oncol. 2009, 27, 2686–2691. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Road, C.; Ka, L.; Centre, S.; Way, R.; Coukos, G. Rethinking Ovarian Cancer: Recommendations for Improving Outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- ArrayExpress. Available online: https://www.ebi.ac.uk/arrayexpress/browse.html (accessed on 20 May 2012).
- Allison, D.B.; Cui, X.; Page, G.P.; Sabripour, M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006, 7, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Lancashire, L.J.; Lemetre, C.; Ball, G.R. An introduction to artificial neural networks in bioinformatics—Application to complex microarray and mass spectrometry datasets in cancer studies. Brief. Bioinform. 2009, 10, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Mukhopadhyay, K. Survival analysis in clinical trials: Basics and must know areas. Perspect Clin. Res. 2011, 2, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Bonome, T.; Levine, D.A.; Shih, J.; Randonovich, M.; Pise-Masison, C.A.; Bogomolniy, F.; Ozbun, L.; Brady, J.; Barrett, J.C.; Birrer, M.J. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008, 68, 5478–5486. [Google Scholar] [CrossRef] [PubMed]
- Crijns, A.P.; Fehrmann, R.S.; de Jong, S.; Gerbens, F.; Meersma, G.J.; Klip, H.G.; Hollema, H.; Hofstra, R.M.W.; te Meerman, G.J.; de Vries, E.G.E.; et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS. Med. 2009, 6, e24. [Google Scholar] [CrossRef] [PubMed]
- Lancashire, L.J.; Rees, R.C.; Ball, G.R. Identification of gene transcript signatures predictive for estrogen receptor and lymph node status using a stepwise forward selection artificial neural network modelling approach. Artif. Intell. Med. 2008, 43, 99–111. [Google Scholar] [CrossRef] [PubMed]
- CompanDX. Available online: http://www.compandx.com (accessed on 16 June 2015).
- STRING: STRING. Available online: http://string-db.org/ (accessed on 10 November 2013).
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, 561–568. [Google Scholar] [CrossRef] [PubMed]
- GeneCards: GeneCards. Available online: http://www.genecards.org/cgi-bin/carddisp.pl?gene=EDNRA&search=68d7ab3090aceaa545e123e269be507a (accessed on 29 September 2014).
- ProteinAtlas: The Human Protein Atlas. Available online: http://www.proteinatlas.org/ (accessed on 29 September 2014).
- Fury, W.; Batliwalla, F.; Gregersen, P.K.; Li, W. Overlapping probabilities of top ranking gene lists, hypergeometric distribution, and stringency of gene selection criterion. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006, 1, 5531–5534. [Google Scholar] [PubMed]
- Biomax: Biomax. Available online: http://www.biomax.us/tissue-arrays/Ovary/OV6161 (accessed on 7 November 2014).
- Devlin, B.; Roeder, K.; Wasserman, L. False discovery or missed discovery? Heredity 2003, 91, 537–538. [Google Scholar] [CrossRef] [PubMed]
- Kanety, H.; Kattan, M.; Goldberg, I.; Kopolovic, J.; Ravia, J.; Menczer, J.; Karasik, A. Increased insulin-like growth factor binding protein-2 (IGFBP-2) gene expression and protein production lead to high IGFBP-2 content in malignant ovarian cyst fluid. Br. J. Cancer 1996, 73, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Mircean, C.; Shmulevich, I.; Wang, H.; Liu, J.; Niemistö, A.; Kavanagh, J.J.; Lee, J.-H.; Zhang, W. Insulin-like growth factor binding protein 2 promotes ovarian cancer cell invasion. Mol. Cancer 2005, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayer, R.A.; Lancaster, J.M.; Pittman, J.; Gray, J.; Whitaker, R.; Marks, J.R.; Berchuck, A. High insulin-like growth factor-2 (IGF-2) gene expression is an independent predictor of poor survival for patients with advanced stage serous epithelial ovarian cancer. Gynecol. Oncol. 2005, 96, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Thériault, B.L.; Shepherd, T.G.; Mujoomdar, M.L.; Nachtigal, M.W. BMP4 induces EMT and Rho GTPase activation in human ovarian cancer cells. Carcinogenesis 2007, 28, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Laatio, L.; Myllynen, P.; Serpi, R.; Rysä, J.; Ilves, M.; Lappi-Blanco, E.; Ruskoaho, H.; Vähäkangas, K.; Puistola, U. BMP-4 expression has prognostic significance in advanced serous ovarian carcinoma and is affected by cisplatin in OVCAR-3 cells. Tumor Biol. 2011, 32, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, E.; Ranki, A.; Sipilä, L.; Karenko, L.; Abdel-Rahman, W.M.; Ovaska, K.; Siggberg, L.; Aapola, U.; Ässämäki, R.; Häyry, V.; et al. Potential role of a navigator gene NAV3 in colorectal cancer. Br. J. Cancer 2012, 106, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Little, N.A.; Hastie, N.D.; Davies, R.C. Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum. Mol. Genet. 2000, 9, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- De Caceres, I.I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguan-Garcia, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; de CastroCarpeño, L.; et al. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Spinella, F.; di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.P.; Mori, S. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coveney, C.; Boocock, D.J.; Rees, R.C.; Deen, S.; Ball, G.R. Data Mining of Gene Arrays for Biomarkers of Survival in Ovarian Cancer. Microarrays 2015, 4, 324-338. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4030324
Coveney C, Boocock DJ, Rees RC, Deen S, Ball GR. Data Mining of Gene Arrays for Biomarkers of Survival in Ovarian Cancer. Microarrays. 2015; 4(3):324-338. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4030324
Chicago/Turabian StyleCoveney, Clare, David J. Boocock, Robert C. Rees, Suha Deen, and Graham R. Ball. 2015. "Data Mining of Gene Arrays for Biomarkers of Survival in Ovarian Cancer" Microarrays 4, no. 3: 324-338. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays4030324