
A Double-Hybridization Approach for the Transcription- and Amplification-Free Detection of Specific mRNA on a Microarray


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Panc-1 Cell Culture
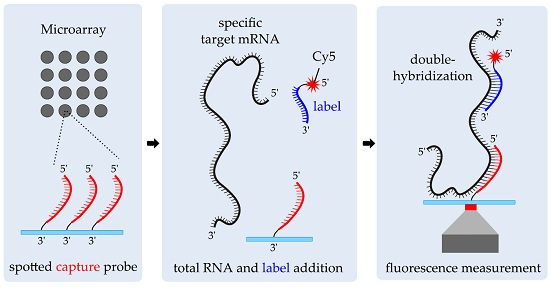
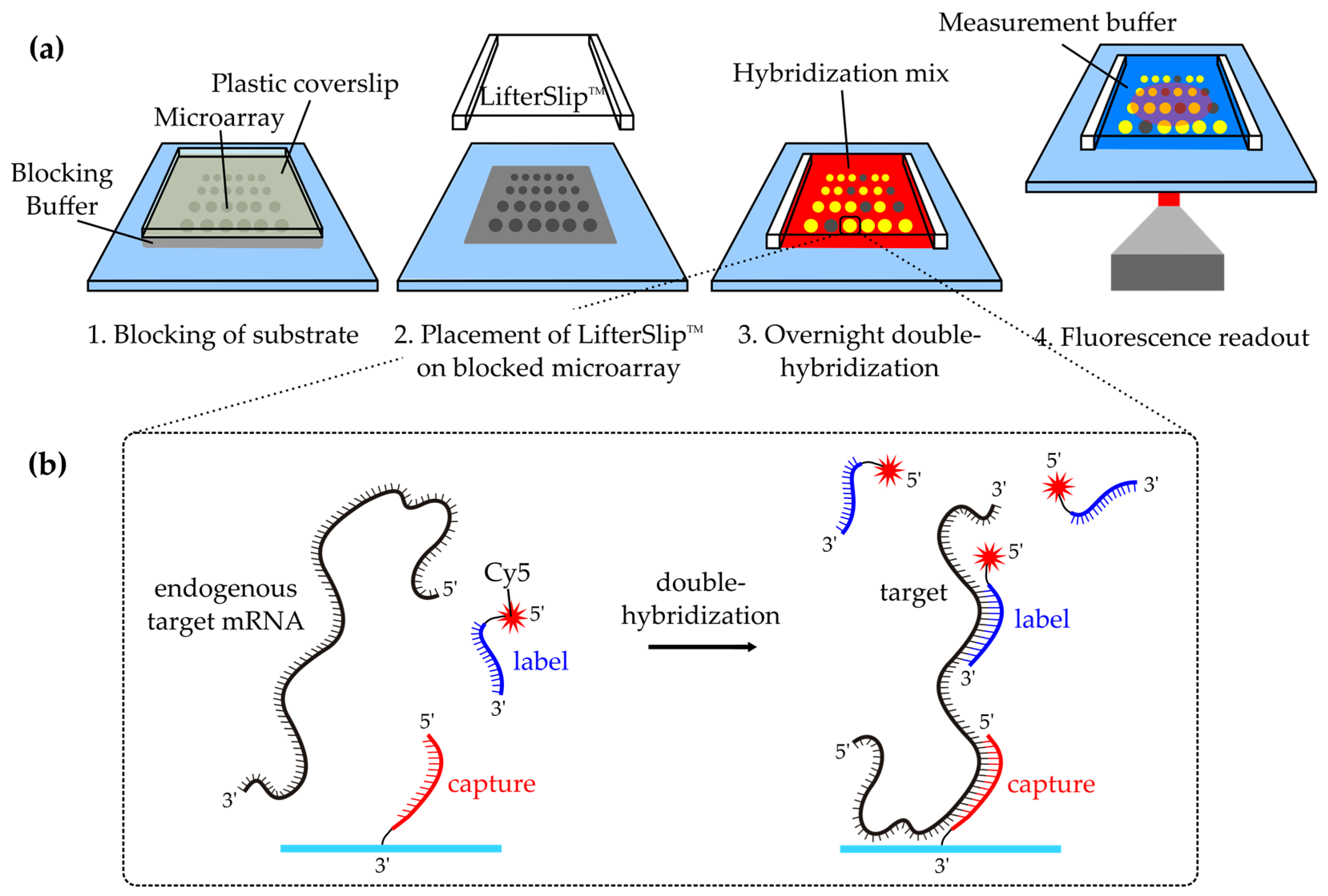
2.2. Double-Hybridization Principle
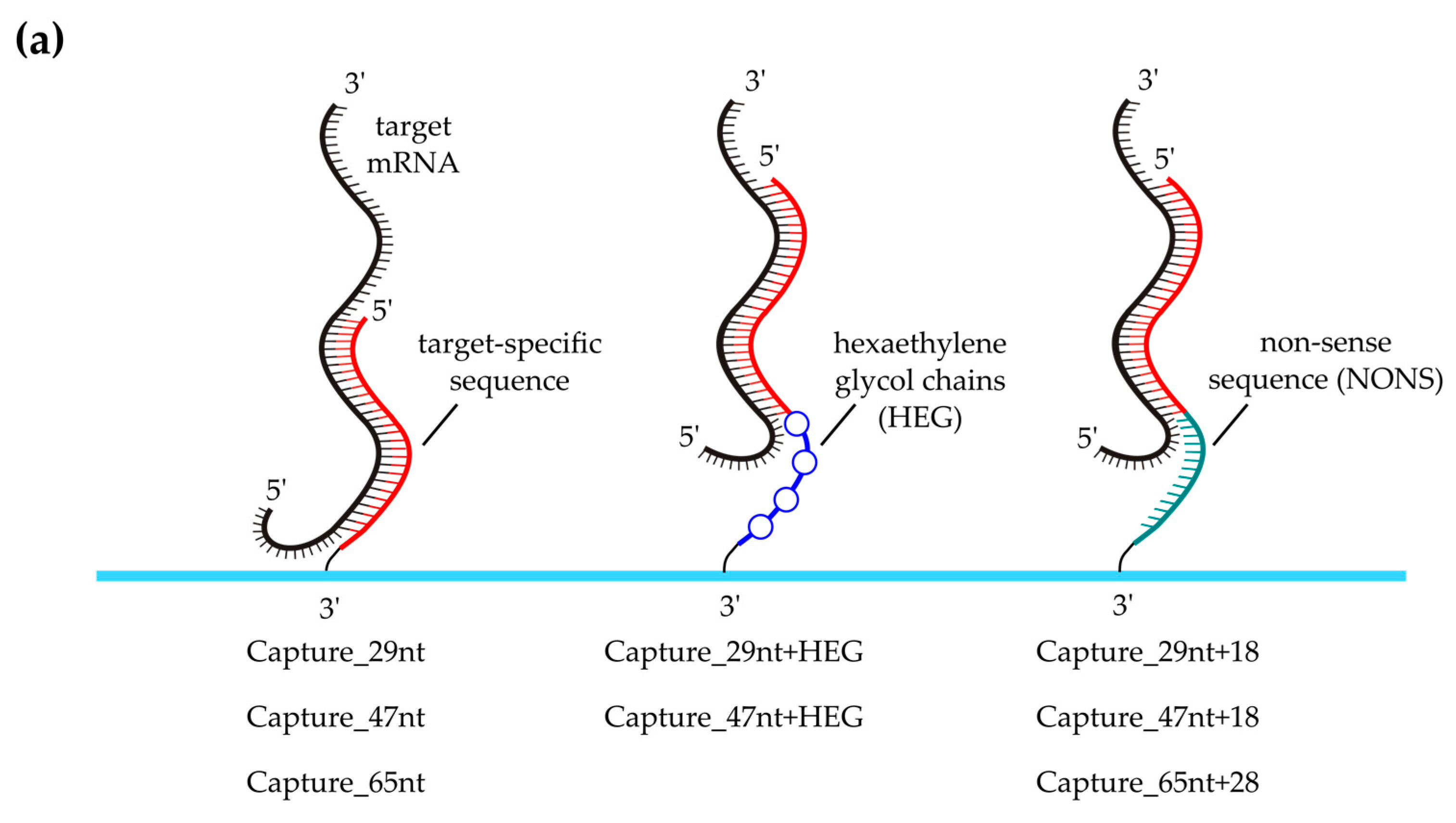
2.3. Probe Design
2.4. Isolation of Total RNA for Microarray Analysis
2.5. DNA Microarray Fabrication
2.6. Double-Hybridization of RPLP0 mRNA
2.7. Fluorescence Microscopy
2.8. Data Analysis
3. Results
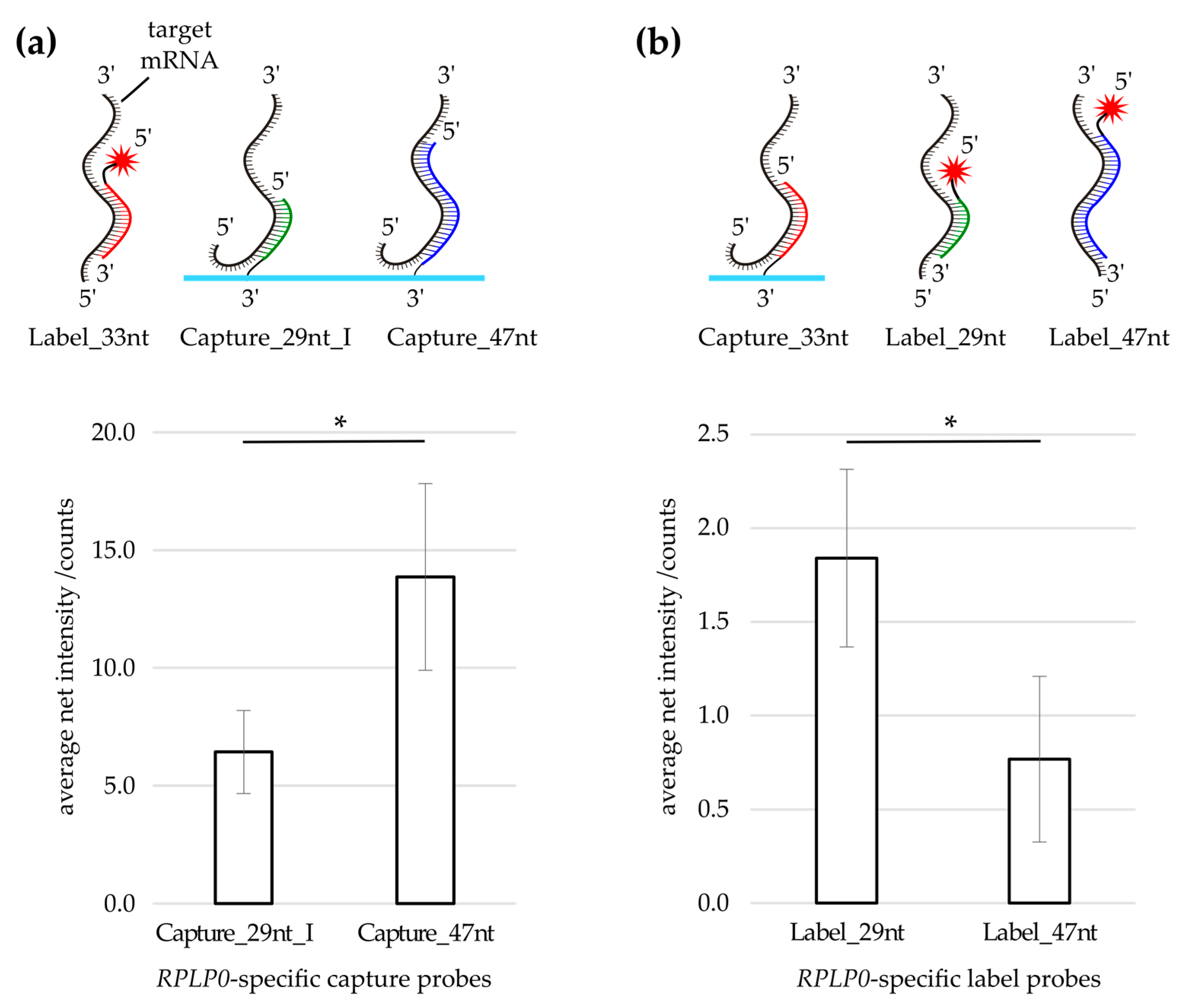
3.1. Label Probe Optimization
3.2. Evaluation of Capture Probe Length
3.3. Longer Specific Capture Probes Increase Specific Signal
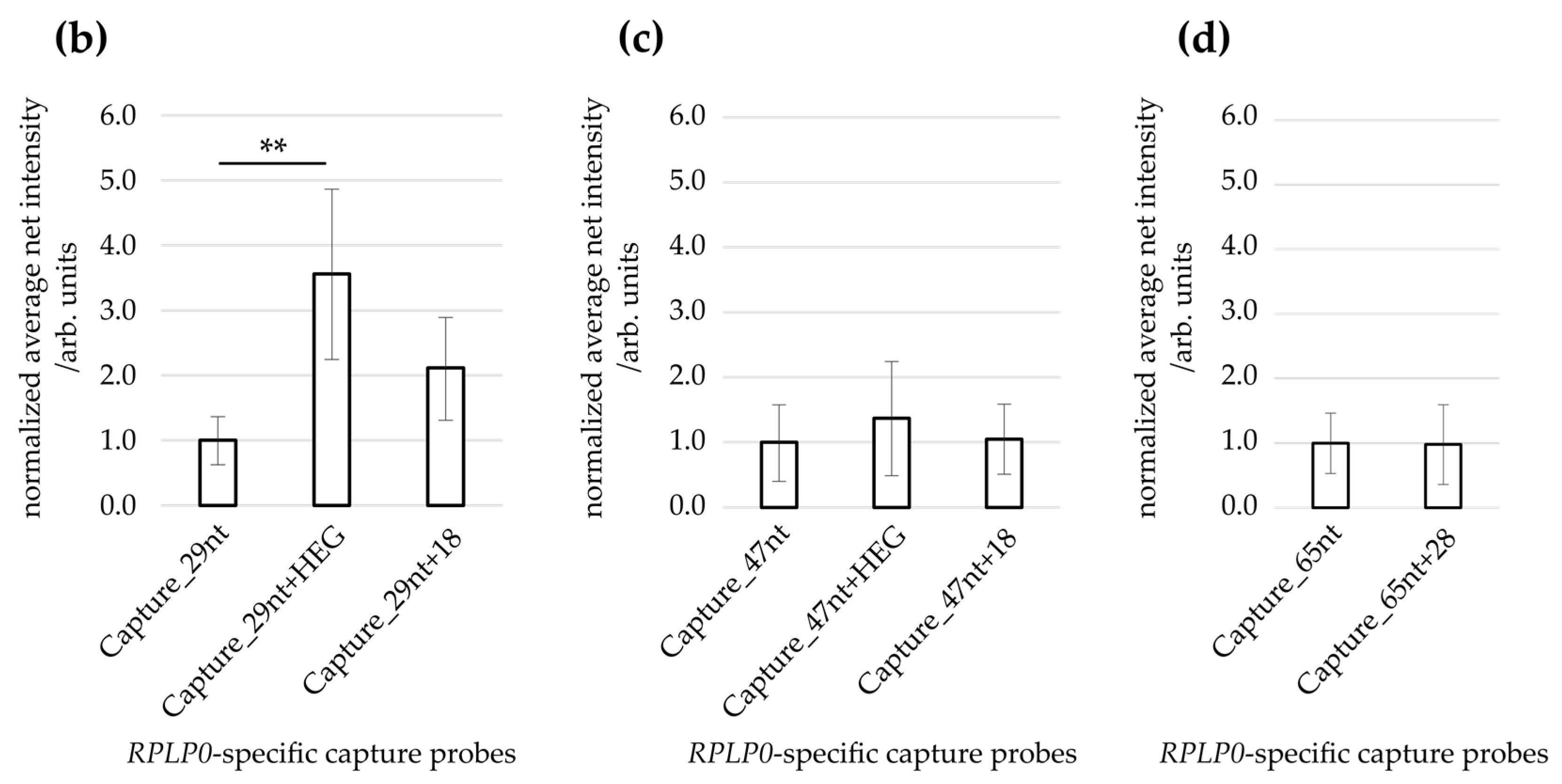
3.4. Effect of Capture Probe Spacers
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.-O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.-G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, A.; Kubista, M.; Pfaffl, M. Comparison of reverse transcriptases in gene expression analysis. Clin. Chem. 2004, 50, 1678–1680. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.-L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.W.; Irimia, M. When good transcripts go bad: Artifactual RT-PCR “splicing” and Genome Analysis. BioEssays News Rev. Mol. Cell. Dev. Biol. 2008, 30, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Cocquet, J.; Chong, A.; Zhang, G.; Veitia, R.A. Reverse transcriptase template switching and false alternative transcripts. Genomics 2006, 88, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Qin, A.X.; Giger, J.M.; Guo, H.; Baldwin, K.M. Potential pitfalls in the accuracy of analysis of natural sense-antisense RNA pairs by reverse transcription-PCR. BMC Biotechnol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Xu, Z.; Clauder-Münster, S.; Steinmetz, L.M. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, M.; Hemberg, M.; Rorsman, P.; Ståhlberg, A. Quantification of mRNA in single cells and modelling of RT-qPCR induced noise. BMC Mol. Biol. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, A.; Håkansson, J.; Xian, X.; Semb, H.; Kubista, M. Properties of the reverse transcription reaction in mRNA quantification. Clin. Chem. 2004, 50, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 2005, 71, 8966–8969. [Google Scholar] [CrossRef] [PubMed]
- Degrelle, S.A.; Hennequet-Antier, C.; Chiapello, H.; Piot-Kaminski, K.; Piumi, F.; Robin, S.; Renard, J.-P.; Hue, I. Amplification biases: Possible differences among deviating gene expressions. BMC Genomics 2008, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subkhankulova, T.; Livesey, F.J. Comparative evaluation of linear and exponential amplification techniques for expression profiling at the single-cell level. Genome Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Itzkovitz, S.; van Oudenaarden, A. Validating transcripts with probes and imaging technology. Nat. Methods 2011, 8, S12–S19. [Google Scholar] [CrossRef] [PubMed]
- Kosman, D.; Mizutani, C.M.; Lemons, D.; Cox, W.G.; McGinnis, W.; Bier, E. Multiplex detection of RNA expression in Drosophila embryos. Science 2004, 305. [Google Scholar] [CrossRef] [PubMed]
- Levsky, J.M.; Shenoy, S.M.; Pezo, R.C.; Singer, R.H. Single-cell gene expression profiling. Science 2002, 297, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Luc, S.; Marco, E.; Lin, T.-W.; Peng, C.; Kerenyi, M.A.; Beyaz, S.; Kim, W.; Xu, J.; Das, P.P.; et al. Mapping cellular hierarchy by single-cell analysis of the cell surface repertoire. Cell. Stem Cell. 2013, 13, 492–505. [Google Scholar] [CrossRef] [PubMed]
- McDavid, A.; Dennis, L.; Danaher, P.; Finak, G.; Krouse, M.; Wang, A.; Webster, P.; Beechem, J.; Gottardo, R. Modeling bi-modality improves characterization of cell cycle on gene expression in single cells. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Mayr, R.; Haider, M.; Thünauer, R.; Haselgrübler, T.; Schütz, G.J.; Sonnleitner, A.; Hesse, J. A microfluidic platform for transcription- and amplification-free detection of zepto-mole amounts of nucleic acid molecules. Biosens. Bioelectron. 2016, 78, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.-C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GenBank. Available online: http://www.ncbi.nlm.nih.gov (accessed on 13 Feburary 2014).
- Hesse, J.; Sonnleitner, M.; Sonnleitner, A.; Freudenthaler, G.; Jacak, J.; Höglinger, O.; Schindler, H.; Schütz, G.J. Single-molecule reader for high-throughput bioanalysis. Anal. Chem. 2004, 76, 5960–5964. [Google Scholar] [CrossRef] [PubMed]
- Freudenthaler, G.; Sonnleitner, M.; Sonnleitner, A. Device for the Microscopic Examination of Samples. WO 2006/066286 A1, 29 June 2006. [Google Scholar]
- Hesch, C.; Hesse, J.; Jacak, J.; Schütz, G.J. Two-stage focus-hold system for rapid ultra-sensitive read-out of large-area biochips. J. Microsc. 2009, 234, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Hesse, J.; Jacak, J.; Kasper, M.; Regl, G.; Eichberger, T.; Winklmayr, M.; Aberger, F.; Sonnleitner, M.; Schlapak, R.; Howorka, S.; Muresan, L.; Frischauf, A.-M.; Schütz, G.J. RNA expression profiling at the single molecule level. Genome Res. 2006, 16, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wolf, L.K.; Georgiadis, R.M. Secondary structure effects on DNA hybridization kinetics: A solution versus surface comparison. Nucleic Acids Res. 2006, 34, 3370–3377. [Google Scholar] [CrossRef] [PubMed]
- Shchepinov, M.S.; Case-Green, S.C.; Southern, E.M. Steric factors influencing hybridisation of nucleic acids to oligonucleotide arrays. Nucleic Acids Res. 1997, 25, 1155–1161. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | RPLP0 a-Specific Label Probe b | ||||
|---|---|---|---|---|---|
| Label_29nt | Label_33nt | Label_47nt | Label_51nt | Label_52nt | |
| Capture/Label swap (Figure 2) | 10 nM | 10 nM | 10 nM | – | – |
| Effect of capture length and spacers (Table 2) | – | 10 nM | – | – | – |
| Spacer effects (Figure 3) | – | 10 nM | – | – | – |
| Label dilution series (Figure S1) | – | 100 pM, 1 nM, 10 nM, 100 nM | – | – | – |
| Label comparison (Figure S2) | – | 10 nM | – | 10 nM | 10 nM |
| Capture length comparison (Figure S3) | – | 10 nM | – | 10 nM | 10 nM |
| Unspecific signal with increasing capture length (Figure S4) | – | 10 nM | – | – | – |
| Compared Capture Probes | Fold-Change ± SD a | n b | ||
|---|---|---|---|---|
| Original Probe | → | Modified Probe | ||
Capture_29nt  | → | Capture_47nt  | 7.0 ± 2.6 | 5 |
Capture_47nt  | → | Capture_65nt  | 1.9 ± 0.8 | 9 |
Capture_65nt  | → | Capture_93nt  | 2.0 ± 0.3 | 4 |
Capture_29nt+18  | → | Capture_47nt  | 3.2 ± 1.9 | 5 |
Capture_47nt+18  | → | Capture_65nt  | 2.3 ± 1.4 | 5 |
Capture_65nt+28  | → | Capture_93nt  | 2.0 ± 0.8 | 4 |
 target-specific capture sequence;
target-specific capture sequence;  target-specific 3′-elongation;
target-specific 3′-elongation;  non-specific (NONS) 3′‑elongation; → indicates compared capture probe-pairs; a For all capture probe-pairs, the fold-change was calculated by dividing the modified probe value by the original probe value; The mean fold-change was obtained by averaging the fold-change values across the technical replicates; SD = Standard deviation of the mean fold-change; b n = Number of technical replicates.
non-specific (NONS) 3′‑elongation; → indicates compared capture probe-pairs; a For all capture probe-pairs, the fold-change was calculated by dividing the modified probe value by the original probe value; The mean fold-change was obtained by averaging the fold-change values across the technical replicates; SD = Standard deviation of the mean fold-change; b n = Number of technical replicates.© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, M.; Haselgrübler, T.; Sonnleitner, A.; Aberger, F.; Hesse, J. A Double-Hybridization Approach for the Transcription- and Amplification-Free Detection of Specific mRNA on a Microarray. Microarrays 2016, 5, 5. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5010005
Haider M, Haselgrübler T, Sonnleitner A, Aberger F, Hesse J. A Double-Hybridization Approach for the Transcription- and Amplification-Free Detection of Specific mRNA on a Microarray. Microarrays. 2016; 5(1):5. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5010005
Chicago/Turabian StyleHaider, Michaela, Thomas Haselgrübler, Alois Sonnleitner, Fritz Aberger, and Jan Hesse. 2016. "A Double-Hybridization Approach for the Transcription- and Amplification-Free Detection of Specific mRNA on a Microarray" Microarrays 5, no. 1: 5. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5010005