
Living Cell Microarrays: An Overview of Concepts


Abstract
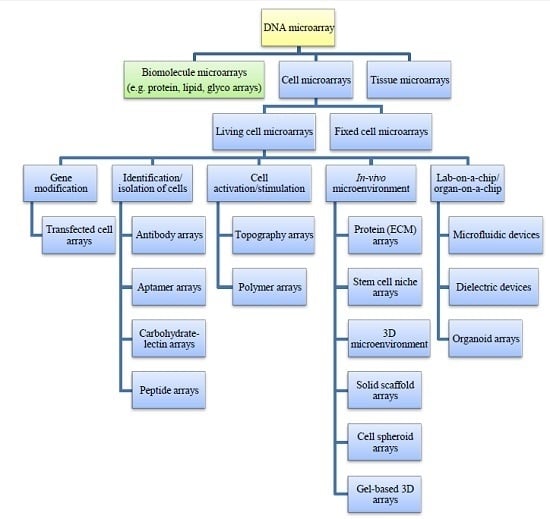
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Literature Review Section
2.1. Historical Background
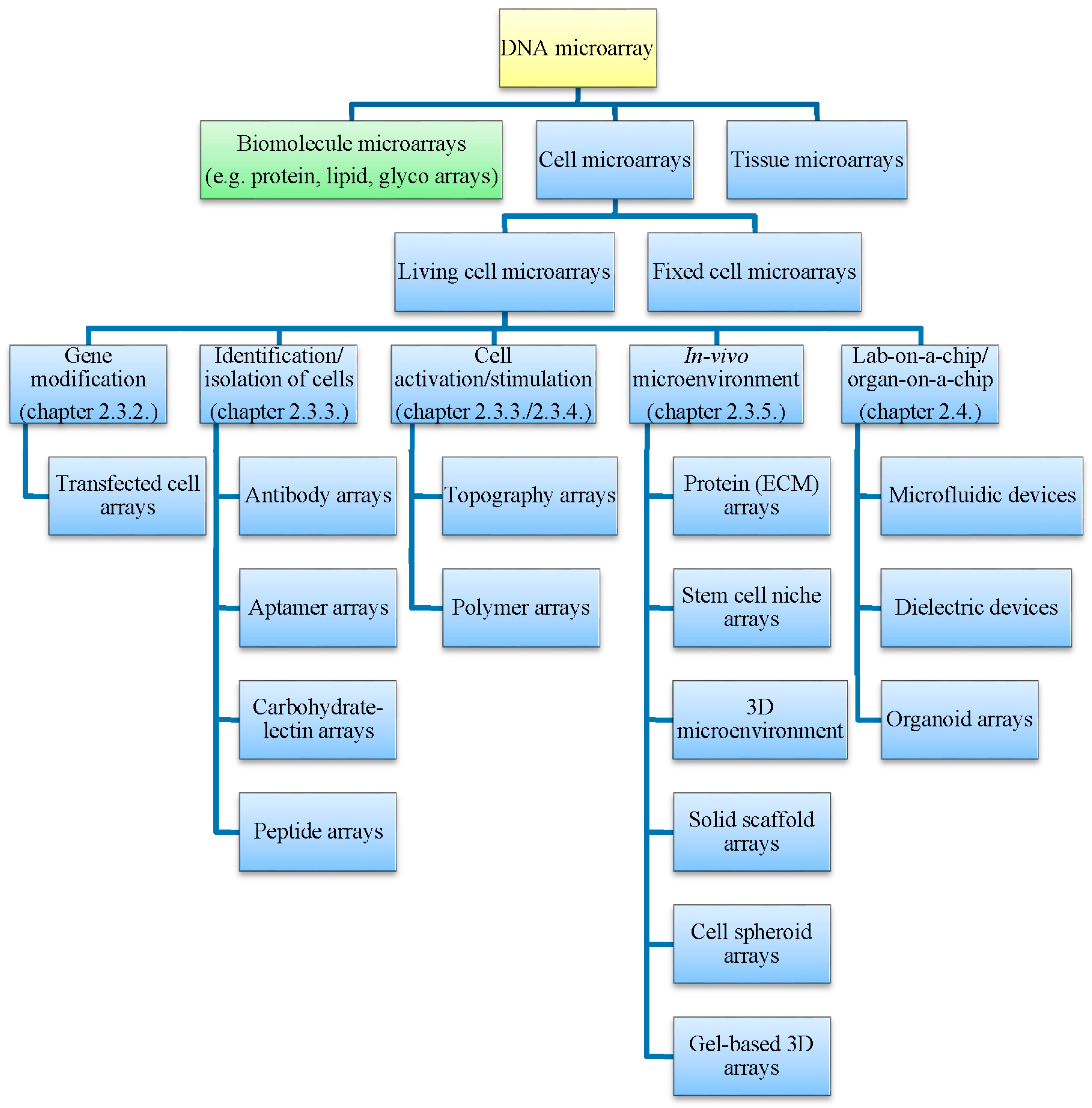
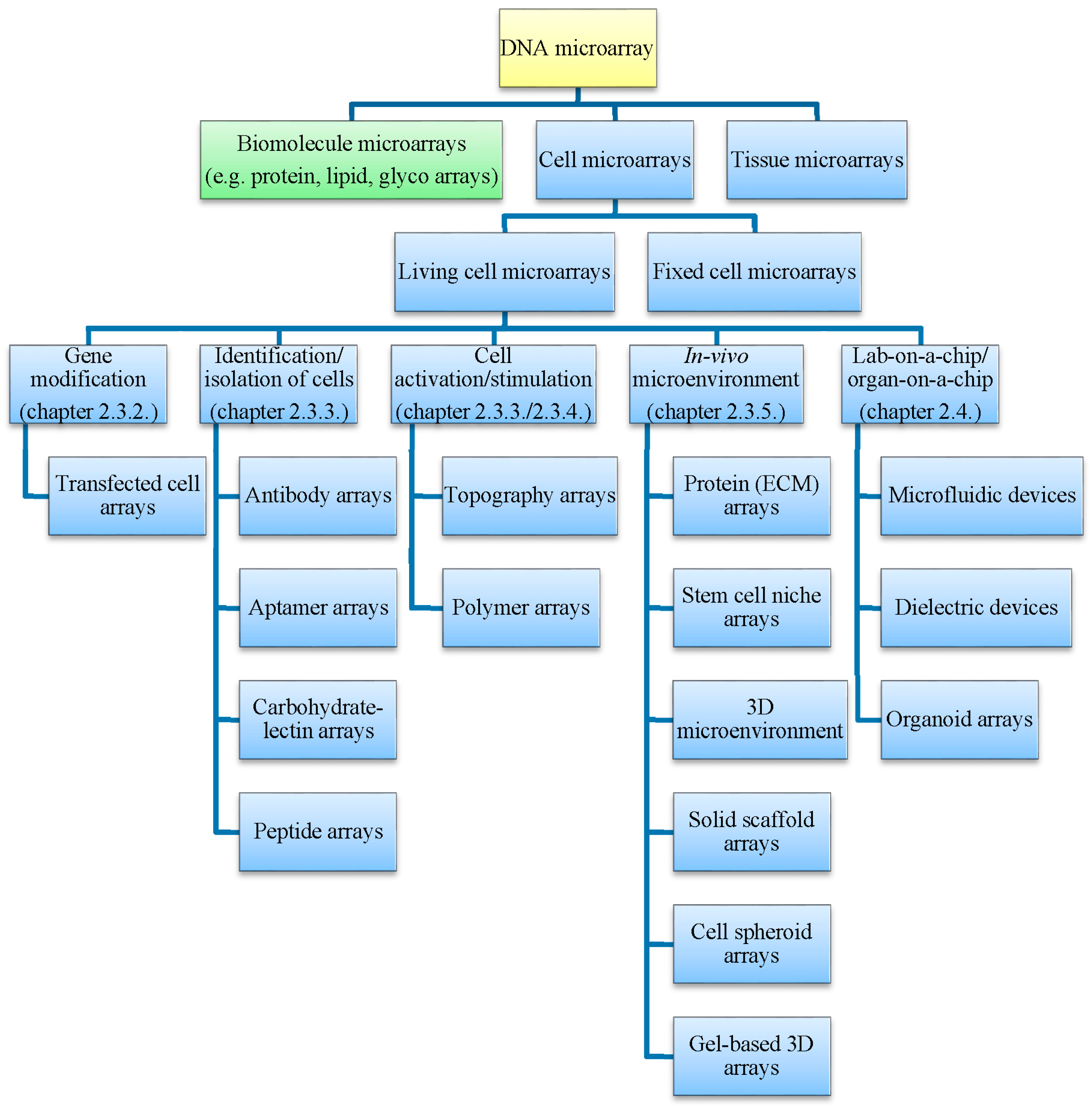
2.2. Tissue and Cell Microarrays
2.3. Living Cell Microarrays
2.3.1. Microarraying of Cells
2.3.2. Transfected Cell Microarrays
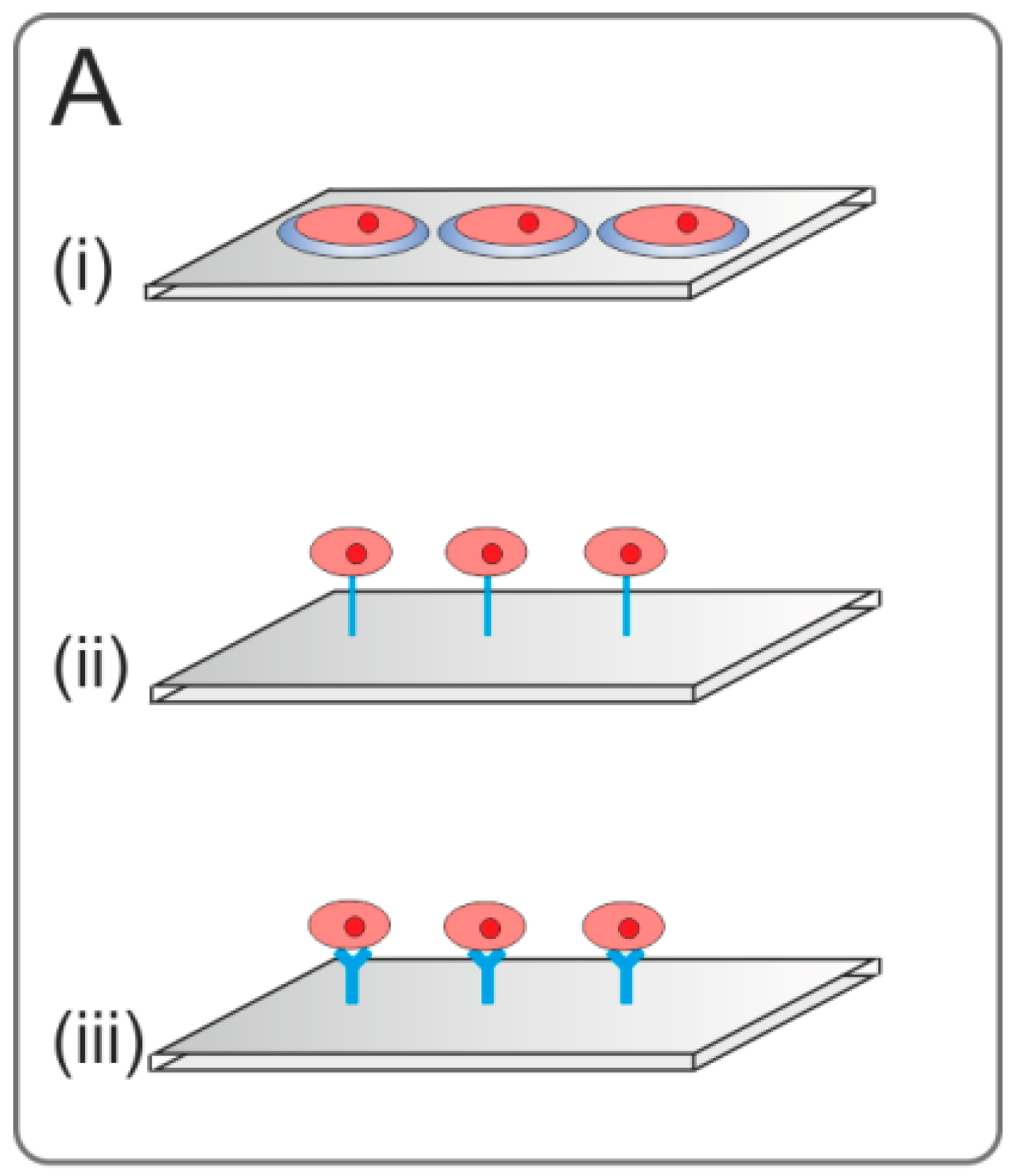
2.3.3. Affinity-Based Immobilization
2.3.4. Adsorption and Entrapment
Structured Surfaces
Biomaterials for Untargeted Cell Attachment and Stimulation
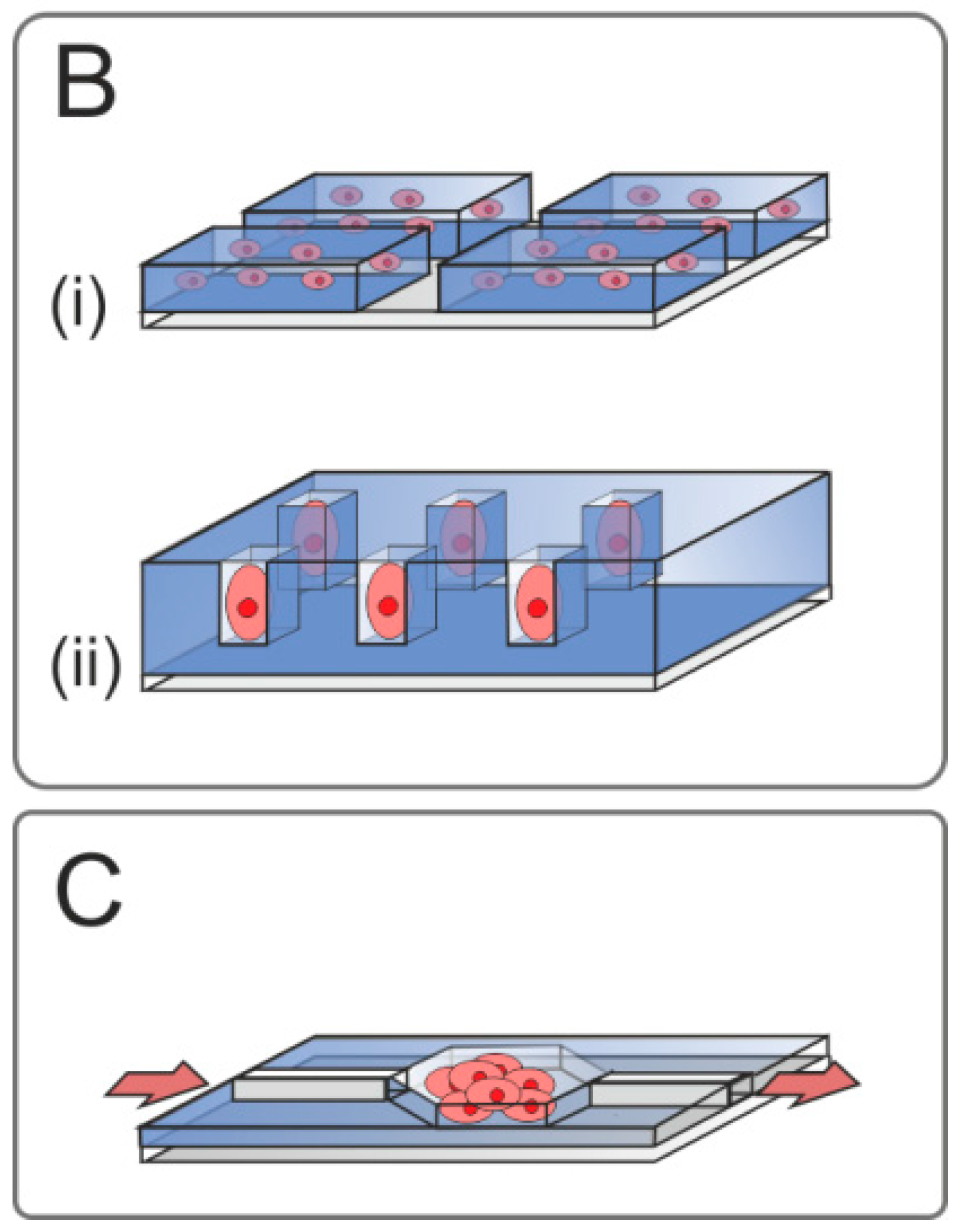
2.3.5. Simulation of In Vivo Microenvironment
Stem Cell Niche
Three-Dimensional Cell Constructs
2.4. Specialized Microarray Systems
2.4.1. Microfluidic Systems
2.4.2. Organ-on-a-Chip Systems
3. Current Limitations
4. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| ADME | Absorption, distribution, metabolism, and excretion |
| APTES | (3-Aminopropyl)triethoxysilane |
| CD | Cluster of Differentiation |
| CHO | Chinese Hamster Ovary |
| CMA | Cell microarray |
| CSC | Cancer stem cell |
| CTC | Circulating tumor cells |
| DIC | Differential interference contrast |
| DNA | Deoxyribonucleic acid |
| ECIS | Electric cell-substrate impedance sensor |
| ECM | Extracellular matrix |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EpCAM | Epithelial Cell Adhesion Molecule |
| ESC | Embryonic stem cell |
| FA-Mont | Folic acid modified montmorillonite clay |
| FDA | Food and Drug Administration |
| GFP | Green fluorescent protein |
| IgG | Immunoglobulin G |
| iPSC | Induced pluripotent stem cell |
| LCMA | Living cell microarray |
| MEA | Multi-electrode array |
| MEMS | Micro-electromechanical systems |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger RNA |
| MSC | Mesenchymal stem cell |
| NHS | N‑Hydroxysuccinimide |
| OOC | Organ-on-a-chip |
| PCR | Polymerase chain reaction |
| PEG | Polyethylene glycol |
| PLA | Polylactic acid |
| PLL | Poly-l-lysine |
| PSMA | Prostate-specific membrane antigen |
| RNA | Ribonucleic acid |
| RNAi | RNA interference |
| SC | Stem cell |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| SPR | Surface plasmon resonance |
| TMA | Tissue microarray |
| UV | Ultraviolet |
References
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, A. DNA microarray technology for the microbiologist: An overview. Appl. Microbiol. Biotechnol. 2006, 73, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar, N. A review on DNA microarray technology. IJCRR 2013, 5, 01–05. [Google Scholar]
- Luo, J.; Zha, S.; Gage, W.R.; Dunn, T.A.; Hicks, J.L.; Bennett, C.J.; Ewing, C.M.; Platz, E.A.; Ferdinandusse, S.; Wanders, R.J.; et al. Α-methylacyl-coa racemase: A new molecular marker for prostate cancer. Cancer Res. 2002, 62, 2220–2226. [Google Scholar] [PubMed]
- Gerhold, D.; Rushmore, T.; Caskey, C.T. DNA chips: Promising toys have become powerful tools. Trends Biochem. Sci. 1999, 24, 168–173. [Google Scholar] [CrossRef]
- Pirrung, M.C. How to make a DNA chip. Angew. Chem. 2002, 41, 1276–1289. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Corrigendum: Global quantification of mammalian gene expression control. Nature 2013, 495, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.G.; Kokpinar, O.; Friehs, K.; Stahl, F.; Scheper, T. Systematic investigation of optimal aptamer immobilization for protein-microarray applications. Anal. Chem. 2008, 80, 7372–7378. [Google Scholar] [CrossRef] [PubMed]
- Lübbecke, M.; Walter, J.G.; Stahl, F.; Scheper, T. Aptamers as detection molecules on reverse phase protein microarrays for the analysis of cell lysates. Eng. Life Sci. 2012, 12, 144–151. [Google Scholar] [CrossRef]
- Witt, M.; Walter, J.-G.; Stahl, F. Aptamer microarrays—Current status and future prospects. Microarrays 2015, 4, 115. [Google Scholar] [CrossRef]
- Okamoto, T.; Suzuki, T.; Yamamoto, N. Microarray fabrication with covalent attachment of DNA using bubble jet technology. Nature Biotechnol. 2000, 18, 438–441. [Google Scholar]
- Rose, S. Application of a novel microarraying system in genomics research and drug discovery. J. Assoc. Lab. Autom. 1998, 3, 53–56. [Google Scholar]
- Battifora, H. The multitumor (sausage) tissue block: Novel method for immunohistochemical antibody testing. Lab. Investig. J. Tech. Methods Pathol. 1986, 55, 244–248. [Google Scholar]
- Kononen, J.; Bubendorf, L.; Kallioniemi, A.; Barlund, M.; Schraml, P.; Leighton, S.; Torhorst, J.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Rimm, D.L.; Camp, R.L.; Charette, L.A.; Olsen, D.A.; Provost, E. Amplification of tissue by construction of tissue microarrays. Exp. Mol. Pathol. 2001, 70, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Barlund, M.; Forozan, F.; Kononen, J.; Bubendorf, L.; Chen, Y.; Bittner, M.L.; Torhorst, J.; Haas, P.; Bucher, C.; Sauter, G.; et al. Detecting activation of ribosomal protein s6 kinase by complementary DNA and tissue microarray analysis. J. Nat. Cancer Inst. 2000, 92, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Sauter, G.; Simon, R.; Hillan, K. Tissue microarrays in drug discovery. Nat. Rev. Drug Dis. 2003, 2, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Vogel, U. Overview on techniques to construct tissue arrays with special emphasis on tissue microarrays. Microarrays 2014, 3, 103. [Google Scholar] [CrossRef]
- Rubin, M.A.; Dunn, R.; Strawderman, M.; Pienta, K.J. Tissue microarray sampling strategy for prostate cancer biomarker analysis. Am. J. Surg. Pathol. 2002, 26, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Mirlacher, M.; Sauter, G. Tissue microarrays in cancer diagnosis. Expert Rev. Mol. Diagn. 2003, 3, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Hukku, B. Cell line characterization and authentication. Methods Cell Biol. 1998, 57, 203–216. [Google Scholar] [PubMed]
- Pipas, J.M. Sv40: Cell transformation and tumorigenesis. Virology 2009, 384, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Poulos, S.P.; Dodson, M.V.; Hausman, G.J. Cell line models for differentiation: Preadipocytes and adipocytes. Exp. Biol. Med. 2010, 235, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, T.; Chung, J.Y.; Hewitt, S.M. Tissue microarrays: Bridging the gap between research and the clinic. Expert Rev. Proteom. 2005, 2, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Waterworth, A.; Hanby, A.; Speirs, V. A novel cell array technique for high-throughput, cell-based analysis. In Vitro Cell. Dev. Biol. Anim. 2005, 41, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, B.; Bermudo, R.; Thomson, T.; Nayach, I.; Soler, M.; Sanchez, M.; Castillo, M.; Calvo, J.; Campo, E.; Fernandez, P.L. Paraffin-embedded cell line microarray (peclima): Development and validation of a high-throughput method for antigen profiling of cell lines. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2005, 72, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.C.; Stromberg, S.; Backvall, H.; Kampf, C.; Uhlen, M.; Wester, K.; Ponten, F. Analysis of protein expression in cell microarrays: A tool for antibody-based proteomics. J. Histochem. Cytochem. 2006, 54, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Kampf, C.; Andersson, A.-C.; Wester, K.; Björling, E.; Uhlen, M.; Ponten, F. Antibody-based tissue profiling as a tool for clinical proteomics. Clin. Proteom. 2004, 1, 285–299. [Google Scholar] [CrossRef]
- La Spada, A.; Rainoldi, B.; De Blasio, A.; Biunno, I. Application of tissue microarray technology to stem cell research. Microarrays 2014, 3, 159–167. [Google Scholar] [CrossRef]
- Hart, T.; Zhao, A.; Garg, A.; Bolusani, S.; Marcotte, E.M. Human cell chips: Adapting DNA microarray spotting technology to cell-based imaging assays. PLoS ONE 2009, 4, e7088. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.M.; Stoll, D.; Templin, M.F.; Joos, T.O. Cell microarrays: An emerging technology for the characterization of antibodies. BioTechniques 2002, 33, S54–S61. [Google Scholar]
- Masuda, N.; Ohnishi, T.; Kawamoto, S.; Monden, M.; Okubo, K. Analysis of chemical modification of rna from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999, 27, 4436–4443. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Chott, A.; Fabiano, A.; Battifora, H. Effect of formalin tissue fixation and processing on immunohistochemistry. Am. J. Surg. Pathol. 2000, 24, 1016–1019. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg Fejzo, M.; Slamon, D.J. Frozen tumor tissue microarray technology for analysis of tumor rna, DNA, and proteins. Am. J. Pathol. 2001, 159, 1645–1650. [Google Scholar] [CrossRef]
- Stephan, J.P.; Schanz, S.; Wong, A.; Schow, P.; Wong, W.L. Development of a frozen cell array as a high-throughput approach for cell-based analysis. Am. J. Pathol. 2002, 161, 787–797. [Google Scholar] [CrossRef]
- Miyaji, T.; Hewitt, S.M.; Liotta, L.A.; Star, R.A. Frozen protein arrays: A new method for arraying and detecting recombinant and native tissue proteins. Proteomics 2002, 2, 1489–1493. [Google Scholar] [CrossRef]
- Ziauddin, J.; Sabatini, D.M. Microarrays of cells expressing defined cDNAs. Nature 2001, 411, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Angres, B. Cell microarrays. Expert Rev. Mol. Diagn. 2005, 5, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Elad, T.; Lee, J.H.; Belkin, S.; Gu, M.B. Microbial whole-cell arrays. Microbial. Biotechnol. 2008, 1, 137–148. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, J.R.; Belkin, S. Where microbiology meets microengineering: Design and applications of reporter bacteria. Nat. Rev. Micro. 2010, 8, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Yarmush, M.L.; King, K.R. Living-cell microarrays. Ann. Rev. Biomed. Eng. 2009, 11, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Szittner, Z.; Prechl, J. Life on a microarray: Assessing live cell functions in a microarray format. Cell. Mol. Life Sci. 2012, 69, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Anglin, E.; Davey, R.; Herrid, M.; Hope, S.; Kurkuri, M.; Pasic, P.; Hor, M.; Fenech, M.; Thissen, H.; Voelcker, N.H. Cell microarrays for the screening of factors that allow the enrichment of bovine testicular cells. Cytom. A J. Int. Soc. Anal. Cytol. 2010, 77, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Kumar, R.A.; Sukumaran, S.M.; Hogg, M.G.; Clark, D.S.; Dordick, J.S. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc. Natl. Acad. Sci. USA 2008, 105, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Seidel, D.; Krinke, D.; Jahnke, H.-G.; Hirche, A.; Kloß, D.; Mack, T.G.; Striggow, F.; Robitzki, A. Induced tauopathy in a novel 3D-culture model mediates neurodegenerative processes: A real-time study on biochips. PLoS ONE 2012, 7, e49150. [Google Scholar] [CrossRef] [PubMed]
- Loessner, D.; Kobel, S.; Clements, J.; Lutolf, M.; Hutmacher, D. Hydrogel microwell arrays allow the assessment of protease-associated enhancement of cancer cell aggregation and survival. Microarrays 2013, 2, 208. [Google Scholar] [CrossRef]
- Mutiu, A.I.; Brandl, C.J. RNA isolation from yeast using silica matrices. J. Biomol. Tech. 2005, 16, 316–317. [Google Scholar] [PubMed]
- Achilles, J.; Stahl, F.; Harms, H.; Muller, S. Isolation of intact rna from cytometrically sorted saccharomyces cerevisiae for the analysis of intrapopulation diversity of gene expression. Nat. Protocols 2007, 2, 2203–2211. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Olson, J.; Zhao, W.; Atala, A.; Zhu, J.-M.; Yoo, J.J. Characterization of cell constructs generated with inkjet printing technology using in vivo magnetic resonance imaging. J. Manuf. Sci. Eng. 2008, 130, 021013–021013. [Google Scholar] [CrossRef]
- Cagnin, S.; Cimetta, E.; Guiducci, C.; Martini, P.; Lanfranchi, G. Overview of micro- and nano-technology tools for stem cell applications: Micropatterned and microelectronic devices. Sensors 2012, 12, 15947. [Google Scholar] [CrossRef] [PubMed]
- Spira, M.E.; Hai, A. Multi-electrode array technologies for neuroscience and cardiology. Nat. Nano. 2013, 8, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.D.; Connor, S.M.O.; Blum, R.A.; Brown, E.A.; DeWeerth, S.P. Multielectrode impedance tuning: Reducing noise and improving stimulation efficacy. In Proceedings of the 26th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Francisco, CA, USA, 1–5 September 2004; pp. 4115–4117.
- Asakura, K.; Hayashi, S.; Ojima, A.; Taniguchi, T.; Miyamoto, N.; Nakamori, C.; Nagasawa, C.; Kitamura, T.; Osada, T.; Honda, Y.; et al. Improvement of acquisition and analysis methods in multi-electrode array experiments with ips cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 2015, 75, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Michelini, E.; Roda, A. Staying alive: New perspectives on cell immobilization for biosensing purposes. Anal. Bioanal. Chem. 2012, 402, 1785–1797. [Google Scholar] [CrossRef] [PubMed]
- Belkin, S.; Gu, M. Whole Cell Sensing Systems I: Reporter Cells and Devices; Springer: Berlin/Heidelberg, Germany, 2010; Volume 117, pp. 1–208. [Google Scholar]
- Belkin, S.; Gu, M.B. Whole Cell Sensing Systems II: Applications; Springer: Berlin/Heidelberg, Germany, 2010; Volume 118, pp. 1–222. [Google Scholar]
- Date, A.; Pasini, P.; Daunert, S. Fluorescent and bioluminescent cell-based sensors: Strategies for their preservation. Adv. Biochem. Eng. Biotechnol. 2010, 117, 57–75. [Google Scholar] [PubMed]
- Tourniaire, G.; Collins, J.; Campbell, S.; Mizomoto, H.; Ogawa, S.; Thaburet, J.F.; Bradley, M. Polymer microarrays for cellular adhesion. Chem. Commun. 2006, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk, R.; Timur, S.; Scheper, T.; Stahl, F. Development of living cell microarrays using non-contact micropipette printing. J. Biotechnol. 2016, 217, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Anglin, E.J.; Salisbury, C.; Bailey, S.; Hor, M.; Macardle, P.; Fenech, M.; Thissen, H.; Voelcker, N.H. Sorted cell microarrays as platforms for high-content informational bioassays. Lab Chip 2010, 10, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Ghaedi, M.; Tuleuova, N.; Zern, M.A.; Wu, J.; Revzin, A. Bottom-up signaling from hgf-containing surfaces promotes hepatic differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2011, 407, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Rasi Ghaemi, S.; Harding, F.; Delalat, B.; Vasani, R.; Voelcker, N.H. Surface engineering for long-term culturing of mesenchymal stem cell microarrays. Biomacromolecules 2013, 14, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.; Singh, A.; Nguyen, A.H.; Bratt-Leal, A.M.; McDevitt, T.C.; Lu, H. Microfluidic-based patterning of embryonic stem cells for in vitro development studies. Lab Chip 2013, 13, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Yahya, W.; Kadri, N.; Ibrahim, F. Cell patterning for liver tissue engineering via dielectrophoretic mechanisms. Sensors 2014, 14, 11714. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Joo, S.; Duhon, M.; Heller, M.; Wallace, B.; Xu, X. Dielectrophoretic cell separation and gene expression profiling on microelectronic chip arrays. Anal. Chem. 2002, 74, 3362–3371. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Jin, J.; Gregory, C.; Hickman, J.J.; Boland, T. Inkjet printing of viable mammalian cells. Biomaterials 2005, 26, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.E.; Gough, J.E.; Derby, B. Delivery of human fibroblast cells by piezoelectric drop-on-demand inkjet printing. Biomaterials 2008, 29, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yusof, A.; Keegan, H.; Spillane, C.D.; Sheils, O.M.; Martin, C.M.; O’Leary, J.J.; Zengerle, R.; Koltay, P. Inkjet-like printing of single-cells. Lab Chip 2011, 11, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zahari, M.S.; Renuse, S.; Jacob, H.K.; Sakamuri, S.; Singal, M.; Gabrielson, E.; Sukumar, S.; Pandey, A. A breast cancer cell microarray (CMA) as a rapid method to characterize candidate biomarkers. Cancer Biol. Ther. 2014, 15, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Boland, T. Human microvasculature fabrication using thermal inkjet printing technology. Biomaterials 2009, 30, 6221–6227. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.J.; Gilmore, K.J.; Beirne, S.; McCallum, D.; Wallace, G.G.; in het Panhuis, M. Bio-ink for on-demand printing of living cells. Biomaterials Sci. 2013, 1, 224–230. [Google Scholar] [CrossRef]
- Berthuy, O.I.; Blum, L.J.; Marquette, C.A. Cells on chip for multiplex screening. Biosens. Bioelectron. 2016, 76, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Berthuy, O.I.; Mandon, C.A.; Corgier, B.P.; Octobre, G.G.; Ceccone, G.; Spampinato, V.; Blum, L.J.; Marquette, C.A. Material surface engineering for multiplex cell culture in microwell. J. Mater. Sci. 2014, 49, 4481–4489. [Google Scholar] [CrossRef]
- Ruedinger, F.; Lavrentieva, A.; Blume, C.; Pepelanova, I.; Scheper, T. Hydrogels for 3d mammalian cell culture: A starting guide for laboratory practice. Appl. Microbiol. Biotechnol. 2015, 99, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Dababneh, A.B.; Ozbolat, I.T. Bioprinting technology: A current state-of-the-art review. J. Manuf. Sci. Eng. 2014, 136, 061016. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Echeverri, C.J.; Perrimon, N. High-throughput rnai screening in cultured cells: A user’s guide. Nat. Rev. Genet. 2006, 7, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Erfle, H.; Simpson, J.C.; Bastiaens, P.I.; Pepperkok, R. Sirna cell arrays for high-content screening microscopy. BioTechniques 2004, 37, 454–458. [Google Scholar] [PubMed]
- Yoshikawa, T.; Uchimura, E.; Kishi, M.; Funeriu, D.P.; Miyake, M.; Miyake, J. Transfection microarray of human mesenchymal stem cells and on-chip sirna gene knockdown. J. Control. Release 2004, 96, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Rantala, J.K.; Mäkelä, R.; Aaltola, A.-R.; Laasola, P.; Mpindi, J.-P.; Nees, M.; Saviranta, P.; Kallioniemi, O. A cell spot microarray method for production of high density sirna transfection microarrays. BMC Genom. 2011, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Erfle, H.; Eskova, A.; Reymann, J.; Starkuviene, V. Cell arrays and high-content screening. In Protein Microarrays; Korf, U., Ed.; Humana Press: New York, NY, USA, 2011; Volume 785, pp. 277–287. [Google Scholar]
- Delehanty, J.B.; Shaffer, K.M.; Lin, B. A comparison of microscope slide substrates for use in transfected cell microarrays. Biosens. Bioelectron. 2004, 20, 773–779. [Google Scholar] [CrossRef] [PubMed]
- McConnell, K.I.; Schweller, R.M.; Diehl, M.R.; Suh, J. Live-cell microarray surface coatings supporting reverse transduction by adeno-associated viruses. BioTechniques 2011, 51, 255–258. [Google Scholar] [PubMed]
- Rajan, S.; Djambazian, H.; Dang, H.C.; Sladek, R.; Hudson, T.J. The living microarray: A high-throughput platform for measuring transcription dynamics in single cells. BMC Genom. 2011, 12, 115. [Google Scholar] [CrossRef] [PubMed]
- Palmer, E.L.; Miller, A.D.; Freeman, T.C. Identification and characterisation of human apoptosis inducing proteins using cell-based transfection microarrays and expression analysis. BMC Genom. 2006, 7, 1–17. [Google Scholar]
- Mannherz, O.; Mertens, D.; Hahn, M.; Lichter, P. Functional screening for proapoptotic genes by reverse transfection cell array technology. Genomics 2006, 87, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on nf-κb activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, M.; Honstein, T.; Kirchhoff, S.; Kreider, R.; Schmidt, H.; Sipos, B.; Gress, T.M. A multistep high-content screening approach to identify novel functionally relevant target genes in pancreatic cancer. PLoS ONE 2015, 10, e0122946. [Google Scholar] [CrossRef] [PubMed]
- Erfle, H.; Lisauskas, T.; Claas, C.; Reymann, J.; Starkuviene, V. Cell arrays for the measurement of organelle dynamics in living cells. In Cell-Based Microarrays; Palmer, E., Ed.; Humana Press: New York, NY, USA, 2011; Volume 706, pp. 73–81. [Google Scholar]
- Neumann, B.; Walter, T.; Hériché, J.K.; Bulkescher, J.; Erfle, H.; Conrad, C.; Rogers, P.; Poser, I.; Held, M.; Liebel, U.; et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Improving the efficiency of rna interference in mammals. Nat. Rev. Genet. 2004, 5, 355–365. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, E.P. siRNAs and shRNAs: Tools for protein knockdown by gene silencing. Word Lab. 2013. [Google Scholar] [CrossRef]
- Fellmann, C.; Lowe, S.W. Stable rna interference rules for silencing. Nat. Cell. Biol. 2014, 16, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Siomi, M.C. On the road to reading the rna-interference code. Nature 2009, 457, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Erfle, H.; Pepperkok, R. Production of sirna and cdna-transfected cell arrays on noncoated chambered coverglass for high-content screening microscopy in living cells. In Target Discovery and Validation Reviews and Protocols; Sioud, M., Ed.; Humana Press: New York, NY, USA, 2007; Volume 360, pp. 155–161. [Google Scholar]
- Mousses, S.; Caplen, N.J.; Cornelison, R.; Weaver, D.; Basik, M.; Hautaniemi, S.; Elkahloun, A.G.; Lotufo, R.A.; Choudary, A.; Dougherty, E.R.; et al. Rnai microarray analysis in cultured mammalian cells. Genome Res. 2003, 13, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Erfle, H.; Neumann, B.; Liebel, U.; Rogers, P.; Held, M.; Walter, T.; Ellenberg, J.; Pepperkok, R. Reverse transfection on cell arrays for high content screening microscopy. Nat. Protocols 2007, 2, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H.; Taxman, D.J. Short hairpin rna (shrna): Design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010, 629, 141–158. [Google Scholar] [PubMed]
- Starkuviene, V.; Pepperkok, R.; Erfle, H. Transfected cell microarrays: An efficient tool for high-throughput functional analysis. Expert Rev. Proteom. 2007, 4, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Fjeldbo, C.S.; Misund, K.; Günther, C.C.; Langaas, M.; Steigedal, T.S.; Thommesen, L.; Laegreid, A.; Bruland, T. Functional studies on transfected cell microarray analysed by linear regression modelling. Nucleic Acids Res. 2008, 36, e97. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Conklin, D.S.; Mittal, V. High-throughput selection of effective rnai probes for gene silencing. Genome Res. 2003, 13, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.C.; Joggerst, B.; Laketa, V.; Verissimo, F.; Cetin, C.; Erfle, H.; Bexiga, M.G.; Singan, V.R.; Heriche, J.-K.; Neumann, B.; et al. Genome-wide rnai screening identifies human proteins with a regulatory function in the early secretory pathway. Nat. Cell Biol. 2012, 14, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Fengler, S.; H Bastiaens, P.I.; Grecco, H.E.; Roda-Navarro, P. Optimizing cell arrays for accurate functional genomics. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Davis, M.M. Molecular and functional analysis using live cell microarrays. Curr. Opin. Chem. Biol. 2006, 10, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Tateno, H.; Uchiyama, N.; Kuno, A.; Togayachi, A.; Sato, T.; Narimatsu, H.; Hirabayashi, J. A novel strategy for mammalian cell surface glycome profiling using lectin microarray. Glycobiology 2007, 17, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Milgram, S.; Bombera, R.; Livache, T.; Roupioz, Y. Antibody microarrays for label-free cell-based applications. Methods 2012, 56, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Lin, J.; Yu, Y.; Pastuovic, M.; Wei, M.; Duan, W. RNA aptamer against a cancer stem cell marker epithelial cell adhesion molecule. Cancer Sci. 2011, 102, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhu, Z.; An, Y.; Zhang, W.; Zhang, H.; Liu, D.; Yu, C.; Duan, W.; Yang, C.J. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal. Chem 2013, 85, 4141–4149. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, J.; Zhang, Y.; Lin, Z.; Lin, J.-M. Targeted isolation and analysis of single tumor cells with aptamer-encoded microwell array on microfluidic device. Lab Chip 2012, 12, 5180–5185. [Google Scholar] [CrossRef] [PubMed]
- Dharmasiri, U.; Balamurugan, S.; Adams, A.A.; Okagbare, P.I.; Obubuafo, A.; Soper, S.A. Highly efficient capture and enumeration of low abundance prostate cancer cells using prostate-specific membrane antigen aptamers immobilized to a polymeric microfluidic device. Electrophoresis 2009, 30, 3289–3300. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bao, K.; Frangioni, J.V.; Choi, H.S. Screening of small molecule microarrays for ligands targeted to the extracellular epitopes of living cells. Microarrays 2015, 4, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Falsey, J.R.; Renil, M.; Park, S.; Li, S.; Lam, K.S. Peptide and small molecule microarray for high throughput cell adhesion and functional assays. Bioconjugate Chem. 2001, 12, 346–353. [Google Scholar] [CrossRef]
- Bongartz, R.; Ag, D.; Seleci, M.; Walter, J.-G.; Yalcinkaya, E.E.; Demirkol, D.O.; Stahl, F.; Timur, S.; Scheper, T. Folic acid-modified clay: Targeted surface design for cell culture applications. J. Mater. Chem. B 2013, 1, 522–528. [Google Scholar] [CrossRef]
- Flaim, C.J.; Chien, S.; Bhatia, S.N. An extracellular matrix microarray for probing cellular differentiation. Nat. Methods 2005, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Nimrichter, L.; Gargir, A.; Gortler, M.; Altstock, R.T.; Shtevi, A.; Weisshaus, O.; Fire, E.; Dotan, N.; Schnaar, R.L. Intact cell adhesion to glycan microarrays. Glycobiology 2004, 14, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Hook, A.L.; Anderson, D.G.; Langer, R.; Williams, P.; Davies, M.C.; Alexander, M.R. High throughput methods applied in biomaterial development and discovery. Biomaterials 2010, 31, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Shadpour, H.; Sims, C.E.; Allbritton, N.L. Enrichment and expansion of cells using antibody-coated micropallet arrays. Cytom. A J. Int. Soc. Anal. Cytol. 2009, 75, 609–618. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, Y.; Xie, X.; Zhu, T.; Soules, M.; DiMeco, F.; Vescovi, A.L.; Fan, X.; Lubman, D.M. Identification of cell surface glycoprotein markers for glioblastoma-derived stem-like cells using a lectin microarray and lc-ms/ms approach. J. Proteome Res. 2010, 9, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Colpo, P.; Ruiz, A.; Ceriotti, L.; Rossi, F. Surface functionalization for protein and cell patterning. In Whole Cell Sensing Systems I: Reporter Cells and Devices; Belkin, S., Gu, B.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 109–130. [Google Scholar]
- Falconnet, D.; Csucs, G.; Grandin, H.M.; Textor, M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials 2006, 27, 3044–3063. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.H.; Lee, S.H.; Lee, S.H.; Ko, H.J.; Park, T.H. Cell-based high-throughput odorant screening system through visualization on a microwell array. Biosens. Bioelectron. 2014, 53, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.C.; Mian, M.K.; Shrivastava, S.; Chung, B.G.; Khademhosseini, A. A microwell array system for stem cell culture. Biomaterials 2008, 29, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cohen, R.E.; Hammond, P.T.; Irvine, D.J. Live lymphocyte arrays for biosensing. Adv. Funct Mater. 2006, 16, 1313–1323. [Google Scholar] [CrossRef]
- Barrett, D.G.; Yousaf, M.N. Rapid patterning of cells and cell co-cultures on surfaces with spatial and temporal control through centrifugation. Angew. Chem. 2007, 46, 7437–7439. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Kishi, H.; Tokimitsu, Y.; Kondo, S.; Honda, R.; Rao, S.R.; Omori, M.; Tamiya, E.; Muraguchi, A. Single-cell microarray for analyzing cellular response. Anal. Chem. 2005, 77, 8050–8056. [Google Scholar] [CrossRef] [PubMed]
- Yatsushiro, S.; Yamamura, S.; Yamaguchi, Y.; Shinohara, Y.; Tamiya, E.; Horii, T.; Baba, Y.; Kataoka, M. Rapid and highly sensitive detection of malaria-infected erythrocytes using a cell microarray chip. PLoS ONE 2010, 5, e13179. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Yatsushiro, S.; Yamaguchi, Y.; Abe, K.; Shinohara, Y.; Tamiya, E.; Baba, Y.; Kataoka, M. Accurate detection of carcinoma cells by use of a cell microarray chip. PLoS ONE 2012, 7, e32370. [Google Scholar] [CrossRef] [PubMed]
- Reymann, J.; Beil, N.; Beneke, J.; Kaletta, P.P.; Burkert, K.; Erfle, H. Next-generation 9216-microwell cell arrays for high-content screening microscopy. BioTechniques 2009, 47, 877–878. [Google Scholar] [PubMed]
- Zawko, S.A.; Schmidt, C.E. Simple benchtop patterning of hydrogel grids for living cell microarrays. Lab Chip 2010, 10, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Ankam, S.; Teo, B.K.K.; Kukumberg, M.; Yim, E.K.F. High throughput screening to investigate the interaction of stem cells with their extracellular microenvironment. Organogenesis 2013, 9, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Moe, A.A.; Suryana, M.; Marcy, G.; Lim, S.K.; Ankam, S.; Goh, J.Z.; Jin, J.; Teo, B.K.; Law, J.B.; Low, H.Y.; et al. Microarray with micro- and nano-topographies enables identification of the optimal topography for directing the differentiation of primary murine neural progenitor cells. Small 2012, 8, 3050–3061. [Google Scholar] [CrossRef] [PubMed]
- Hook, A.L.; Thissen, H.; Voelcker, N.H. Surface manipulation of biomolecules for cell microarray applications. Trends Biotechnol. 2006, 24, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.G.; Levenberg, S.; Langer, R. Nanoliter-scale synthesis of arrayed biomaterials and application to human embryonic stem cells. Nat. Biotechnol. 2004, 22, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Hook, A.L.; Thissen, H.; Voelcker, N.H. Advanced substrate fabrication for cell microarrays. Biomacromolecules 2009, 10, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Lavrentieva, A.; Majore, I.; Kasper, C.; Hass, R. Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell Commun. Signal. 2010, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collet, G.; El Hafny-Rahbi, B.; Nadim, M.; Tejchman, A.; Klimkiewicz, K.; Kieda, C. Hypoxia-shaped vascular niche for cancer stem cells. Contemp. Oncol. 2015, 19, A39–A43. [Google Scholar] [CrossRef] [PubMed]
- Eiselleova, L.; Peterkova, I.; Neradil, J.; Slaninova, I.; Hampl, A.; Dvorak, P. Comparative study of mouse and human feeder cells for human embryonic stem cells. Int. J. Dev. Biol. 2008, 52, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, Y.K.; Yang, M.T.; Desai, R.A.; Yu, X.; Liu, Z.; Chen, C.S. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat. Methods 2010, 7, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Gobaa, S.; Hoehnel, S.; Roccio, M.; Negro, A.; Kobel, S.; Lutolf, M.P. Artificial niche microarrays for probing single stem cell fate in high throughput. Nat. Methods 2011, 8, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Kuschel, C.; Steuer, H.; Maurer, A.N.; Kanzok, B.; Stoop, R.; Angres, B. Cell adhesion profiling using extracellular matrix protein microarrays. BioTechniques 2006, 40, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhao, L.; Zhang, X. In-channel printing-device opening assay for micropatterning multiple cells and gene analysis. Anal. Chem. 2015, 87, 2048–2053. [Google Scholar] [CrossRef] [PubMed]
- Reticker-Flynn, N.E.; Braga Malta, D.F.; Winslow, M.M.; Lamar, J.M.; Xu, M.J.; Underhill, G.H.; Hynes, R.O.; Jacks, T.E.; Bhatia, S.N. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat. Commun 2012, 3, 1122. [Google Scholar] [CrossRef] [PubMed]
- Zschenker, O.; Streichert, T.; Hehlgans, S.; Cordes, N. Genome-wide gene expression analysis in cancer cells reveals 3d growth to affect ecm and processes associated with cell adhesion but not DNA repair. PLoS ONE 2012, 7, e34279. [Google Scholar] [CrossRef] [PubMed]
- Page, H.; Flood, P.; Reynaud, E.G. Three-dimensional tissue cultures: Current trends and beyond. Cell. Tissue Res. 2013, 352, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A.; Kruuv, J. A multi-component radiation survival curve using an in vitro tumour model. Int J. Radiat Biol Relat Stud. Phys. Chem Med. 1970, 18, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A. Phytohemagglutinin (pha)-induced transformation of lymphocytes from patients with cancer. Cancer 1971, 27, 574–578. [Google Scholar] [CrossRef]
- Lee, J.M.; Mhawech-Fauceglia, P.; Lee, N.; Parsanian, L.C.; Lin, Y.G.; Gayther, S.A.; Lawrenson, K. A three-dimensional microenvironment alters protein expression and chemosensitivity of epithelial ovarian cancer cells in vitro. Lab. Investig. 2013, 93, 528–542. [Google Scholar] [PubMed]
- Friedrich, J.; Ebner, R.; Kunz-Schughart, L.A. Experimental anti-tumor therapy in 3-D: Spheroids—old hat or new challenge? Int J. Radiat. Biol. 2007, 83, 849–871. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.T.; Hughes-Fulford, M. Monolayer and spheroid culture of human liver hepatocellular carcinoma cell line cells demonstrate distinct global gene expression patterns and functional phenotypes. Tissue Eng. A 2009, 15, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lasher, C.D.; Milford, L.M.; Murali, T.M.; Rajagopalan, P. A comparative study of genome-wide transcriptional profiles of primary hepatocytes in collagen sandwich and monolayer cultures. Tissue Eng. Part. C Methods 2010, 16, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.Y.; Liu, B.H.; Sieber, M.; Hsu, S.H. Substrate-dependent gene regulation of self-assembled human msc spheroids on chitosan membranes. BMC Genom. 2014, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schafer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3D microenvironment on phenotype, gene expression, and egfr inhibition of colorectal cancer cell lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef] [PubMed]
- Schmeichel, K.L.; Bissell, M.J. Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 2003, 116, 2377–2388. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C. Extracellular matrix remodelling and cellular differentiation. Curr. Opin. Cell Biol. 1999, 11, 634–640. [Google Scholar] [CrossRef]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Auk-Emblem, P.; Dornish, M. 3D cell culture in alginate hydrogels. Microarrays 2015, 4, 133. [Google Scholar] [CrossRef]
- Lin, R.Z.; Chang, H.Y. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol. J. 2008, 3, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Motta, A.; Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol. Biol. 2011, 695, 17–39. [Google Scholar] [PubMed]
- Gidrol, X.; Fouque, B.; Ghenim, L.; Haguet, V.; Picollet-D’hahan, N.; Schaack, B. 2D and 3D cell microarrays in pharmacology. Curr. Opin. Pharmacol. 2009, 9, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Ock, J.; Li, W. Fabrication of a three-dimensional tissue model microarray using laser foaming of a gas-impregnated biodegradable polymer. Biofabrication 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Karp, J.M.; Yeh, J.; Eng, G.; Fukuda, J.; Blumling, J.; Suh, K.Y.; Cheng, J.; Mahdavi, A.; Borenstein, J.; Langer, R.; et al. Controlling size, shape and homogeneity of embryoid bodies using poly(ethylene glycol) microwells. Lab Chip 2007, 7, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Itaka, K.; Ohba, S.; Nishiyama, N.; Chung, U.I.; Yamasaki, Y.; Kataoka, K. 3D spheroid culture system on micropatterned substrates for improved differentiation efficiency of multipotent mesenchymal stem cells. Biomaterials 2009, 30, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.M.; Zhang, C.; Toh, Y.C.; Kim, S.H.; Foo, H.L.; Tan, C.H.; van Noort, D.; Park, S.; Yu, H. A gel-free 3D microfluidic cell culture system. Biomaterials 2008, 29, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, T.G.; Kwon, S.J.; Bale, S.S.; Lee, M.Y.; Diogo, M.M.; Clark, D.S.; Cabral, J.M.; Dordick, J.S. Three-dimensional cell culture microarray for high-throughput studies of stem cell fate. Biotechnol. Bioeng. 2010, 106, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Meli, L.; Barbosa, H.S.; Hickey, A.M.; Gasimli, L.; Nierode, G.; Diogo, M.M.; Linhardt, R.J.; Cabral, J.M.; Dordick, J.S. Three dimensional cellular microarray platform for human neural stem cell differentiation and toxicology. Stem Cell Res. 2014, 13, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Meli, L.; Jordan, E.T.; Clark, D.S.; Linhardt, R.J.; Dordick, J.S. Influence of a three-dimensional, microarray environment on human cell culture in drug screening systems. Biomaterials 2012, 33, 9087–9096. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.G.; Purwada, A.; Cerchietti, L.; Inghirami, G.; Melnick, A.; Gaharwar, A.K.; Singh, A. Microscale bioadhesive hydrogel arrays for cell engineering applications. Cell. Mol. Bioeng. 2014, 7, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Ranga, A.; Gobaa, S.; Okawa, Y.; Mosiewicz, K.; Negro, A.; Lutolf, M.P. 3D niche microarrays for systems-level analyses of cell fate. Nat. Commun. 2014, 5, 4324. [Google Scholar] [CrossRef] [PubMed]
- Akselrod, G.M.; Timp, W.; Mirsaidov, U.; Zhao, Q.; Li, C.; Timp, R.; Timp, K.; Matsudaira, P.; Timp, G. Laser-guided assembly of heterotypic three-dimensional living cell microarrays. Biophys J. 2006, 91, 3465–3473. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.G.; Itle, L.J.; Pishko, M.V. Molding of hydrogel microstructures to create multiphenotype cell microarrays. Anal. Chem. 2003, 75, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Ino, K.; Arai, T.; Ramon-Azcon, J.; Takahashi, Y.; Shiku, H.; Matsue, T. Alginate gel microwell arrays using electrodeposition for three-dimensional cell culture. Lab Chip 2013, 13, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Charwat, V.; Schütze, K.; Holnthoner, W.; Lavrentieva, A.; Gangnus, R.; Hofbauer, P.; Hoffmann, C.; Angres, B.; Kasper, C. Potential and limitations of microscopy and raman spectroscopy for live-cell analysis of 3D cell cultures. J. Biotechnol. 2015, 205, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Figallo, E.; Cannizzaro, C.; Elvassore, N.; Vunjak-Novakovic, G. Micro-bioreactor arrays for controlling cellular environments: Design principles for human embryonic stem cell applications. Methods 2009, 47, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.R.; Song, H.; Sung, J.H.; Kim, D.; Kim, K. Microfluidic assay-based optical measurement techniques for cell analysis: A review of recent progress. Biosens. Bioelectron. 2016, 77, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Loufakis, D.N.; Cao, Z.; Chang, Y.W.; Achenie, L.E.K.; Lu, C. Diffusion-based microfluidic PCR for “one-pot” analysis of cells. Lab Chip 2014, 14, 2905–2909. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lv, X.; Zhang, X.; Saeed, O.; Deng, Y. Microfluidics for cell-cell interactions: A review. Front. Chem. Sci. Eng. 2016, 10, 90–98. [Google Scholar] [CrossRef]
- Sia, S.K.; Whitesides, G.M. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis 2003, 24, 3563–3576. [Google Scholar] [CrossRef] [PubMed]
- Businaro, L.; De Ninno, A.; Schiavoni, G.; Lucarini, V.; Ciasca, G.; Gerardino, A.; Belardelli, F.; Gabriele, L.; Mattei, F. Cross talk between cancer and immune cells: Exploring complex dynamics in a microfluidic environment. Lab Chip 2013, 13, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, Q.; Jafarpoorchekab, H.; Huang, C.B.; Silacci, P.; Carrara, S.; Koklu, G.; Ghaye, J.; Ramsden, J.; Ruffert, C.; Vergeres, G.; et al. Nutrichip: Nutrition analysis meets microfluidics. Lab Chip 2013, 13, 196–203. [Google Scholar] [CrossRef] [PubMed]
- U.S., F.D.A. Innovation or stagnation: Challenge and Opportunity on the Critical Path to New Medical Products; Challenges and Opportunities Report—March 2004; FDA: Silver Spring, MD, USA, 2004. [Google Scholar]
- Bhise, N.S.; Ribas, J.; Manoharan, V.; Zhang, Y.S.; Polini, A.; Massa, S.; Dokmeci, M.R.; Khademhosseini, A. Organ-on-a-chip platforms for studying drug delivery systems. J. Control. Release 2014, 190, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Ng, C.P.; Lanz, H.L.; Vulto, P.; Suter-Dick, L.; Masereeuw, R. Kidney-on-a-chip technology for drug-incuced nephrotoxiciy screening. Trends Biotechnol. 2016, 34, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Doryab, A.; Amoabediny, G.; Salehi-Najafabadi, A. Advances in pulmonary therapy and drug development: Lung tissue engineering to lung-on-a-chip. Biotechnol. Adv. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Samanipour, R.; Koo, K.I.; Kim, K. Organ-on-a-chip platforms for drug delivery and cell characterization: A review. Sens. Mater. 2015, 27, 487–506. [Google Scholar] [CrossRef]
- Ghaemmaghami, A.M.; Hancock, M.J.; Harrington, H.; Kaji, H.; Khademhosseini, A. Biomimetic tissues on a chip for drug discovery. Drug Discov. Today 2012, 17, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hubner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Sung, J.H. Organ-on-a-chip technology and microfluidic whole-body models for pharmacokinetic drug toxicity screening. Biotechnol. J. 2013, 8, 1258–1266. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonczyk, R.; Kurth, T.; Lavrentieva, A.; Walter, J.-G.; Scheper, T.; Stahl, F. Living Cell Microarrays: An Overview of Concepts. Microarrays 2016, 5, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020011
Jonczyk R, Kurth T, Lavrentieva A, Walter J-G, Scheper T, Stahl F. Living Cell Microarrays: An Overview of Concepts. Microarrays. 2016; 5(2):11. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020011
Chicago/Turabian StyleJonczyk, Rebecca, Tracy Kurth, Antonina Lavrentieva, Johanna-Gabriela Walter, Thomas Scheper, and Frank Stahl. 2016. "Living Cell Microarrays: An Overview of Concepts" Microarrays 5, no. 2: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/microarrays5020011