Comprehensive Review of Methodology to Detect Reactive Oxygen Species (ROS) in Mammalian Species and Establish Its Relationship with Antioxidants and Cancer


 , ,
, ,  , , ,
, , ,
Abstract
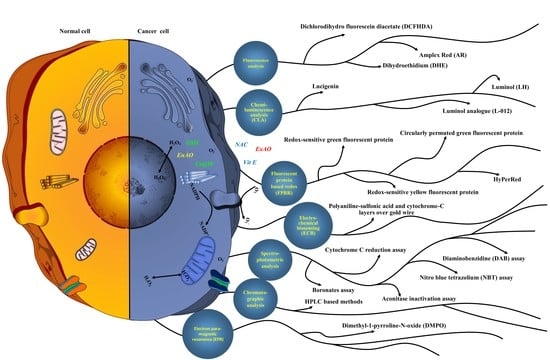
:
1. Introduction
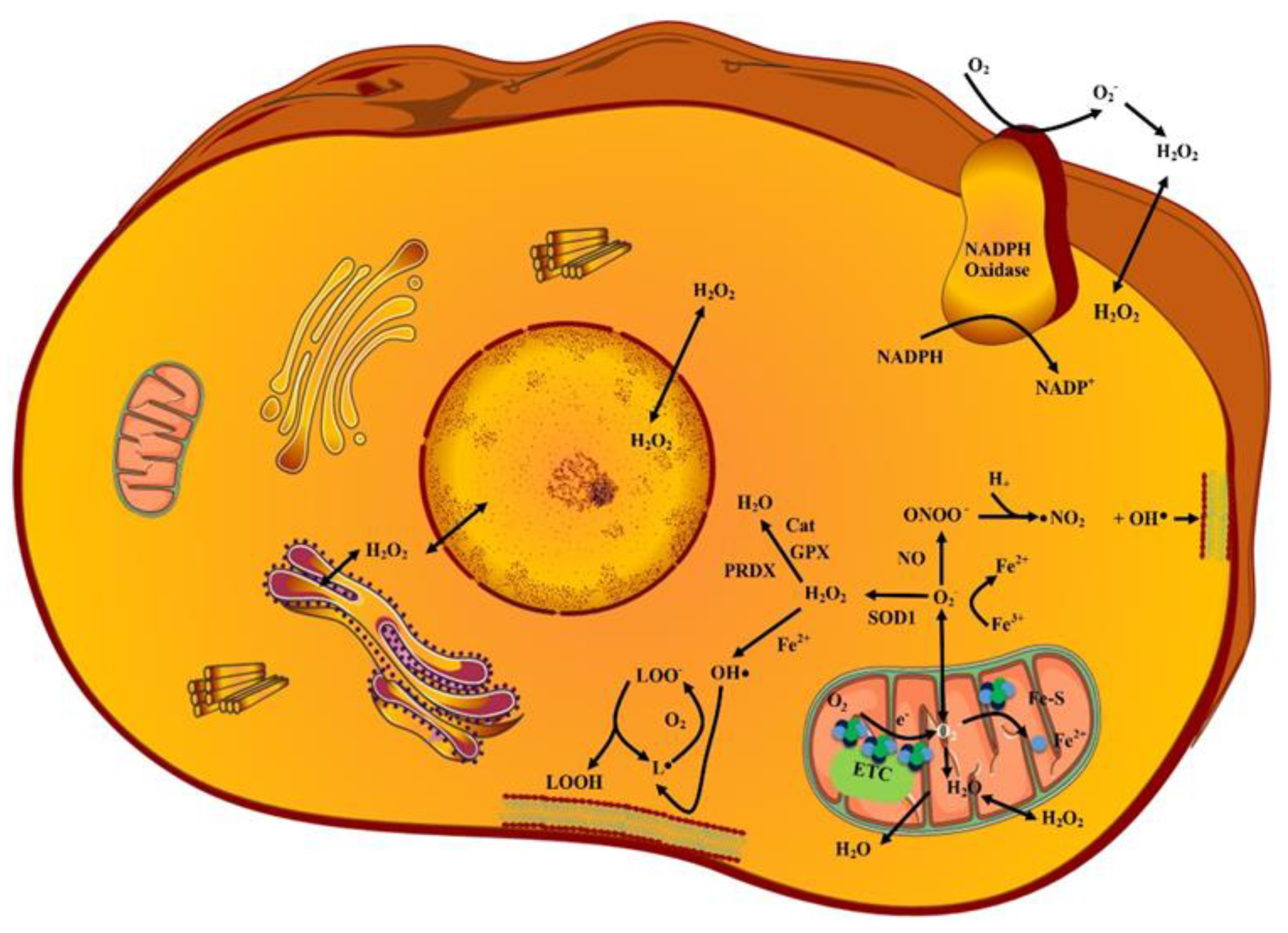
2. ROS Sources and Types
2.1. Superoxides (O2˙−)
2.2. Hydrogen Peroxides (H2O2)
2.3. Hydroxy Radicals (˙OH)
2.4. Lipid Peroxides (LP)
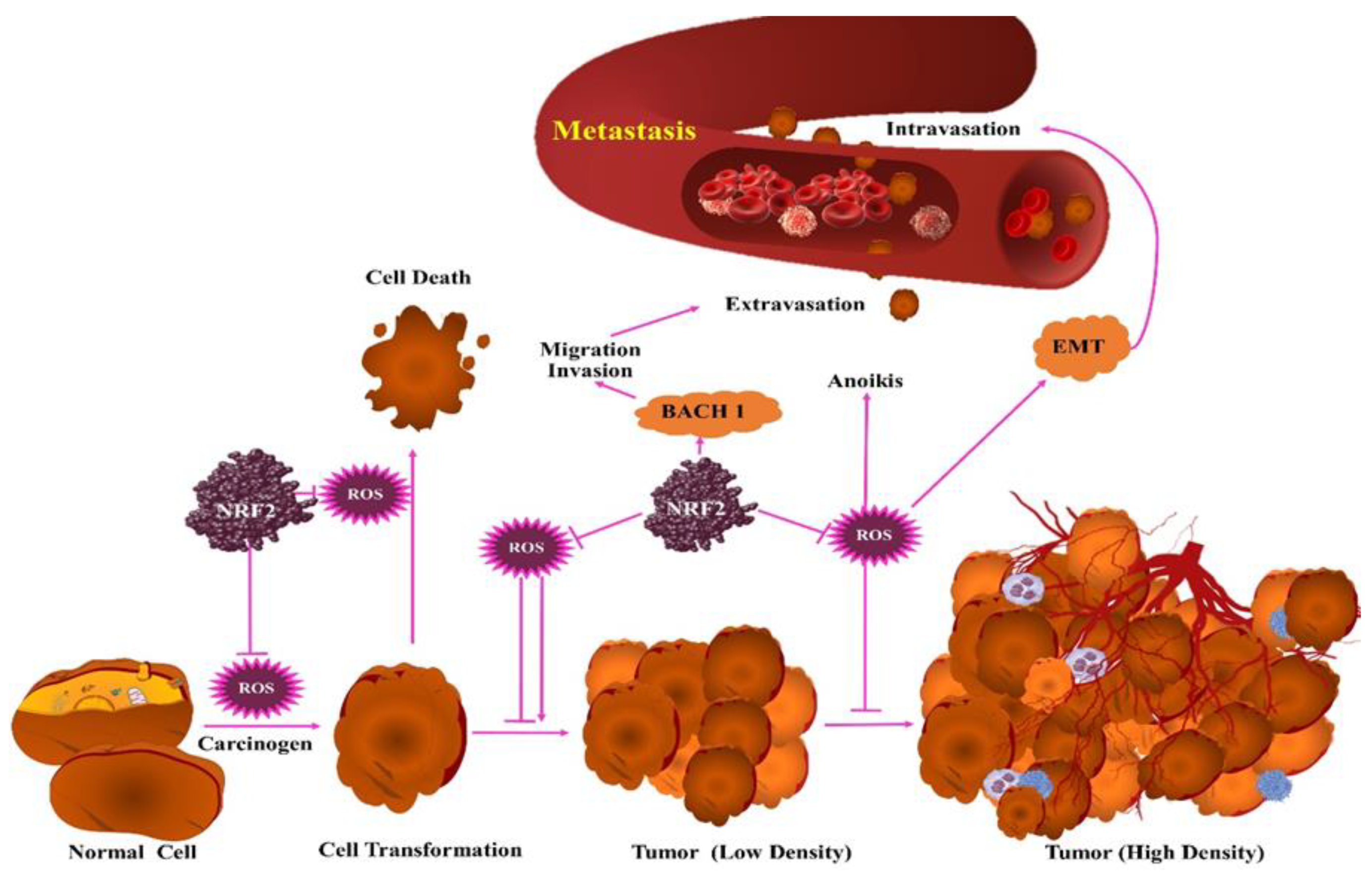
3. Protumorigenic Effects of ROS
4. Antitumorigenic Effects of ROS
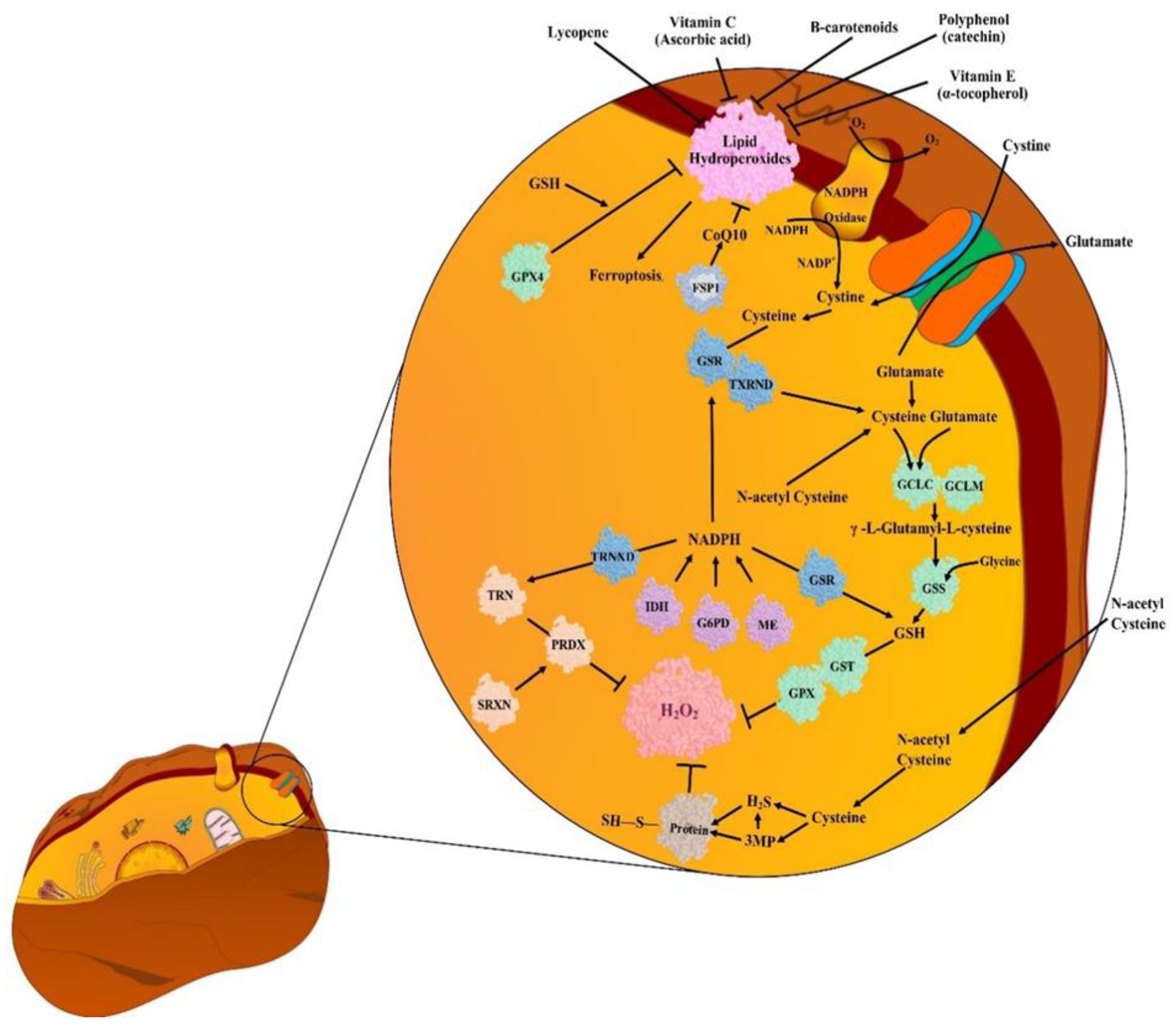
5. Antioxidants (AOs)
5.1. Endogenous Antioxidants (EnAOs)
5.1.1. Tumor Prevention by EnAOs
5.1.2. Tumor Progression by EnAOs
5.2. Exogenous Antioxidants (ExAOs)
6. Unusual Detoxification of ROS in Cancer
7. Methods to Detect ROS
7.1. Fluorescent Chemicals-Based ROS Detection
7.1.1. Dihydroethidium (DHE)
7.1.2. Dichlorodihydro Fluorescein Diacetate (DCFHDA)
7.1.3. Amplex Red (AR)
7.2. Fluorescent Protein-Based Redox (FPBR) Analysis
7.2.1. Redox-Sensitive Green Fluorescent Protein (roGFP)
roGFP1 and roGFP2
roGFP1-iX
roGFP1-iL and roGFP1-RX
Grx1-roGFP2-iL and roGFP2-Orp1
7.2.2. Redox-Sensitive Yellow Fluorescent Protein (rxYFP)
rxYFP-Grx1P
rxYFP 3R
7.2.3. HyPer
HyPer-2 and HyPer-3
7.2.4. Circularly Permuted Green Fluorescent Protein (cpYFP)
7.2.5. HyPerRed (rxRFP)
TrxRFP1 and cpRFP
7.3. Chemiluminescence Analysis (CLA)
7.3.1. Lucigenin
7.3.2. Luminol (LH)
7.3.3. Luminol Analogue (L012)
7.4. Electro-Chemical Biosensing (ECB)
7.5. Chromatographic Analysis
7.6. Spectro-Photometric Analysis
7.6.1. Cytochrome c Reduction Assay
7.6.2. Nitro Blue Tetrazolium (NBT) Assay
7.6.3. Aconitase Inactivation Assay
7.6.4. Boronates Assay
7.6.5. Diaminobenzidine (DAB) Assay
7.7. Electron Paramagnetic Resonance (EPR) or Electron Spin Resonance Assay
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer IV, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y. ROS Systems Are a New Integrated Network for Sensing Homeostasis and Alarming Stresses in Organelle Metabolic Processes. Redox Biol. 2020, 101696. [Google Scholar] [CrossRef]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020. [Google Scholar] [CrossRef]
- Fuloria, S.; Subramaniyan, V.; Karupiah, S.; Kumari, U.; Sathasivam, K.; Meenakshi, D.U.; Wu, Y.S.; Guad, R.M.; Udupa, K.; Fuloria, N.K. A Comprehensive Review on Source, Types, Effects, Nanotechnology, Detection, and Therapeutic Management of Reactive Carbonyl Species Associated with Various Chronic Diseases. Antioxidants 2020, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Ann. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The Role of Cellular Reactive Oxygen Species in Cancer Chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Jakubczyk, K.; Dec, K.; Ka\lduńska, J.; Kawczuga, D.; Kochman, J.; Janda, K. Reactive Oxygen Species-Sources, Functions, Oxidative Damage. Pol. Merkur. Lek. Organ Pol. Tow. Lek. 2020, 48, 124. [Google Scholar]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic Signaling Mediated by Oxidants in Ras-Transformed Fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Bauer, G. HOCl-Dependent Singlet Oxygen and Hydroxyl Radical Generation Modulate and Induce Apoptosis of Malignant Cells. Anticancer Res. 2013, 33, 3589–3602. [Google Scholar] [PubMed]
- Bauer, G. Targeting Extracellular ROS Signaling of Tumor Cells. Anticancer Res. 2014, 34, 1467–1482. [Google Scholar] [PubMed]
- Keyer, K.; Imlay, J.A. Superoxide Accelerates DNA Damage by Elevating Free-Iron Levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, B.P.; Weissman, J.S. The FAD-and O2-Dependent Reaction Cycle of Ero1-Mediated Oxidative Protein Folding in the Endoplasmic Reticulum. Mol. Cell 2002, 10, 983–994. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane Transport of Hydrogen Peroxide. Biochim. Biophys. Acta BBA-Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G. HOCl and the Control of Oncogenesis. J. Inorg. Biochem. 2018, 179, 10–23. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of Iron and Hydrogen Peroxide: The Fenton Reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Figge, F.H.J. Cosmic Radiation and Cancer. Science 1947, 323–325. [Google Scholar] [CrossRef]
- Droge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Zhyvoloup, A.; Baković, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G. Protein CoAlation and Antioxidant Function of Coenzyme A in Prokaryotic Cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O.; Kim, M.-O.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kim, G.-Y. Butein Induces G2/M Phase Arrest and Apoptosis in Human Hepatoma Cancer Cells through ROS Generation. Cancer Lett. 2010, 288, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Pottgiesser, S.J.; Heinzelmann, S.; Bauer, G. Intercellular HOCl-Mediated Apoptosis Induction in Malignant Cells: Interplay between NOX1-Dependent Superoxide Anion Generation and DUOX-Related HOCl-Generating Peroxidase Activity. Anticancer Res. 2015, 35, 5927–5943. [Google Scholar] [PubMed]
- Heigold, S.; Sers, C.; Bechtel, W.; Ivanovas, B.; Schäfer, R.; Bauer, G. Nitric Oxide Mediates Apoptosis Induction Selectively in Transformed Fibroblasts Compared to Nontransformed Fibroblasts. Carcinogenesis 2002, 23, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G. Nitric Oxide’s Contribution to Selective Apoptosis Induction in Malignant Cells through Multiple Reaction Steps. Crit. Rev. Oncog. 2016, 21. [Google Scholar] [CrossRef]
- Heinzelmann, S.; Bauer, G. Multiple Protective Functions of Catalase against Intercellular Apoptosis-Inducing ROS Signaling of Human Tumor Cells. Biol. Chem. 2010, 391, 675–693. [Google Scholar] [CrossRef]
- Böhm, B.; Heinzelmann, S.; Motz, M.; Bauer, G. Extracellular Localization of Catalase Is Associated with the Transformed State of Malignant Cells. Biol. Chem. 2015, 396, 1339–1356. [Google Scholar] [CrossRef] [Green Version]
- Scheit, K.; Bauer, G. Direct and Indirect Inactivation of Tumor Cell Protective Catalase by Salicylic Acid and Anthocyanidins Reactivates Intercellular ROS Signaling and Allows for Synergistic Effects. Carcinogenesis 2015, 36, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Riethmüller, M.; Burger, N.; Bauer, G. Singlet Oxygen Treatment of Tumor Cells Triggers Extracellular Singlet Oxygen Generation, Catalase Inactivation and Reactivation of Intercellular Apoptosis-Inducing Signaling. Redox Biol. 2015, 6, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G. Signal Amplification by Tumor Cells: Clue to the Understanding of the Antitumor Effects of Cold Atmospheric Plasma and Plasma-Activated Medium. IEEE Trans. Radiat. Plasma Med. Sci. 2017, 2, 87–98. [Google Scholar] [CrossRef]
- Girard, P.-M.; Arbabian, A.; Fleury, M.; Bauville, G.; Puech, V.; Dutreix, M.; Sousa, J.S. Synergistic Effect of H2O2and NO2 in Cell Death Induced by Cold Atmospheric He Plasma. Sci. Rep. 2016, 6, 29098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, G.; Sersenová, D.; Graves, D.B.; Machala, Z. Cold Atmospheric Plasma and Plasma-Activated Medium Trigger RONS-Based Tumor Cell Apoptosis. Sci. Rep. 2019, 9, 1–28. [Google Scholar] [CrossRef]
- Machala, Z.; Tarabova, B.; Hensel, K.; Spetlikova, E.; Sikurova, L.; Lukes, P. Formation of ROS and RNS in Water Electro-S Prayed through Transient Spark Discharge in Air and Their Bactericidal Effects. Plasma Process. Polym. 2013, 10, 649–659. [Google Scholar] [CrossRef]
- Metelmann, H.-R.; Seebauer, C.; Miller, V.; Fridman, A.; Bauer, G.; Graves, D.B.; Pouvesle, J.-M.; Rutkowski, R.; Schuster, M.; Bekeschus, S. Clinical Experience with Cold Plasma in the Treatment of Locally Advanced Head and Neck Cancer. Clin. Plasma Med. 2018, 9, 6–13. [Google Scholar] [CrossRef]
- Bauer, G. Cold Atmospheric Plasma and Plasma-Activated Medium: Antitumor Cell Effects with Inherent Synergistic Potential. Plasma Med. 2019, 9. [Google Scholar] [CrossRef]
- Verkman, A.S.; Hara-Chikuma, M.; Papadopoulos, M.C. Aquaporins—New Players in Cancer Biology. J. Mol. Med. 2008, 86, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Talbot, A.; Nourmohammadi, N.; Sherman, J.H.; Cheng, X.; Keidar, M. Toward Understanding the Selective Anticancer Capacity of Cold Atmospheric Plasma—A Model Based on Aquaporins. Biointerphases 2015, 10, 040801. [Google Scholar] [CrossRef]
- Deichman, G.I. Natural Selection and Early Changes of Phenotype of Tumor Cells in Vivo: Acquisition of New Defense Mechanisms. Biochem. CC BIOKHIMIIA 2000, 65, 78–94. [Google Scholar]
- Deichman, G.I. Early Phenotypic Changes of in Vitro Transformed Cells during in Vivo Progression: Possible Role of the Host Innate Immunity. Semin. Cancer Biol. 2002, 12, 317–326. [Google Scholar] [CrossRef]
- Bauer, G. Tumor Cell-Protective Catalase as a Novel Target for Rational Therapeutic Approaches Based on Specific Intercellular ROS Signaling. Anticancer Res. 2012, 32, 2599–2624. [Google Scholar] [PubMed]
- Bauer, G. Targeting Protective Catalase of Tumor Cells with Cold Atmospheric Plasma-Activated Medium (PAM). Anti-Cancer Agents Med. Chem. Former. Curr. Med. Chem.-Anti-Cancer Agents 2018, 18, 784–804. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Motz, M. The Antitumor Effect of Single-Domain Antibodies Directed towards Membrane-Associated Catalase and Superoxide Dismutase. Anticancer Res. 2016, 36, 5945–5956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Jiang, G.; Wu, Y.; Antony, S.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Roy, K.; Makhlouf, H.; Chuaqui, R. NADPH Oxidase 1 Is Highly Expressed in Human Large and Small Bowel Cancers. PLoS ONE 2020, 15, e0233208. [Google Scholar] [CrossRef]
- Di Mascio, P.; Bechara, E.J.; Medeiros, M.H.; Briviba, K.; Sies, H. Singlet Molecular Oxygen Production in the Reaction of Peroxynitrite with Hydrogen Peroxide. FEBS Lett. 1994, 355, 287–289. [Google Scholar] [CrossRef] [Green Version]
- Bauer, G. The Synergistic Effect between Hydrogen Peroxide and Nitrite, Two Long-Lived Molecular Species from Cold Atmospheric Plasma, Triggers Tumor Cells to Induce Their Own Cell Death. Redox Biol. 2019, 26, 101291. [Google Scholar] [CrossRef]
- Lukes, P.; Dolezalova, E.; Sisrova, I.; Clupek, M. Aqueous-Phase Chemistry and Bactericidal Effects from an Air Discharge Plasma in Contact with Water: Evidence for the Formation of Peroxynitrite through a Pseudo-Second-Order Post-Discharge Reaction of H2O2 and HNO2. Plasma Sources Sci. Technol. 2014, 23, 015019. [Google Scholar] [CrossRef]
- Deng, K.; Wu, B.; Wang, C.-X.; Wang, Q.; Yu, H.; Li, J.-M.; Li, K.-H.; Zhao, H.-Y.; Huang, S.-W. An Oxidation-Enhanced Magnetic Resonance Imaging Probe for Visual and Specific Detection of Singlet Oxygen Generated in Photodynamic Cancer Therapy In Vivo. Adv. Healthc. Mater. 2020, 9, 2000533. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J.; Goldstein, S.; Czapski, G. Peroxynitrous Acid Homolyzes Into ˙OH And ˙NO2 Radicals. Chem. Res. Toxicol. 1998, 11, 712–713. [Google Scholar] [CrossRef]
- Christensen, H.; Sehested, K.; Corfitzen, H. Reactions of Hydroxyl Radicals with Hydrogen Peroxide at Ambient and Elevated Temperatures. J. Phys. Chem. 1982, 86, 1588–1590. [Google Scholar] [CrossRef]
- Goldstein, S.; Lind, J.; Merényi, G. Chemistry of Peroxynitrites as Compared to Peroxynitrates. Chem. Rev. 2005, 105, 2457–2470. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Ronsein, G.E.; Corrêa, T.C.; Martinez, G.R.; Medeiros, M.H.; Di Mascio, P. Direct Evidence of Singlet Molecular Oxygen Generation from Peroxynitrate, a Decomposition Product of Peroxynitrite. Dalton Trans. 2009, 5720–5729. [Google Scholar] [CrossRef] [PubMed]
- Escobar, J.A.; Rubio, M.A.; Lissi, E.A. SOD and Catalase Inactivation by Singlet Oxygen and Peroxyl Radicals. Free Radic. Biol. Med. 1996, 20, 285–290. [Google Scholar] [CrossRef]
- Yan, D.; Xiao, H.; Zhu, W.; Nourmohammadi, N.; Zhang, L.G.; Bian, K.; Keidar, M. The Role of Aquaporins in the Anti-Glioblastoma Capacity of the Cold Plasma-Stimulated Medium. J. Phys. Appl. Phys. 2017, 50, 055401. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and P38 Signaling Pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 Is Required for Sustained Activations of JNK/P38 MAP Kinases and Apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Han, J.; Sun, P. The Pathways to Tumor Suppression via Route P38. Trends Biochem. Sci. 2007, 32, 364–371. [Google Scholar] [CrossRef]
- Kennedy, N.J.; Davis, R.J. Role of JNK in Tumor Development. Cell Cycle Georget. Tex 2003, 2, 199. [Google Scholar]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. P38α MAP Kinase as a Sensor of Reactive Oxygen Species in Tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Thornton, T.M.; Rincon, M. Non-Classical P38 Map Kinase Functions: Cell Cycle Checkpoints and Survival. Int. J. Biol. Sci. 2009, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, Á.R. Signal Integration by JNK and P38 MAPK Pathways in Cancer Development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and Oncogene Rescue of Metabolic Defects Caused by Loss of Matrix Attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B. Reductive Carboxylation Supports Redox Homeostasis during Anchorage-Independent Growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Adhikary, A.; Mohanty, S.; Lahiry, L.; Hossain, D.M.S.; Chakraborty, S.; Das, T. Theaflavins Retard Human Breast Cancer Cell Migration by Inhibiting NF-ΚB via P53-ROS Cross-Talk. FEBS Lett. 2010, 584, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding Roles of Superoxide Dismutases in Cell Regulation and Cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Zámockỳ, M.; Koller, F. Understanding the Structure and Function of Catalases: Clues from Molecular Evolution and in Vitro Mutagenesis. Prog. Biophys. Mol. Biol. 1999, 72, 19–66. [Google Scholar] [CrossRef]
- Rashdan, N.A.; Shrestha, B.; Pattillo, C.B. S-Glutathionylation, Friend or Foe in Cardiovascular Health and Disease. Redox Biol. 2020, 101693. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Aspects Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione Peroxidase Family–an Evolutionary Overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The Thioredoxin Antioxidant System. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol Chemistry and Specificity in Redox Signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Synthesis. Biochim. Biophys. Acta BBA-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, Q.; Yang, Y.; Lei, C.; Yang, F.; Liang, L.; Chen, C.; Xia, J.; Wang, K.; Tang, N. GSTZ1 Deficiency Promotes Hepatocellular Carcinoma Proliferation via Activation of the KEAP1/NRF2 Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef]
- Henderson, C.J.; Ritchie, K.J.; McLaren, A.; Chakravarty, P.; Wolf, C.R. Increased Skin Papilloma Formation in Mice Lacking Glutathione Transferase GSTP. Cancer Res. 2011, 71, 7048–7060. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T. Tumor Suppressor Function of the Plasma Glutathione Peroxidase Gpx3 in Colitis-Associated Carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.-T. Life-Long Reduction in MnSOD Activity Results in Increased DNA Damage and Higher Incidence of Cancer but Does Not Accelerate Aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A. Mice Deficient in Both Mn Superoxide Dismutase and Glutathione Peroxidase-1 Have Increased Oxidative Damage and a Greater Incidence of Pathology but No Reduction in Longevity. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2009, 64, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 Inhibits Tumorigenesis via Regulating PTEN/AKT Activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Neumann, C.A.; Krause, D.S.; Carman, C.V.; Das, S.; Dubey, D.P.; Abraham, J.L.; Bronson, R.T.; Fujiwara, Y.; Orkin, S.H.; Van Etten, R.A. Essential Role for the Peroxiredoxin Prdx1 in Erythrocyte Antioxidant Defence and Tumour Suppression. Nature 2003, 424, 561–565. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, M.R.; Orlicky, D.J.; Prigge, J.R.; Krishna, P.; Talago, E.A.; Cavigli, I.R.; Eriksson, S.; Miller, C.G.; Kundert, J.A.; Sayin, V.I. TrxR1, Gsr, and Oxidative Stress Determine Hepatocellular Carcinoma Malignancy. Proc. Natl. Acad. Sci. USA 2019, 116, 11408–11417. [Google Scholar] [CrossRef] [Green Version]
- Sakumi, K.; Tominaga, Y.; Furuichi, M.; Xu, P.; Tsuzuki, T.; Sekiguchi, M.; Nakabeppu, Y. Ogg1 Knockout-Associated Lung Tumorigenesis and Its Suppression by Mth1 Gene Disruption. Cancer Res. 2003, 63, 902–905. [Google Scholar]
- Müller, M.F.; Florian, S.; Pommer, S.; Osterhoff, M.; Esworthy, R.S.; Chu, F.-F.; Brigelius-Flohé, R.; Kipp, A.P. Deletion of Glutathione Peroxidase-2 Inhibits Azoxymethane-Induced Colon Cancer Development. PLoS ONE 2013, 8, e72055. [Google Scholar]
- Mishra, M.; Jiang, H.; Chawsheen, H.A.; Gerard, M.; Toledano, M.B.; Wei, Q. Nrf2-Activated Expression of Sulfiredoxin Contributes to Urethane-Induced Lung Tumorigenesis. Cancer Lett. 2018, 432, 216–226. [Google Scholar] [CrossRef]
- Wu, L.; Jiang, H.; Chawsheen, H.A.; Mishra, M.; Young, M.R.; Gerard, M.; Toledano, M.B.; Colburn, N.H.; Wei, Q. Tumor Promoter-Induced Sulfiredoxin Is Required for Mouse Skin Tumorigenesis. Carcinogenesis 2014, 35, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Yang, J.H.; Huang, F.Z.; Nie, W.P.; Liu, C.P.; Mao, X.H.; Yin, X.M.; Shen, X.B.; Peng, C.; Chen, M.F. Glutathione-s-Transferase A 4 (GSTA4) Suppresses Tumor Growth and Metastasis of Human Hepatocellular Carcinoma by Targeting AKT Pathway. Am. J. Transl. Res. 2017, 9, 301. [Google Scholar]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Hendricks, J.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Trésaugues, L.; Dexheimer, T.S.; Coussens, N.P. Irreversible Inhibition of Cytosolic Thioredoxin Reductase 1 as a Mechanistic Basis for Anticancer Therapy. Sci. Transl. Med. 2018, 10, eaaf7444. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Wu, L.; Mishra, M.; Chawsheen, H.A.; Wei, Q. Expression of Peroxiredoxin 1 and 4 Promotes Human Lung Cancer Malignancy. Am. J. Cancer Res. 2014, 4, 445. [Google Scholar]
- Rolfs, F.; Huber, M.; Gruber, F.; Böhm, F.; Pfister, H.J.; Bochkov, V.N.; Tschachler, E.; Dummer, R.; Hohl, D.; Schäfer, M. Dual Role of the Antioxidant Enzyme Peroxiredoxin 6 in Skin Carcinogenesis. Cancer Res. 2013, 73, 3460–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 Reduces Experimental Non–Small-Cell Lung Cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J. Copper Is Required for Oncogenic BRAF Signalling and Tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative Flux Analysis Reveals Folate-Dependent NADPH Production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-Phosphate Dehydrogenase Deficiency. Lancet 2008, 371, 64–74. [Google Scholar] [CrossRef]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M. The Metabolic Function of Cyclin D3–CDK6 Kinase in Cancer Cell Survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Dore, M.P.; Davoli, A.; Longo, N.; Marras, G.; Pes, G.M. Glucose-6-Phosphate Dehydrogenase Deficiency and Risk of Colorectal Cancer in Northern Sardinia: A Retrospective Observational Study. Medicine (Baltim.) 2016, 95, e5254. [Google Scholar] [CrossRef]
- Carocho, M.; Ferreira, I.C. A Review on Antioxidants, Prooxidants and Related Controversy: Natural and Synthetic Compounds, Screening and Analysis Methodologies and Future Perspectives. Food Chem. Toxicol. 2013, 51, 15–25. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouysegu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef]
- Graf, B.A.; Milbury, P.E.; Blumberg, J.B. Flavonols, Flavones, Flavanones, and Human Health: Epidemiological Evidence. J. Med. Food 2005, 8, 281–290. [Google Scholar] [CrossRef]
- Mut-Salud, N.; Álvarez, P.J.; Garrido, J.M.; Carrasco, E.; Aránega, A.; Rodríguez-Serrano, F. Antioxidant Intake and Antitumor Therapy: Toward Nutritional Recommendations for Optimal Results. Oxid. Med. Cell. Longev. 2016, 2016, 6719534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rodríguez, L.; Mougeot, F.; Alonso-Alvarez, C. Carotenoid-Based Coloration Predicts Resistance to Oxidative Damage during Immune Challenge. J. Exp. Biol. 2010, 213, 1685–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial Effects from a Mechanistic Perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, L.L.; Rojas, S.; Seefeldt, T. Role of Antioxidants in the Prevention of Cancer. J. Exp. Clin. Med. 2012, 4, 215–222. [Google Scholar] [CrossRef]
- Giammanco, M.; Di Majo, D.; Leto, G.; Flandina, C.; Di Piazza, M.; La Guardia, M. The Role of Vitamin k in Bone Remodeling and Osteoporosis. J. Food Res. 2012, 1, 106. [Google Scholar] [CrossRef] [Green Version]
- Puspitasari, I.M.; Abdulah, R.; Yamazaki, C.; Kameo, S.; Nakano, T.; Koyama, H. Updates on Clinical Studies of Selenium Supplementation in Radiotherapy. Radiat. Oncol. 2014, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.S. Zinc: An Antioxidant and Anti-Inflammatory Agent: Role of Zinc in Degenerative Disorders of Aging. J. Trace Elem. Med. Biol. 2014, 28, 364–371. [Google Scholar] [CrossRef]
- Bairati, I.; Meyer, F.; Jobin, E.; Gélinas, M.; Fortin, A.; Nabid, A.; Brochet, F.; Têtu, B. Antioxidant Vitamins Supplementation and Mortality: A Randomized Trial in Head and Neck Cancer Patients. Int. J. Cancer 2006, 119, 2221–2224. [Google Scholar] [CrossRef]
- Suhail, N.; Bilal, N.; Khan, H.Y.; Hasan, S.; Sharma, S.; Khan, F.; Mansoor, T.; Banu, N. Effect of Vitamins C and E on Antioxidant Status of Breast-Cancer Patients Undergoing Chemotherapy. J. Clin. Pharm. Ther. 2012, 37, 22–26. [Google Scholar] [CrossRef]
- Vollbracht, C.; Schneider, B.; Leendert, V.; Weiss, G.; Auerbach, L.; Beuth, J. Intravenous Vitamin C Administration Improves Quality of Life in Breast Cancer Patients during Chemo-/Radiotherapy and Aftercare: Results of a Retrospective, Multicentre, Epidemiological Cohort Study in Germany. In Vivo 2011, 25, 983–990. [Google Scholar]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A Review of Anti-Cancer Properties and Therapeutic Activity in Head and Neck Squamous Cell Carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khafif, A.; Lev-Ari, S.; Vexler, A.; Barnea, I.; Starr, A.; Karaush, V.; Haif, S.; Ben-Yosef, R. Curcumin: A Potential Radio-Enhancer in Head and Neck Cancer. Laryngoscope 2009, 119, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Lecumberri, E.; Dupertuis, Y.M.; Miralbell, R.; Pichard, C. Green Tea Polyphenol Epigallocatechin-3-Gallate (EGCG) as Adjuvant in Cancer Therapy. Clin. Nutr. 2013, 32, 894–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Wang, Y.; Zhang, Y.; Wan, X.; Li, J.; Liu, K.; Wang, F.; Liu, Q.; Yang, C.; Yu, P. Anti-Cancer Activities of Tea Epigallocatechin-3-Gallate in Breast Cancer Patients under Radiotherapy. Curr. Mol. Med. 2012, 12, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.J.; Meltz, M.L.; Herman, T.S. Melatonin: Possible Mechanisms Involved in Its ‘Radioprotective’Effect. Mutat. Res. Mol. Mech. Mutagen. 1998, 404, 187–189. [Google Scholar]
- Shirazi, A.; Ghobadi, G.; Ghazi-Khansari, M. A Radiobiological Review on Melatonin: A Novel Radioprotector. J. Radiat. Res. (Tokyo) 2007, 48, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, E.; Álvarez, P.J.; Melguizo, C.; Prados, J.; Alvarez-Manzaneda, E.; Chahboun, R.; Messouri, I.; Vázquez-Vázquez, M.I.; Aranega, A.; Rodriguez-Serrano, F. Novel Merosesquiterpene Exerts a Potent Antitumor Activity against Breast Cancer Cells in Vitro and in Vivo. Eur. J. Med. Chem. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Hall, S.R.; Blundon, H.L.; Ladda, M.A.; Robertson, A.W.; Martinez-Farina, C.F.; Jakeman, D.L.; Goralski, K.B. Jadomycin Breast Cancer Cytotoxicity Is Mediated by a Copper-Dependent, Reactive Oxygen Species–Inducing Mechanism. Pharmacol. Res. Perspect. 2015, 3, e00110. [Google Scholar] [CrossRef]
- Thorburn, A.; Frankel, A.E. Apoptosis and Anthracycline Cardiotoxicity. Mol. Cancer Ther. 2006, 5, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.P.; Chau, S.P.; Kong, S.K.; Fung, K.P.; Kwok, T.T. Reactive Oxygen Species Mediate Doxorubicin Induced P53-Independent Apoptosis. Life Sci. 2003, 73, 2047–2058. [Google Scholar] [CrossRef]
- Štěrba, M.; Popelová, O.; Vávrová, A.; Jirkovskỳ, E.; Kovaříková, P.; Geršl, V.; Šim\uunek, T. Oxidative Stress, Redox Signaling, and Metal Chelation in Anthracycline Cardiotoxicity and Pharmacological Cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.S.; Burgeiro, A.; Garcia, R.; Moreno, A.J.; Carvalho, R.A.; Oliveira, P.J. Doxorubicin-Induced Cardiotoxicity: From Bioenergetic Failure and Cell Death to Cardiomyopathy. Med. Res. Rev. 2014, 34, 106–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Y.; Luo, A.-Y.; Zhou, Y.-R.; Ren, J.-H. N-Acetylcysteine Reduces Oxidative Stress, Nuclear Factor-ΚB Activity and Cardiomyocyte Apoptosis in Heart Failure. Mol. Med. Rep. 2014, 10, 615–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaney, M.L.; Gardner, J.R.; Karasavvas, N.; Golde, D.W.; Scheinberg, D.A.; Smith, E.A.; O’Connor, O.A. Vitamin C Antagonizes the Cytotoxic Effects of Antineoplastic Drugs. Cancer Res. 2008, 68, 8031–8038. [Google Scholar] [CrossRef] [Green Version]
- Conklin, K.A. Chemotherapy-Associated Oxidative Stress: Impact on Chemotherapeutic Effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Santos Araújo, M.d.C.; Farias, I.L.; Gutierres, J.; Dalmora, S.L.; Flores, N.; Farias, J.; de Cruz, I.; Chiesa, J.; Morsch, V.M.; Chitolina Schetinger, M.R. Uncaria Tomentosa—Adjuvant Treatment for Breast Cancer: Clinical Trial. Evid. Based Complement. Alternat. Med. 2012, 2012, 676984. [Google Scholar]
- Liang, G.; Tang, A.; Lin, X.; Li, L.; Zhang, S.; Huang, Z.; Tang, H.; Li, Q.Q. Green Tea Catechins Augment the Antitumor Activity of Doxorubicin in an in Vivo Mouse Model for Chemoresistant Liver Cancer. Int. J. Oncol. 2010, 37, 111–123. [Google Scholar]
- Qiao, J.; Gu, C.; Shang, W.; Du, J.; Yin, W.; Zhu, M.; Wang, W.; Han, M.; Lu, W. Effect of Green Tea on Pharmacokinetics of 5-Fluorouracil in Rats and Pharmacodynamics in Human Cell Lines in Vitro. Food Chem. Toxicol. 2011, 49, 1410–1415. [Google Scholar] [CrossRef]
- Sahin, K.; Tuzcu, M.; Gencoglu, H.; Dogukan, A.; Timurkan, M.; Sahin, N.; Aslan, A.; Kucuk, O. Epigallocatechin-3-Gallate Activates Nrf2/HO-1 Signaling Pathway in Cisplatin-Induced Nephrotoxicity in Rats. Life Sci. 2010, 87, 240–245. [Google Scholar] [CrossRef]
- Harisa, G.I. Blood Viscosity as a Sensitive Indicator for Paclitaxel Induced Oxidative Stress in Human Whole Blood. Saudi Pharm. J. 2015, 23, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Mizumachi, T.; Suzuki, S.; Naito, A.; Carcel-Trullols, J.; Evans, T.T.; Spring, P.M.; Oridate, N.; Furuta, Y.; Fukuda, S.; Higuchi, M. Increased Mitochondrial DNA Induces Acquired Docetaxel Resistance in Head and Neck Cancer Cells. Oncogene 2008, 27, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, D.L.; Gray, H.; Payne, C.M.; Gillies, R.J. Docetaxel Induces Cell Death through Mitotic Catastrophe in Human Breast Cancer Cells. Mol. Cancer Ther. 2005, 4, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zheng, L.; Tian, Y.; Zhang, Z.; Dong, W.; Wang, X.; Zhang, X.; Cao, C. C6 Ceramide Dramatically Enhances Docetaxel-Induced Growth Inhibition and Apoptosis in Cultured Breast Cancer Cells: A Mechanism Study. Exp. Cell Res. 2015, 332, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Sun, C.; Zhou, B.; Xing, H.; Ma, D.; Chen, G.; Weng, D. Low Concentration of Quercetin Antagonizes the Cytotoxic Effects of Anti-Neoplastic Drugs in Ovarian Cancer. PLoS ONE 2014, 9, e100314. [Google Scholar] [CrossRef]
- Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. The Chemoprotective Agent N-Acetylcysteine Blocks Cisplatin-Induced Apoptosis through Caspase Signaling Pathway. J. Pharmacol. Exp. Ther. 2005, 312, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Das, T.; Pereira, S.; Yang, Z.; Zhao, M.; Mukerji, P.; Hoffman, R.M. Efficacy of Dietary Antioxidants Combined with a Chemotherapeutic Agent on Human Colon Cancer Progression in a Fluorescent Orthotopic Mouse Model. Anticancer Res. 2009, 29, 2421–2426. [Google Scholar]
- Fuchs-Tarlovsky, V.; Bejarano-Rosales, M.; Gutiérrez-Salmeán, G.; Casillas, M.; López-Alvarenga, J.C.; Ceballos-Reyes, G.M. Efecto de La Suplementación Con Antioxidantes Sobre El Estrés Oxidativo y La Calidad de Vida Durante El Tratamiento Oncológico En Pacientes Con Cáncer Cérvico Uterino. Nutr. Hosp. 2011, 26, 819–826. [Google Scholar]
- Liu, X.; Wu, J.; Liu, H.; Zong, N.; Zhao, J. RoGFP1 Is a Quantitative Biosensor in Maize Cells for Cellular Redox Changes Caused by Environmental and Endogenous Stimuli. Biochem. Biophys. Res. Commun. 2014, 452, 503–508. [Google Scholar] [CrossRef]
- Dickey, D.T.; Muldoon, L.L.; Kraemer, D.F.; Neuwelt, E.A. Protection against Cisplatin-Induced Ototoxicity by N-Acetylcysteine in a Rat Model. Hear. Res. 2004, 193, 25–30. [Google Scholar] [CrossRef]
- Dickey, D.T.; Muldoon, L.L.; Doolittle, N.D.; Peterson, D.R.; Kraemer, D.F.; Neuwelt, E.A. Effect of N-Acetylcysteine Route of Administration on Chemoprotection against Cisplatin-Induced Toxicity in Rat Models. Cancer Chemother. Pharmacol. 2008, 62, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atessahin, A.; Yilmaz, S.; Karahan, I.; Ceribasi, A.O.; Karaoglu, A. Effects of Lycopene against Cisplatin-Induced Nephrotoxicity and Oxidative Stress in Rats. Toxicology 2005, 212, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Al-Tonbary, Y.; Al-Haggar, M.; El-Ashry, R.; El-Dakroory, S.; Azzam, H.; Fouda, A. Vitamin e and N-Acetylcysteine as Antioxidant Adjuvant Therapy in Children with Acute Lymphoblastic Leukemia. Adv. Hematol. 2009, 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lissoni, P. Biochemotherapy with Standard Chemotherapies plus the Pineal Hormone Melatonin in the Treatment of Advanced Solid Neoplasms. Pathol. Biol. 2007, 55, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Sookprasert, A.; Johns, N.P.; Phunmanee, A.; Pongthai, P.; Cheawchanwattana, A.; Johns, J.; Konsil, J.; Plaimee, P.; Porasuphatana, S.; Jitpimolmard, S. Melatonin in Patients with Cancer Receiving Chemotherapy: A Randomized, Double-Blind, Placebo-Controlled Trial. Anticancer Res. 2014, 34, 7327–7337. [Google Scholar] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The Antioxidant Function of the P53 Tumor Suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J. HIF-Dependent Antitumorigenic Effect of Antioxidants in Vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Aitio, M.L. N-Acetylcysteine–Passe-Partout or Much Ado about Nothing? Br. J. Clin. Pharmacol. 2006, 61, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J. Antioxidants Can Increase Melanoma Metastasis in Mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.R.; Albanes, D. Selenium, Vitamin E, and Prostate Cancer—Ready for Prime Time? Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar]
- Lippman, S.M.; Goodman, P.J.; Klein, E.A.; Parnes, H.L.; Thompson, I.M.; Kristal, A.R.; Santella, R.M.; Probstfield, J.L.; Moinpour, C.M.; Albanes, D. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J. Natl. Cancer Inst. 2005, 97, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Caroppo, F.; Alaibac, M. Vitamins and Melanoma. Cancers 2015, 7, 1371–1387. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C. Vitamin C Selectively Kills KRAS and BRAF Mutant Colorectal Cancer Cells by Targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W. Ascorbate Regulates Haematopoietic Stem Cell Function and Leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Miljkovic, J.L.; Bostelaar, T.; Adhikari, B.; Yadav, P.K.; Steiger, A.K.; Torregrossa, R.; Pluth, M.D.; Whiteman, M.; Banerjee, R. Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem. Biol. 2018, 13, 2300–2307. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 Mutations: Permanent Activation of an Adaptive Response in Cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Kenneth, H.Y.; Yeo, C.J.; Calhoun, E.S. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K. Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E–Driven Lung Tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E. The Oncogenic Action of NRF2 Depends on De-Glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat. Med. 2017, 23, 1362. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E. NRF2 Regulates Serine Biosynthesis in Non–Small Cell Lung Cancer. Nat. Genet. 2015, 47, 1475. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Kemper, E.K.; Suciu, R.M.; Vinogradova, E.V.; Backus, K.M.; Horning, B.D.; Paul, T.A.; Ichu, T.-A.; Svensson, R.U.; Olucha, J. Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171, 696–709. [Google Scholar] [CrossRef] [Green Version]
- Arnandis, T.; Monteiro, P.; Adams, S.D.; Bridgeman, V.L.; Rajeeve, V.; Gadaleta, E.; Marzec, J.; Chelala, C.; Malanchi, I.; Cutillas, P.R. Oxidative Stress in Cells with Extra Centrosomes Drives Non-Cell-Autonomous Invasion. Dev. Cell 2018, 47, 409–424. [Google Scholar] [CrossRef] [Green Version]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Shimosegawa, T.; Taguchi, K.; Nabeshima, T.; Yamamoto, M.; Masamune, A. Simultaneous K-Ras Activation and Keap1 Deletion Cause Atrophy of Pancreatic Parenchyma. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 314, G65–G74. [Google Scholar] [CrossRef] [PubMed]
- Rogers, Z.N.; McFarland, C.D.; Winters, I.P.; Naranjo, S.; Chuang, C.-H.; Petrov, D.; Winslow, M.M. A Quantitative and Multiplexed Approach to Uncover the Fitness Landscape of Tumor Suppression in Vivo. Nat. Methods 2017, 14, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine Dioxygenase 1 Is a Metabolic Liability for Non-Small Cell Lung Cancer. eLife 2019, 8, e45572. [Google Scholar] [CrossRef]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The Influence of Reactive Oxygen Species on Cell Cycle Progression in Mammalian Cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Kehrer, I.; Kozlov, A.V.; Haller, M.; Redl, H.; Hermann, M.; Grimm, M.; Troppmair, J. Mitochondrial ROS Production under Cellular Stress: Comparison of Different Detection Methods. Anal. Bioanal. Chem. 2011, 400, 2383–2390. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, M.; Yuan, Z. Methods for the Detection of Reactive Oxygen Species. Anal. Methods 2018, 10, 4625–4638. [Google Scholar] [CrossRef]
- Zhao, H.; Joseph, J.; Fales, H.M.; Sokoloski, E.A.; Levine, R.L.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection and Characterization of the Product of Hydroethidine and Intracellular Superoxide by HPLC and Limitations of Fluorescence. Proc. Natl. Acad. Sci. USA 2005, 102, 5727. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Zhao, H.; Xu, Y.; Kalyanaraman, B. Mechanistic Similarities between Oxidation of Hydroethidine by Fremy’s Salt and Superoxide: Stopped-Flow Optical and EPR Studies. Free Radic. Biol. Med. 2005, 39, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, R.; Leen, R.; Tytgat, G.A.; Caron, H.N.; Van Kuilenburg, A.B. Fenretinide Induces Mitochondrial ROS and Inhibits the Mitochondrial Respiratory Chain in Neuroblastoma. Cell. Mol. Life Sci. 2010, 67, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Hardy, M.; Kalyanaraman, B. HPLC Study of Oxidation Products of Hydroethidine in Chemical and Biological Systems: Ramifications in Superoxide Measurements. Free Radic. Biol. Med. 2009, 46, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiou, C.D.; Papapostolou, I.; Grintzalis, K. Superoxide Radical Detection in Cells, Tissues, Organisms (Animals, Plants, Insects, Microorganisms) and Soils. Nat. Protoc. 2008, 3, 1679. [Google Scholar] [CrossRef]
- Wang, H.; Joseph, J.A. Quantifying Cellular Oxidative Stress by Dichlorofluorescein Assay Using Microplate Reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Glebska, J.; Koppenol, W.H. Peroxynitrite-Mediated Oxidation of Dichlorodihydrofluorescein and Dihydrorhodamine. Free Radic. Biol. Med. 2003, 35, 676–682. [Google Scholar] [CrossRef]
- Afzal, M.; Matsugo, S.; Sasai, M.; Xu, B.; Aoyama, K.; Takeuchi, T. Method to Overcome Photoreaction, a Serious Drawback to the Use of Dichlorofluorescin in Evaluation of Reactive Oxygen Species. Biochem. Biophys. Res. Commun. 2003, 304, 619–624. [Google Scholar] [CrossRef]
- Vejražka, M.; Míček, R.; Štípek, S. Apocynin Inhibits NADPH Oxidase in Phagocytes but Stimulates ROS Production in Non-Phagocytic Cells. Biochim. Biophys. Acta BBA-Gen. Subj. 2005, 1722, 143–147. [Google Scholar] [CrossRef]
- Karlsson, M.; Kurz, T.; Brunk, U.T.; Nilsson, S.E.; Frennesson, C.I. What Does the Commonly Used DCF Test for Oxidative Stress Really Show? Biochem. J. 2010, 428, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Possel, H.; Noack, H.; Augustin, W.; Keilhoff, G.; Wolf, G. 2, 7-Dihydrodichlorofluorescein Diacetate as a Fluorescent Marker for Peroxynitrite Formation. FEBS Lett. 1997, 416, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Kalyanaraman, B. “ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis”—A Critical Commentary. Free Radic. Biol. Med. 2008, 45, 1217–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkes, L.K.; Patel, K.B.; Wardman, P.; Wrona, M. Kinetics of Reaction of Nitrogen Dioxide with Dihydrorhodamine and the Reaction of the Dihydrorhodamine Radical with Oxygen: Implications for Quantifying Peroxynitrite Formation in Cells. Arch. Biochem. Biophys. 2009, 484, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for Detection of Reactive Metabolites of Oxygen and Nitrogen: In Vitro and in Vivo Considerations. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 286, R431–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Yadav, S.; Shah, P.G.; Liu, T.; Tian, B.; Pukszta, S.; Villaluna, N.; Kutejová, E.; Newlon, C.S.; Santos, J.H. Roles for the Human ATP-Dependent Lon Protease in Mitochondrial DNA Maintenance. J. Biol. Chem. 2007, 282, 17363–17374. [Google Scholar] [CrossRef] [Green Version]
- Dębski, D.; Smulik, R.; Zielonka, J.; Michałowski, B.; Jakubowska, M.; Dębowska, K.; Adamus, J.; Marcinek, A.; Kalyanaraman, B.; Sikora, A. Mechanism of Oxidative Conversion of Amplex® Red to Resorufin: Pulse Radiolysis and Enzymatic Studies. Free Radic. Biol. Med. 2016, 95, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Votyakova, T.V.; Reynolds, I.J. Detection of Hydrogen Peroxide with Amplex Red: Interference by NADH and Reduced Glutathione Auto-Oxidation. Arch. Biochem. Biophys. 2004, 431, 138–144. [Google Scholar] [CrossRef]
- Dikalov, S.; Griendling, K.K.; Harrison, D.G. Measurement of Reactive Oxygen Species in Cardiovascular Studies. Hypertension 2007, 49, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Miwa, S.; Treumann, A.; Bell, A.; Vistoli, G.; Nelson, G.; Hay, S.; von Zglinicki, T. Carboxylesterase Converts Amplex Red to Resorufin: Implications for Mitochondrial H2O2 Release Assays. Free Radic. Biol. Med. 2016, 90, 173–183. [Google Scholar] [CrossRef]
- Dooley, C.T.; Dore, T.M.; Hanson, G.T.; Jackson, W.C.; Remington, S.J.; Tsien, R.Y. Imaging Dynamic Redox Changes in Mammalian Cells with Green Fluorescent Protein Indicators. J. Biol. Chem. 2004, 279, 22284–22293. [Google Scholar] [CrossRef] [Green Version]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating Mitochondrial Redox Potential with Redox-Sensitive Green Fluorescent Protein Indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [Green Version]
- Yano, T.; Oku, M.; Akeyama, N.; Itoyama, A.; Yurimoto, H.; Kuge, S.; Fujiki, Y.; Sakai, Y. A Novel Fluorescent Sensor Protein for Visualization of Redox States in the Cytoplasm and in Peroxisomes. Mol. Cell. Biol. 2010, 30, 3758–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, J.R.; Remington, S.J. Development of a Family of Redox-Sensitive Green Fluorescent Protein Indicators for Use in Relatively Oxidizing Subcellular Environments. Biochemistry 2008, 47, 8678–8688. [Google Scholar] [CrossRef] [PubMed]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic Reticulum: Reduced and Oxidized Glutathione Revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lith, M.V.; Tiwari, S.; Pediani, J.; Milligan, G.; Bulleid, N.J. Real-Time Monitoring of Redox Changes in the Mammalian Endoplasmic Reticulum. J. Cell Sci. 2011, 124, 2349–2356. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.B.; Remington, S.J. Re-Engineering Redox-Sensitive Green Fluorescent Protein for Improved Response Rate. Protein Sci. 2006, 15, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-Based Protein Thiol Oxidation by H2O2-Scavenging Peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Schneider, J.F.; Degrossoli, A.; Lupilova, N.; Dick, T.P.; Leichert, L.I. Systematic in Vitro Assessment of Responses of RoGFP2-Based Probes to Physiologically Relevant Oxidant Species. Free Radic. Biol. Med. 2017, 106, 329–338. [Google Scholar] [CrossRef]
- Maulucci, G.; Labate, V.; Mele, M.; Panieri, E.; Arcovito, G.; Galeotti, T.; Østergaard, H.; Winther, J.R.; De Spirito, M.; Pani, G. High-Resolution Imaging of Redox Signaling in Live Cells through an Oxidation-Sensitive Yellow Fluorescent Protein. Sci. Signal. 2008, 1, pl3. [Google Scholar] [CrossRef]
- Dardalhon, M.; Kumar, C.; Iraqui, I.; Vernis, L.; Kienda, G.; Banach-Latapy, A.; He, T.; Chanet, R.; Faye, G.; Outten, C.E. Redox-Sensitive YFP Sensors Monitor Dynamic Nuclear and Cytosolic Glutathione Redox Changes. Free Radic. Biol. Med. 2012, 52, 2254–2265. [Google Scholar] [CrossRef] [Green Version]
- Björnberg, O.; Østergaard, H.; Winther, J.R. Measuring Intracellular Redox Conditions Using GFP-Based Sensors. Antioxid. Redox Signal. 2006, 8, 354–361. [Google Scholar] [CrossRef]
- Hansen, R.E.; Østergaard, H.; Winther, J.R. Increasing the Reactivity of an Artificial Dithiol- Disulfide Pair through Modification of the Electrostatic Milieu. Biochemistry 2005, 44, 5899–5906. [Google Scholar] [CrossRef]
- Østergaard, H.; Henriksen, A.; Hansen, F.G.; Winther, J.R. Shedding Light on Disulfide Bond Formation: Engineering a Redox Switch in Green Fluorescent Protein. EMBO J. 2001, 20, 5853–5862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Barrera, A.; Quinto, C.; Johnson, E.A.; Wu, H.-M.; Cheung, A.Y.; Cardenas, L. Using hyper as a molecular probe to visualize hydrogen peroxide in living plant cells: A method with virtually unlimited potential in plant biology. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 527, pp. 275–290. [Google Scholar]
- Labuschagne, C.F.; Brenkman, A.B. Current Methods in Quantifying ROS and Oxidative Damage in Caenorhabditis Elegans and Other Model Organism of Aging. Ageing Res. Rev. 2013, 12, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically Encoded Fluorescent Indicator for Intracellular Hydrogen Peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, K.A.; Belousov, V.V. Genetically Encoded Fluorescent Redox Sensors. Biochim. Biophys. Acta BBA-Gen. Subj. 2014, 1840, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Markvicheva, K.N.; Bilan, D.S.; Mishina, N.M.; Gorokhovatsky, A.Y.; Vinokurov, L.M.; Lukyanov, S.; Belousov, V.V. A Genetically Encoded Sensor for H2O2 with Expanded Dynamic Range. Bioorg. Med. Chem. 2011, 19, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A Genetically Encoded H2O2 Probe with Improved Performance for Ratiometric and Fluorescence Lifetime Imaging. ACS Chem. Biol. 2013, 8, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Pouvreau, S. Superoxide Flashes in Mouse Skeletal Muscle Are Produced by Discrete Arrays of Active Mitochondria Operating Coherently. PLoS ONE 2010, 5, e13035. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M. Superoxide Flashes in Single Mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Quatresous, E.; Legrand, C.; Pouvreau, S. Mitochondria-Targeted CpYFP: PH or Superoxide Sensor? J. Gen. Physiol. 2012, 140, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Ermakova, Y.G.; Bilan, D.S.; Matlashov, M.E.; Mishina, N.M.; Markvicheva, K.N.; Subach, O.M.; Subach, F.V.; Bogeski, I.; Hoth, M.; Enikolopov, G. Red Fluorescent Genetically Encoded Indicator for Intracellular Hydrogen Peroxide. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Makar, M.; Wang, M.X.; Ai, H. Monitoring Thioredoxin Redox with a Genetically Encoded Red Fluorescent Biosensor. Nat. Chem. Biol. 2017, 13, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Chen, Z.; Ai, H. Monitoring Redox Dynamics in Living Cells with a Redox-Sensitive Red Fluorescent Protein. Anal. Chem. 2015, 87, 2802–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-Time Imaging of the Intracellular Glutathione Redox Potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef]
- Pasin, F.; Kulasekaran, S.; Natale, P.; Simón-Mateo, C.; García, J.A. Rapid Fluorescent Reporter Quantification by Leaf Disc Analysis and Its Application in Plant-Virus Studies. Plant Methods 2014, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Afanas’ ev, I.B.; Kleschyov, A.L.; Harrison, D.G. Detection of Superoxide in Vascular Tissue. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1761–1768. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, S.; Merenyi, G. The chemistry of peroxynitrite: Implications for biological activity. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 436, pp. 49–61. [Google Scholar]
- Li, Y.; Zhu, H.; Kuppusamy, P.; Roubaud, V.; Zweier, J.L.; Trush, M.A. Validation of Lucigenin (Bis-N-Methylacridinium) as a Chemilumigenic Probe for Detecting Superoxide Anion Radical Production by Enzymatic and Cellular Systems. J. Biol. Chem. 1998, 273, 2015–2023. [Google Scholar] [CrossRef] [Green Version]
- Maskiewicz, R.; Sogah, D.; Bruice, T.C. Chemiluminescent Reactions of Lucigenin. 1. Reactions of Lucigenin with Hydrogen Peroxide. J. Am. Chem. Soc. 1979, 101, 5347–5354. [Google Scholar] [CrossRef]
- Tarpey, M.M.; White, C.R.; Suarez, E.; Richardson, G.; Radi, R.; Freeman, B.A. Chemiluminescent Detection of Oxidants in Vascular Tissue: Lucigenin but Not Coelenterazine Enhances Superoxide Formation. Circ. Res. 1999, 84, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Merenyi, G.; Lind, J.; Eriksen, T.E. The Equilibrium Reaction of the Luminol Radical with Oxygen and the One-Electron-Reduction Potential of 5-Aminophthalazine-1, 4-Dione. J. Phys. Chem. 1984, 88, 2320–2323. [Google Scholar] [CrossRef]
- Kielland, A.; Blom, T.; Nandakumar, K.S.; Holmdahl, R.; Blomhoff, R.; Carlsen, H. In Vivo Imaging of Reactive Oxygen and Nitrogen Species in Inflammation Using the Luminescent Probe L-012. Free Radic. Biol. Med. 2009, 47, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ ev, I.B. Lucigenin Chemiluminescence Assay for Superoxide Detection. Circ. Res. 2001, 89, e46. [Google Scholar]
- Beissenhirtz, M.K.; Scheller, F.W.; Lisdat, F. A Superoxide Sensor Based on a Multilayer Cytochrome c Electrode. Anal. Chem. 2004, 76, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Ganesana, M.; Erlichman, J.S.; Andreescu, S. Real-Time Monitoring of Superoxide Accumulation and Antioxidant Activity in a Brain Slice Model Using an Electrochemical Cytochrome c Biosensor. Free Radic. Biol. Med. 2012, 53, 2240–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decher, G.; Lehr, B.; Lowack, K.; Lvov, Y.; Schmitt, J. New Nanocomposite Films for Biosensors: Layer-by-Layer Adsorbed Films of Polyelectrolytes, Proteins or DNA. Biosens. Bioelectron. 1994, 9, 677–684. [Google Scholar] [CrossRef]
- Grebe, S.K.; Singh, R.J. LC-MS/MS in the Clinical Laboratory–Where to from Here? Clin. Biochem. Rev. 2011, 32, 5. [Google Scholar]
- Floyd, R.A.; Henderson, R.; Watson, J.J.; Wong, P.K. Use of Salicylate with High Pressure Liquid Chromatography and Electrochemical Detection (LCE) as a Sensitive Measure of Hydroxyl Free Radicals in Adriamycin Treated Rats. J. Free Radic. Biol. Med. 1986, 2, 13–18. [Google Scholar] [CrossRef]
- Piszcz, P.; Żurawski, K.; Głód, B.K. Application of HPLC to Study the Reaction of Free Radicals with Antioxidants and/or Toxins. J. Chem. 2014, 2014. [Google Scholar] [CrossRef]
- Kolbeck, R.C.; She, Z.-W.; Callahan, L.A.; Nosek, M.T. Increased Superoxide Production during Fatigue in the Perfused Rat Diaphragm. Am. J. Respir. Crit. Care Med. 1997, 156, 140–145. [Google Scholar] [CrossRef]
- Thomson, L.; Trujillo, M.; Telleri, R.; Radi, R. Kinetics of Cytochrome C2+ Oxidation by Peroxynitrite: Implications for Superoxide Measurements in Nitric Oxide-Producing Biological-Systems. Arch. Biochem. Biophys. 1995, 319, 491–497. [Google Scholar] [CrossRef]
- Jones, O.T.; Hancock, J.T. [22] Assays of plasma membrane NADPH oxidase. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 233, pp. 222–229. [Google Scholar]
- Kuthan, H.; Ullrich, V.; Estabrook, R.W. A Quantitative Test for Superoxide Radicals Produced in Biological Systems. Biochem. J. 1982, 203, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim Choi, H.; Woo Kim, J.; Cha, Y.-N.; Kim, C. A Quantitative Nitroblue Tetrazolium Assay for Determining Intracellular Superoxide Anion Production in Phagocytic Cells. J. Immunoassay Immunochem. 2006, 27, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Senft, A.P.; Dalton, T.P.; Nebert, D.W.; Genter, M.B.; Hutchinson, R.J.; Shertzer, H.G. Dioxin Increases Reactive Oxygen Production in Mouse Liver Mitochondria. Toxicol. Appl. Pharmacol. 2002, 178, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Fridovich, I. Inactivation-Reactivation of Aconitase in Escherichia Coli. A Sensitive Measure of Superoxide Radical. J. Biol. Chem. 1992, 267, 8757–8763. [Google Scholar] [CrossRef]
- Hausladen, A.; Fridovich, I. [4] Measuring nitric oxide and superoxide: Rate constants for aconitase reactivity. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 269, pp. 37–41. [Google Scholar]
- Melov, S.; Coskun, P.; Patel, M.; Tuinstra, R.; Cottrell, B.; Jun, A.S.; Zastawny, T.H.; Dizdaroglu, M.; Goodman, S.I.; Huang, T.-T. Mitochondrial Disease in Superoxide Dismutase 2 Mutant Mice. Proc. Natl. Acad. Sci. USA 1999, 96, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Castro, L.; Rodriguez, M.; Radi, R. Aconitase Is Readily Inactivated by Peroxynitrite, but Not by Its Precursor, Nitric Oxide. J. Biol. Chem. 1994, 269, 29409–29415. [Google Scholar] [CrossRef]
- Sikora, A.; Zielonka, J.; Lopez, M.; Joseph, J.; Kalyanaraman, B. Direct Oxidation of Boronates by Peroxynitrite: Mechanism and Implications in Fluorescence Imaging of Peroxynitrite. Free Radic. Biol. Med. 2009, 47, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Sikora, A.; Hardy, M.; Joseph, J.; Dranka, B.P.; Kalyanaraman, B. Boronate Probes as Diagnostic Tools for Real Time Monitoring of Peroxynitrite and Hydroperoxides. Chem. Res. Toxicol. 2012, 25, 1793–1799. [Google Scholar] [CrossRef] [Green Version]
- Albers, A.E.; Dickinson, B.C.; Miller, E.W.; Chang, C.J. A Red-Emitting Naphthofluorescein-Based Fluorescent Probe for Selective Detection of Hydrogen Peroxide in Living Cells. Bioorg. Med. Chem. Lett. 2008, 18, 5948–5950. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, B.C.; Chang, C.J. A Targetable Fluorescent Probe for Imaging Hydrogen Peroxide in the Mitochondria of Living Cells. J. Am. Chem. Soc. 2008, 130, 9638–9639. [Google Scholar] [CrossRef] [Green Version]
- Thordal-Christensen, H.; Zhang, Z.; Wei, Y.; Collinge, D.B. Subcellular Localization of H2O2 in Plants. H2O2 Accumulation in Papillae and Hypersensitive Response during the Barley—Powdery Mildew Interaction. Plant J. 1997, 11, 1187–1194. [Google Scholar] [CrossRef]
- Stockert, J.C.; Blazquez-Castro, A. Establishing the Subcellular Localization of Photodynamically-Induced ROS Using 3, 3′-Diaminobenzidine: A Methodological Proposal, with a Proof-of-Concept Demonstration. Methods 2016, 109, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Li, L.; Abe, Y.; Ogata, M.; Ishimoto, Y.; Gonda, R.; Mashino, T.; Mochizuki, M.; Uemoto, M.; Miyata, N. DMPO-OH Radical Formation from 5, 5-Dimethyl-1-Pyrroline N-Oxide (DMPO) in Hot Water. Anal. Sci. 2007, 23, 219–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villamena, F.A.; Zweier, J.L. Detection of Reactive Oxygen and Nitrogen Species by EPR Spin Trapping. Antioxid. Redox Signal. 2004, 6, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.; Jiang, J.; Mason, R.P. Characterization of the High-Resolution ESR Spectra of Superoxide Radical Adducts of 5-(Diethoxyphosphoryl)-5-Methyl-1-Pyrroline N-Oxide (DEPMPO) and 5, 5-Dimethyl-1-Pyrroline N-Oxide (DMPO). Analysis of Conformational Exchange. Free Radic. Res. 2005, 39, 825–836. [Google Scholar] [CrossRef]
- Turner, M.J., III; Rosen, G.M. Spin Trapping of Superoxide and Hydroxyl Radicals with Substituted Pyrroline 1-Oxides. J. Med. Chem. 1986, 29, 2439–2444. [Google Scholar] [CrossRef]
- Zhao, H.; Joseph, J.; Zhang, H.; Karoui, H.; Kalyanaraman, B. Synthesis and Biochemical Applications of a Solid Cyclic Nitrone Spin Trap: A Relatively Superior Trap for Detecting Superoxide Anions and Glutathiyl Radicals. Free Radic. Biol. Med. 2001, 31, 599–606. [Google Scholar] [CrossRef]
- Dikalov, S.; Skatchkov, M.; Bassenge, E. Spin Trapping of Superoxide Radicals and Peroxynitrite by 1-Hydroxy-3-Carboxy-Pyrrolidine and 1-Hydroxy-2, 2, 6, 6-Tetramethyl-4-Oxo-Piperidine and the Stability of Corresponding Nitroxyl Radicals towards Biological Reductants. Biochem. Biophys. Res. Commun. 1997, 231, 701–704. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Polienko, Y.F.; Kirilyuk, I. Electron Paramagnetic Resonance Measurements of Reactive Oxygen Species by Cyclic Hydroxylamine Spin Probes. Antioxid. Redox Signal. 2018, 28, 1433–1443. [Google Scholar] [CrossRef]
- Berliner, L.J. The Evolution of Biomedical EPR (ESR). Biomed. Spectrosc. Imaging 2016, 5, 5–26. [Google Scholar] [CrossRef] [Green Version]
- Wardman, P. Fluorescent and Luminescent Probes for Measurement of Oxidative and Nitrosative Species in Cells and Tissues: Progress, Pitfalls, and Prospects. Free Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X. Targeting Mitochondrial Reactive Oxygen Species as Novel Therapy for Inflammatory Diseases and Cancers. J. Hematol. Oncol.J Hematol Oncol 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, B.C.; Tang, Y.; Chang, Z.; Chang, C.J. A Nuclear-Localized Fluorescent Hydrogen Peroxide Probe for Monitoring Sirtuin-Mediated Oxidative Stress Responses in Vivo. Chem. Biol. 2011, 18, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambertucci, R.H.; Hirabara, S.M.; Silveira, L.d.R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate Increases Superoxide Production through Mitochondrial Electron Transport Chain and NADPH Oxidase Activity in Skeletal Muscle Cells. J. Cell. Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detection Methods | Probes | Target ROS | Pros | Cons |
|---|---|---|---|---|
| Fluorescent Chemicals |
| O2˙− and H2O2 |
|
|
| Fluorescent Proteins |
| H2O2 and Variation in redox level |
|
|
| Chemi-luminescence |
| H2O2 and O2˙− |
|
|
| Electro-chemical Biosensing |
| O2˙− |
|
|
| Chromatography |
| ˙OH |
|
|
| Spectro-photometry |
| O2˙− and H2O2 |
|
|
| EPR/ESR | Spin traps | ROS and RNS |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuloria, S.; Subramaniyan, V.; Karupiah, S.; Kumari, U.; Sathasivam, K.; Meenakshi, D.U.; Wu, Y.S.; Sekar, M.; Chitranshi, N.; Malviya, R.; et al. Comprehensive Review of Methodology to Detect Reactive Oxygen Species (ROS) in Mammalian Species and Establish Its Relationship with Antioxidants and Cancer. Antioxidants 2021, 10, 128. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010128
Fuloria S, Subramaniyan V, Karupiah S, Kumari U, Sathasivam K, Meenakshi DU, Wu YS, Sekar M, Chitranshi N, Malviya R, et al. Comprehensive Review of Methodology to Detect Reactive Oxygen Species (ROS) in Mammalian Species and Establish Its Relationship with Antioxidants and Cancer. Antioxidants. 2021; 10(1):128. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010128
Chicago/Turabian StyleFuloria, Shivkanya, Vetriselvan Subramaniyan, Sundram Karupiah, Usha Kumari, Kathiresan Sathasivam, Dhanalekshmi Unnikrishnan Meenakshi, Yuan Seng Wu, Mahendran Sekar, Nitin Chitranshi, Rishabha Malviya, and et al. 2021. "Comprehensive Review of Methodology to Detect Reactive Oxygen Species (ROS) in Mammalian Species and Establish Its Relationship with Antioxidants and Cancer" Antioxidants 10, no. 1: 128. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10010128