Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer


Department of Food Science and Engineering, Ewha Womans University, Seoul 03760, Korea
Antioxidants 2021, 10(2), 169; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020169
Submission received: 13 January 2021
/
Revised: 21 January 2021
/
Accepted: 21 January 2021
/
Published: 24 January 2021
(This article belongs to the Section Health Outcomes of Antioxidants and Oxidative Stress)
Abstract
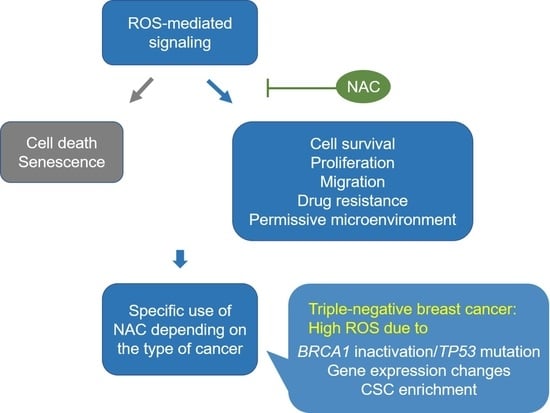
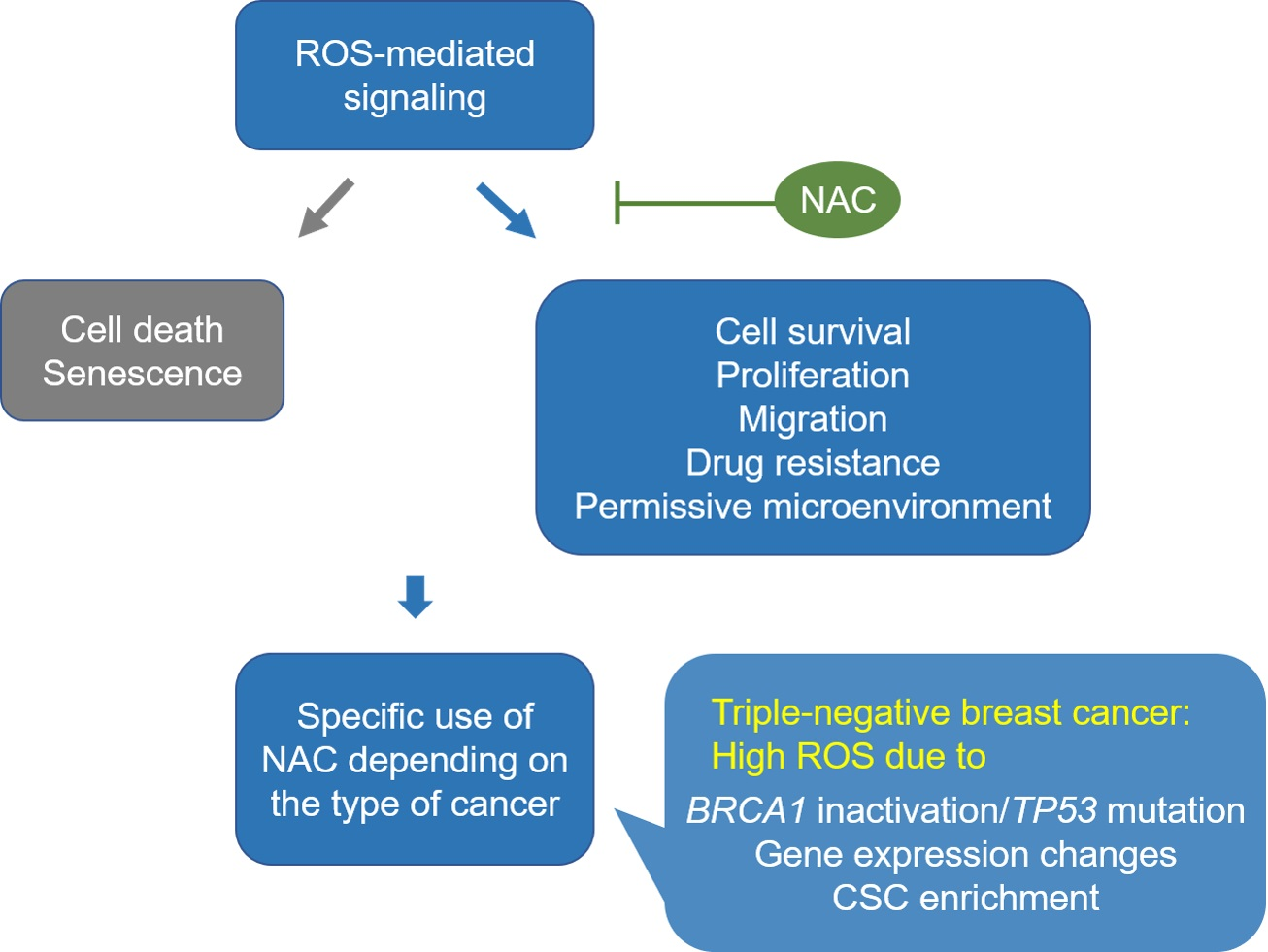
:N-acetylcysteine (NAC) is a widely used antioxidant with therapeutic potential. However, the cancer-promoting effect of NAC observed in some preclinical studies has raised concerns regarding its clinical use. Reactive oxygen species (ROS) can mediate signaling that results in both cancer-promoting and cancer-suppressing effects. The beneficial effect of NAC may depend on whether the type of cancer relies on ROS signaling for its survival and metastasis. Triple-negative breast cancer (TNBC) has aggressive phenotypes and is currently treated with standard chemotherapy as the main systemic treatment option. Particularly, basal-like TNBC cells characterized by inactivated BRCA1 and mutated TP53 produce high ROS levels and rely on ROS signaling for their survival and malignant progression. In addition, the high ROS levels in TNBC cells can mediate the interplay between cancer cells and the tissue microenvironment (TME) to trigger the recruitment and conversion of stromal cells and induce hypoxic responses, thus leading to the creation of cancer-supportive TMEs and increased cancer aggressiveness. However, NAC treatment effectively reduces the ROS production and ROS-mediated signaling that contribute to cell survival, metastasis, and drug resistance in TNBC cells. Therefore, the inclusion of NAC in standard chemotherapy could probably provide additional benefits for TNBC patients.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
N-acetylcysteine (NAC) is widely used as a medication and a dietary supplement. It functions as a mucolytic due to its ability to break the disulfide bonds in the glycoproteins of the mucus, thus resulting in a decrease in viscosity [1]. In addition, NAC is used for the treatment of acetaminophen overdose because it can restore the depleted glutathione (GSH) reserves in the hepatocytes during the process of detoxification [2]. Moreover, NAC has been widely studied for its antioxidant effects [1,3,4]. This antioxidant activity is mainly attributed to its ability to act as a precursor of cysteine, which is itself a component of the primary intracellular antioxidant, GSH. Because the availability of cysteine can limit the rate of GSH synthesis under conditions of oxidative stress, the administration of NAC can replenish GSH stores [2,4]. By contrast, the direct antioxidant activity of NAC was thought to be irrelevant because the thiol group has inherently low reactivity toward oxidants [1]. However, an alternative mechanism has recently been suggested involving its ability to break the disulfide bonds of the cysteinylated proteins to release free thiols and regenerate reduced proteins, which can have direct antioxidant activity in certain cases (e.g., mercaptoalbumin) [1,5]. Another proposed mechanism involves the metabolic conversion of NAC-derived thiols into hyperactivated thiols (hydropersulfides) that act as direct oxidant scavengers and/or protective caps for critical protein thiols [6]. Therefore, NAC is a potent antioxidant due to both its direct antioxidant activity and the supply of a GHS precursor. Hence, NAC has been recommended for use in treating diseases where oxidative stress is involved at the onset and/or progression of the disease state [4]. The therapeutic use of NAC has been reported in several cancers, including breast cancer. For example, a pilot study has encouragingly suggested that NAC may be effective as a single agent in the inhibition of cancer cell proliferation in breast cancer patients [7].
Both the incidence of cancer and the number of cancer-related deaths are rapidly increasing worldwide, with breast cancer being the most frequently diagnosed. Approximately 2.1 million women were newly diagnosed with breast cancer in 2018, accounting for almost one in four cancer cases among women [8]. Due to early detection and effective treatment, the prognosis of breast cancer is relatively favorable. However, it is estimated that 627,000 women still died from breast cancer in 2018, which accounts for approximately 15% of all cancer-related deaths among women in that year [8]. Moreover, breast cancer is a highly heterogeneous disease in which certain types (e.g., triple-negative breast cancer) exhibit worse prognosis due to the lack of a targeted therapy [9]. Although NAC is well-tolerated and available at a low cost, the mechanisms underlying its efficacy in cancer treatment are complex and unclear. Moreover, NAC has been shown to exhibit cancer-promoting effects in some cancer types, including lung cancer [10] and melanoma [11], thus raising concerns regarding the clinical use of NAC.
Antioxidants are generally regarded as cancer-preventive agents because they protect biomolecules from the detrimental effects of reactive oxygen species (ROS). However, antioxidants are also thought to protect cancer cells from ROS-induced cell death, thereby promoting their proliferation and malignancy. Nevertheless, this view may be an over-simplification of the functions of ROS [12,13]. ROS are known to trigger diverse responses that range from homeostasis to cell death. Therefore, it is important to define the types of cancers that rely on ROS for their survival and malignant behavior in order to facilitate the safe and efficacious use of NAC as a clinical treatment. Moreover, cancer also involves a complex interplay between the tumor microenvironment (TME) and the cancer cells. In this context, NAC may act on both the cancer cells and the TME. Hence, an understanding of the responses of both cancer cells and the TME to both ROS and NAC will be important for the therapeutic use of NAC. Hence, the present review briefly summarizes the functions of ROS as signaling molecules in order to discuss their roles in cancer development and progression. In addition, the current status of antioxidant use in cancer treatment is examined in order to highlight the importance of the selective use of NAC for specific types of cancer. Finally, the discussion focuses on those types of cancer that rely on ROS for cancer cell survival and the creation of a permissive environment that promotes the progression to malignant cancer. These are the types of cancer that can potentially benefit from the use of NAC.
2. The Formation and Elimination of ROS
Cellular ROS levels exist in a steady, dynamic equilibrium between their formation via diverse cellular processes and their elimination via the antioxidant system. As described in the following paragraphs, ROS are produced during a variety of cellular reactions and upon exposure to various extraneous agents including toxicants, drugs, and xenobiotics, or ultraviolet light and other forms of radiation [14]. All of these factors can influence the intracellular ROS levels. Moreover, complex interactions between multiple cellular sources of ROS can be involved in the regulation of ROS homeostasis. To appreciate the resulting dynamic changes in redox states, the various ROS species and their reaction products are briefly described, along with the major cellular sources of ROS formation.
While the major ROS include the superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) [15], other ROS include reactive nitrogen species (RNS) such as nitric oxide (NO•), peroxynitrite (ONOO−), and dinitrogen trioxide (N2O3). Among these, NO• is generated by the family of nitric oxide synthetases [16]. Subsequently, NO⋅ can undergo autooxidation to form N2O3 and react with O2•− to form ONOOˉ [17]. Individual ROS have distinct characteristics regarding their half-lives, localization, inherent chemical properties that confer reactivity to various biological targets, and elimination by antioxidant enzymes [18]. Although NO• is relatively inert, its derivative, ONOO− is a powerful oxidant that can damage many biological molecules [19]. The O2•− is also highly reactive but short-lived. The O2•− generated within cells is rapidly converted into H2O2 by local superoxide dismutases (SODs) [20,21]. By comparison, H2O2 is more stable than O2•− but can be reduced to water in a reaction catalyzed by catalase (CAT) or glutathione peroxidase (GPX) [22]. The reaction involving GPX requires the oxidation of GSH to glutathione disulfide, which is subsequently reduced back to GSH by glutathione reductase, thioredoxin, and glutaredoxin [23,24]. Thus, the cell contains an enzyme system for preventing the build-up of H2O2 and maintaining a steady-state concentration (1–10 nM) [25].
Nevertheless, the presence of trace metals can catalyze the formation of the extremely reactive •OH from H2O2 via the Fenton reaction. This •OH cannot be eliminated enzymatically and can lead to cellular damage or genomic instability due to the indiscriminate oxidation of nearby lipids, proteins, and DNA [18,26]. In particular, ROS such as •OH and ONOOˉ can oxidize nucleic acid bases or the deoxyribose backbone to potentially form oxidation products, strand breaks, and DNA-protein crosslinks [27]. The ONOOˉ reacts primarily with deoxyguanosine to cause modifications such as 8-oxo-2’-deoxyguanosine and 8-nitro-2’-deoxyguanosine [17]. In addition, ROS can modify protein structure and function via the nitration of tyrosine residues and the oxidation of critical cysteine residues [28]. Further, ROS can initiate lipid peroxidation via the formation of the fatty acid radical, which is highly unstable and readily reacts with molecular oxygen to produce the lipid peroxyl radical (LOO•). This unstable radical species reacts in turn with another free fatty acid to produce a different fatty acid radical and a lipid peroxide (LOOH) [29]. Thus, a free radical chain reaction generates lipid peroxides that can be decomposed to form reactive lipid electrophiles such as 4-hydroxy-2-nonenal (HNE). These breakdown products (e.g., HNE) can form covalent adducts with nucleophilic functional groups in proteins, nucleic acids and membrane lipids [29,30]. Moreover, their prolonged half-lives may allow them to serve as second-messengers of oxidative stress [31]. Therefore, ROS can react with cellular macromolecules and lead to biological and pathological effects. However, it should be also noted that the functional significance of many ROS reaction products in vivo still needs to be validated and is under active study [17].
ROS are generated in multiple cellular organelles including mitochondria, peroxisomes, the endoplasmic reticulum (ER), and the plasma membrane. A major source of ROS is the mitochondrial respiratory chain, a normal metabolic process that converts the energy stored in macronutrients into adenosine triphosphate [26]. During mitochondrial respiration, electrons are transferred from electron donors to electron acceptors via oxidation-reduction reactions. The last electron acceptor in this chain is an oxygen molecule, which is normally reduced to water. However, a small fraction of the consumed molecular oxygen undergoes incomplete reduction, leading to O2•− formation [15].
Peroxisomes play a central role in various metabolic pathways including fatty acid oxidation, anaplerotic reactions, ether phospholipid synthesis, and hydrogen peroxide metabolism [32]. Many enzymes involved in these metabolic pathways produce specific ROS or RNS during their normal catalysis [33]. Peroxisomes also contain catalase and other antioxidant enzymes that effectively eliminate ROS [33]. Therefore, peroxisomes have the ability to rapidly produce and decompose ROS, thus allowing dynamic changes in ROS levels. Furthermore, peroxisomes and the mitochondria are metabolically interconnected and intimately related in redox regulation [34,35].
ROS are also produced in the ER during the metabolism of xenobiotics, the synthesis of unsaturated fatty acids, and protein folding [36]. A major source of ROS in the ER is oxidative protein folding that involves intramolecular and intermolecular disulfide bond formation [37]. For example, protein disulfide isomerase (PDI) is an ER oxidoreductase that catalyzes oxidative folding of protein. During disulfide bond formation, cysteine residues in the active sites of PDI accept two electrons from the cysteine thiol groups of nascent polypeptide substrates, leading to the reduction of PDI and oxidation of the substrate [38]. Subsequently, PDI can be regenerated in its oxidized form by the transfer of electrons to ER oxidoreductin 1 (Ero1), a flavin adenine dinucleotide (FAD)-binding protein [37]. After accepting electrons from PDI, Ero1 is re-oxidized by the transfer of electrons to molecular oxygen, but incomplete reduction results in the production of O2•−, which can be converted to H2O2 or other ROS [39,40].
In addition, NADPH oxidases (NOXs), a group of transmembrane proteins found in the plasma membrane, are recognized as major sources of cellular ROS [41]. Seven types of NOX are found in humans [42]. ROS production by NOXs is well recognized as a host defense function [43]. In addition to phagocytic cells, NOXs are expressed in non-phagocytic cells in a wide variety of tissues, albeit at much lower levels [44]. However, NOX activity becomes rapidly activated upon their assembly in membranes in response to various ligands such as cytokines, hormones, and growth factors [24,42]. The NOXs transfer electrons sequentially from cytosolic NADPH to FAD, then to each of two hemes, and finally to molecular oxygen on the opposite side of the cell membrane, thus producing O2•− (NOX1–3 and NOX5) or H2O2 (NOX4 and dual oxidase 1–2) [41,42]. Several NOXs (e.g., NOX4) are also located in the ER membrane, where they catalyze the generation of ROS [45]. Therefore, a variety of ROS are produced from multiple cellular sources which cumulatively contribute to the physiological or pathological effects of ROS.
3. The Function of ROS as Signaling Molecules
In addition to pathophysiological responses, ROS-mediated signal transduction plays a role in various basal and adaptive physiological responses for organismal homeostasis [14]. Compared to other ROS molecules, H2O2 is relatively stable and has both reducing and oxidizing properties, making it a central redox signaling molecule [46]. Protein modification by H2O2 yields instrumental signaling intermediates and is specific to certain target proteins [20]. The best-studied mechanism by which H2O2 achieves specificity in signaling mediation occurs via the oxidation of cysteine (Cys) residues on target proteins [20,47]. At physiological pH, Cys residues exist in the form of thiolate anions (Cys-S−), which are more susceptible to oxidation than the protonated cysteine thiol (Cys-SH) [46]. The thiolate anion can be oxidized to the sulfenic form (Cys-SOH) by H2O2, leading to functional changes in target proteins. The sulfenic form can be reduced back to the thiolate anion by disulfide reductases such as thioredoxin and glutaredoxin to restore function [48]. Hence, a steady-state physiological flux of H2O2 (in the nM range) toward specific target proteins leads to reversible oxidation, thereby serving as a reversible signal transduction mechanism that results in the alteration of protein activity, localization, and interaction with other biomolecules [47]. However, persistence of the H2O2-mediated sulfenic form can lead to further oxidation to sulfinic (SO2H) or sulfonic (SO3H) forms. Unlike the sulfenic modifications, the formation of sulfinic and sulfonic species can be irreversible, thus leading to permanent protein damage [49]. The O2•− has also been found to interact directly with specific intracellular targets and trigger signal transduction through the inactivation of specific proteins that are sensitive to O2•− levels although it has poor reactivity due to a short half-life [50].
This ROS-mediated signaling can be physically and functionally compartmentalized within the cell [51]. For example, NOXs localize in specific subcellular compartments [42]. Their ROS production is activated by various ligands, and the resulting ROS serve to modulate proximate redox-sensitive targets in intracellular signaling pathways. In addition, antioxidant enzymes are localized in specific cellular locations. Both ROS production and elimination by antioxidant enzymes occur at specific sites in cells and are associated with specific stimuli. This spatial and temporal organization of ROS production and removal can determine the outcomes of ROS signaling [51]. However, ROS are also believed to rapidly diffuse across membranes via certain aquaporins (AQPs) or other specific channels [52,53]. Therefore, ROS generated in one compartment can trigger responses in another [54]. For example, ROS produced by mitochondria facilitate disulfide formation in cell surface proteins, which may regulate protein function [55]. In addition, peroxisome-derived H2O2 could oxidize redox-sensitive cysteine residues in multiple proteins including the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and phosphatase and tensin homolog (PTEN) within as well as outside peroxisomes [56]. The production of peroxisomal ROS could also trigger mitochondrial apoptosis pathways [35]. Thus, ROS readily cross peroxisomal and mitochondrial membranes and can mediate signaling both within and outside the compartments in which they originate.
In addition, ROS produced in one cell can affect neighboring cells. For instance, H2O2 diffuses through the AQP family, in a similar fashion to water [53]. By diffusing through these channels, H2O2 acts as a signaling molecule not only in an autocrine, but also in a paracrine fashion. Notably, H2O2 produced in myofibroblasts induces cell death in adjacent lung epithelial cells [57]. Upon co-culture with fibroblasts derived from psoriatic plaque lesions, the intracellular accumulation of ROS and the activation of ERK have been shown to result in the proliferation of keratinocyte [58]. The same study further revealed that ROS over-produced by NOX4 of the fibroblasts in psoriatic plaques act as mitogens for keratinocytes. Thus, intercellular signaling has been demonstrated between H2O2 donor and recipient cells. The O2•− has a short lifetime due to rapid conversion to H2O2 and cannot diffuse across biological membranes due to its negative charge. Nevertheless, O2•− has been shown to initiate intracellular signaling by its passage through anion channels in the cell membrane [59]. Thus, ROS generated in one cell can stimulate redox signaling in adjacent cells.
4. The Effect of Antioxidant Supplementation upon Cancer Development and Progression
Excessive ROS can react with biomolecules including DNA, which is considered to be a possible cause for the increase in oncogenic mutations and the development of cancer [60]. This notion led to the expectation that dietary antioxidants might effectively prevent cancer development and has been a theoretical basis and explanation for the cancer-preventive effects of various antioxidant-rich plant foods observed in epidemiological studies. However, cancer-promoting effects have also been observed for antioxidants in some preclinical studies [10,11]. For example, supplementation with NAC or vitamin E after the development of cancer was found to increase cancer progression and reduce survival in mouse models of both BRAF- and KRAS-induced lung cancer [10]. Similarly, the oral administration of NAC after small nevi formation was shown to increase lymph node metastases without affecting the number and size of the primary tumors in a transgenic mouse model of melanoma [11]. These cancer-promoting effects of dietary antioxidants have resulted in a change in the perspective on the role of antioxidants in cancer. While antioxidants do prevent ROS-mediated cellular damage, they also protect cancer cells from ROS-mediated death. Therefore, antioxidants can effectively prevent cancer initiation, but can promote the growth of established cancers.
Although the cancer preventive effect of dietary antioxidants has been demonstrated in some studies [61,62], their use has also been shown to be ineffective in decreasing the incidence of cancer [63,64]. Moreover, some antioxidants were found to increase the risk of cancer. For instance, a large phase III randomized placebo-controlled trial demonstrated that dietary supplementation with vitamin E for over 7 years significantly increased the risk of prostate cancer [65]. In addition, some preclinical studies have indicated that supplementation with antioxidants can increase the risk of cancer development [66,67]. Chronic NAC treatment was shown to increase cancer initiation both under abnormal conditions associated with lung oxidative stress (JunD deletion) and during normal aging in mice [66]. In this study, although NAC treatment decreased oxidative stress and cell senescence in the lungs, this led to increased cancer initiation. In addition, GSH synthesis driven by the glutamate cysteine ligase modifier subunit (GCLM) was shown to be necessary for cancer initiation in a transgenic mammary tumor virus-polyoma middle tumor-antigen (MMTV-PyMT) mouse model of breast cancer [67]. In contrast, genetic loss of Gclm significantly delayed the onset of mammary tumor and reduced the mammary tumor burden [67]. The same study further demonstrated that the mammary tumor burden was dramatically reduced by treatment with buthionine sulfoximine (BSO), a potent inhibitor of GSH synthesis, prior to the onset of cancer development, but not upon the onset of mammary tumor growth, thus suggesting the need for GSH in breast cancer development. These results mitigate against the use of antioxidants for the prevention of cancer and reveal an inconsistency in the anticipated beneficial effects of antioxidants in cancer initiation.
Contradictory results have also been obtained with respect to the use of antioxidants in cancer progression. In a melanoma mouse model, cancer cells in metastatic sites (blood and viscera) had higher ROS levels than those in primary sites [68]. This high oxidative stress in metastatic cells was found to suppress distant metastasis, and treatment with an antioxidant (NAC, 200 mg/kg/day) increased the frequency of metastasis without significantly affecting the growth of established primary tumors [68]. However, in another study, the increased ROS levels in highly metastatic cells activated v-Src avian sarcoma (Schimidt-Ruppin A-2) viral oncogene homolog (Src), which led to increased cell migration and metastases. In contrast, mitochondrial superoxide scavenging by rotenone was shown to prevent ROS-mediated Src activation and consequently inhibit both the migration of cancer cells in vitro and spontaneous cancer metastasis in mouse xenograft models using melanoma and breast cancer cell lines [69]. Thus, the roles of ROS and antioxidants in cancer progression cannot be generalized.
A recent study has clearly demonstrated a change in ROS effects depending on the stage of the cancer. Suppressing ROS production during high expression of the antioxidant TP53-inducible glycolysis and apoptosis regulator (TIGAR) promoted premalignant cancer initiation, but limited metastasis in a pancreatic ductal adenocarcinoma (PDAC) model [12]. In contrast, increased ROS production following the loss of TIGAR in PDAC cells drove a phenotype switch that increased invasive and metastatic capacity [12]. However, this might be related to a particular scenario where ROS production was under the control of TIGAR, whose expression changed during the progression of cancer. Overall, the role of ROS (or antioxidants) is inconsistent, both in cancer initiation and progression, and their effects cannot be generalized on the basis of the cancer stage.
Cellular ROS can mediate cancer-promoting signaling and, thus, facilitate cancer cell proliferation and survival. For example, membrane-associated ROS generated via NOXs (e.g., NOX4) are important contributors to the activation of signaling pathways that drive proliferation and metastasis of cancer cells [70]. Mitochondrial ROS have also been shown to be necessary for KRAS-induced cancer development [71]. In contrast, ROS can induce cellular senescence and cell death, thereby acting as cancer-suppressing agents. Furthermore, cancer cells are believed to produce higher levels of ROS and hence, rely more on antioxidant activity to reduce their ROS burden than do non-cancerous cells [72]. Due to the potentially higher sensitivity of cancer cells to ROS-mediated cell death compared to normal cells, exogenous ROS generation has been proposed as a therapy for selectively killing cancer cells without affecting normal cells [73]. Therefore, ROS are considered to be a double-edged sword in cancer development and cancer treatment [74]. Consequently, treatment with antioxidants including NAC can be beneficial or detrimental in cancer management.
5. The Therapeutic Potetial of NAC in TNBC
Breast cancer is a heterogeneous disease [75,76], and this diversity is attributed to distinct genetic, epigenetic, and transcriptomic changes [77,78]. Molecular characteristics based on gene expression data led to the classification of the following molecular breast cancer subtypes: luminal A, luminal B, HER2 over-expression, and basal-like breast cancer [79,80]. These molecular subtypes are relatively well represented by their estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth receptor 2 (HER2) statuses, which can be identified by immunohistochemistry. As such, luminal A is ER- and/or PR-positive and HER2-negative; luminal B is ER- and/or PR-positive and either HER2-positive or -negative; and HER2 over-expression or amplification is hormone receptor-negative and HER2-positive [78]. Basal-like cancers constitute an extremely heterogeneous group, and the majority (80%) of this type, which lack of ER, PR, and HER2 expression, are referred to as triple-negative breast cancers (TNBCs) [81,82].
The above breast cancer classification scheme provides guidance for patient treatment [83]. For example, luminal types depend on estrogen for their growth and can be treated with drugs to reduce either the estrogen action (e.g., tamoxifen) or the actual level of estrogen (e.g., letrozole) [83]. Breast cancer with HER2 amplification can be effectively treated with humanized anti-HER2 monoclonal antibodies or small molecule kinase inhibitors (e.g., trastuzumab) [84]. However, no molecular-based targeted therapy is available for TNBCs [9]. Thus, breast cancer is comprised of various subtypes associated with distinct molecular characteristics and treatment responses. Moreover, the content and reliance of cancer cells on ROS for their survival and progression may vary among the various cancer types. For some cancers, high levels of ROS may be necessary to support their growth and malignant behavior, whereas other cancers may be more effectively treated by induction of ROS-mediated death [72,85]. In addition, the development and progression of a cancer toward the malignant phenotype is known to be highly dependent on interactions with the TME [86,87,88,89]. The ROS can mediate TME-induced signal transduction to promote cancer progression [90]. High ROS levels derived from cancer cells may also result in the creation of a permissive TME by altering adjacent stromal cells to acquire cancer-associated phenotypes and by recruiting immune cells [91,92,93]. Therefore, breast cancers with high ROS production in either the cancer cells or the TME would benefit from a safe and efficacious NAC treatment that is tailored toward the specific cancer type.
5.1. The Dependence on ROS for the Survival and Malignant Progression in TNBC
Otto Warburg was the first to describe a propensity for cancer cells to metabolize glucose anaerobically even in the presence of oxygen (the Warburg effect), and suggested an explanation for this abnormal glucose metabolism in terms of impaired bioenergetic activity of the mitochondria [94]. This led to the belief that oxidative phosphorylation (OXPHOS) is downregulated in cancer cells [95]. However, accumulating evidence suggests that aerobic glycolysis does not predict the loss of oxidative metabolism [96], and impairment of mitochondrial metabolism cannot be generalized across all cancers [97,98]. Moreover, increased OXPHOS and mitochondrial mass have been linked to the invasive and migratory potential of cancer cells [99].
As noted above, TNBC comprises a very heterogeneous subgroup of cancers that accounts for 10–20% of all breast cancers [100,101]. Moreover, due to a lack of effective treatment options, TNBC is often associated with an aggressive phenotype and worse disease-specific outcomes than other breast cancer subtypes [102,103]. Significantly, one of the mechanisms that promote cell survival and drive aggressiveness in TNBC has been linked to high levels of ROS production resulting from gene mutations, gene expression changes and the attainment of stem cell-like properties [91,104,105]. This high ROS content induces multiple signaling which, in turn, leads to highly proliferative, migratory, and drug-resistant phenotypes in TNBC (Figure 1).
5.1.1. High ROS Production Derived from BRCA1 Inactivation and TP53 Mutation in TNBC
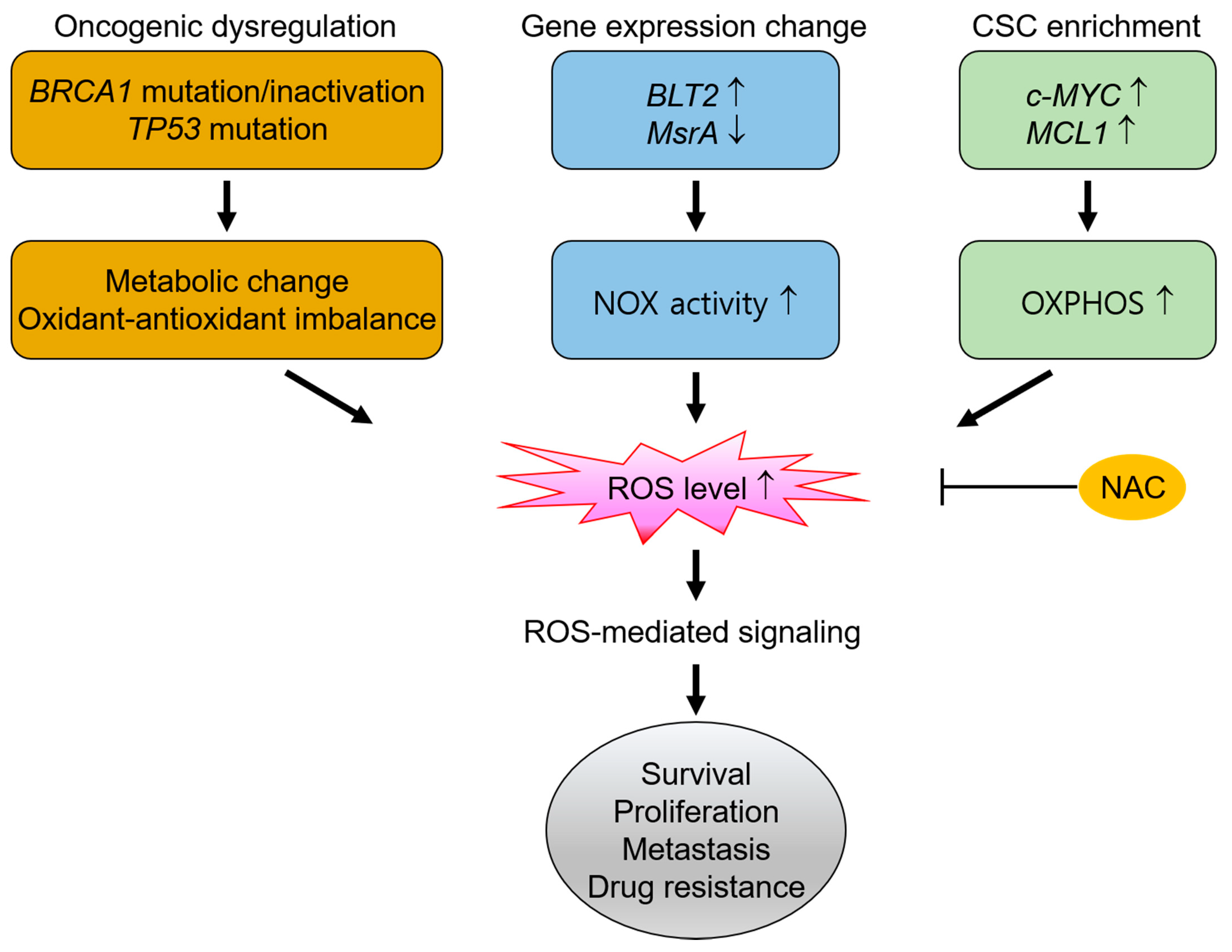
High ROS production contributes to cancer cell survival and growth, particularly in basal-like TNBC [106]. Basal-like TNBC tumor samples from the Cancer Genome Atlas dataset exhibit distinctly different mitochondrial gene expression, thus suggesting differences in mitochondrial function in this subtype of breast cancer [106]. Basal-like TNBC cell lines (MDA-MB-231, MDA-MB-468, and BT-549) contain higher ROS levels than luminal (MCF-7) or non-cancerous (MCF-10A) cell lines, the majority of which are located in the mitochondria [106]. Treatment with NAC (15 or 30 mM) has been shown to induce cell death in TNBC cell lines but not in luminal cells, thus indicating the dependence of basal-like TNBC cells on high levels of ROS for their survival and proliferation [106]. In addition, increased ROS level have been found necessary for the migratory and invasive properties of MDA-MB-231 cells [99]. Elevated levels of mitochondrial DNA have been detected in circulating cancer cells from mice with tumors derived from orthotopically implanted MDA-MB-231 cells compared to primary tumors [99]. This high DNA content in metastatic cancer cells was attributed to high mitochondrial biogenesis and OXPHOS due to high expression of peroxisome proliferator-activated receptor gamma coactivator 1-α. The perturbation of mitochondrial respiratory function in breast cancer cells has also been shown to result in ROS generation [107]. This increased ROS was shown to promote breast cancer cell motility via a pathway mediated by activator protein-1 and C-X-C motif chemokine ligand (CXCL) 14, while pretreatment with NAC (5 μM) was shown to effectively inhibit this ROS-induced cell motility [107]. Thus, metabolic alteration induced by changes in mitochondrial function in basal-like TNBC cells accumulates high ROS levels that contribute to high proliferative and metastatic properties.
The inactivation of breast cancer susceptibility gene 1 (BRCA1) may also be related to high ROS content in basal-like TNBC. BRCA1 dysfunction is regarded as a drivers of basal-like breast cancer as well as a subgroup of TNBC [77,100,108]. Although BRCA1 is infrequently mutated in sporadic TNBC, low expression or inactivation of BRCA1 may occur in wild-type TNBC due to epigenetic gene silencing, miRNA-mediated post-transcriptional suppression, and other nongenetic alterations [109,110,111]. In addition to its established function as a tumor suppressor, BRCA1 has been shown to regulate oxidative stress [112]. The overexpression of wild type, but not the cancer-associated mutant form, of BRCA1 was shown to significantly reduce the cellular levels of ROS and protein damage caused by H2O2, whereas the knockdown of BRCA1 increased the ROS levels in MCF-7 and MCF-10A cells [113]. Thus, the protective role of BRCA1 against oxidative stress, in combination with the oxidative DNA-damaging ability of estrogen metabolites, may partially explain the specificity of BRCA1- and BRCA2-mutant cancers in the breast and ovary, where estrogen levels are relatively high [114]. This has been further demonstrated in a mouse mammary tumor model with specific knock-out of Brca1 in the mammary gland [115]. The DNA double-strand breaks induced by oxidative estrogen metabolites could not be repaired without Brca1, thus resulting in genomic instability and cancer development. By contrast, antioxidant treatment (4-hydroxy-2,2,6,6-tetrametylpeperidine-N-oxyl, tempol) suppressed the levels of estrogen-induced oxidative DNA lesions, and significantly delayed the onset of Brca1-deficiency-induced cancer development [115]. Thus, increased ROS levels in basal-like subtypes of TNBCs may be associated with BRCA1 mutation/inactivation and may be essential for the survival of this type of cancer.
In addition, TNBC exhibits high levels of genomic instability, with TP53 being the most frequently mutated somatic gene (over 80%) [91,100,108]. The TP53 gene regulates cellular ROS levels by either controlling the expression of antioxidant and prooxidant genes or by modulating the metabolic pathways [116,117]. Cancer cells containing mutant TP53 have been shown to exhibit increased mitochondrial biogenesis and OXPHOS, which resulted in increased metastasis [118]. Moreover, low p53 levels result in decreased transcription of antioxidant genes (e.g., Mn-SOD and GPX1), whereas high levels induce an imbalance of antioxidant gene transcription (i.e., upregulation of Mn-SOD and GPX, but not of CAT) and transactivation of prooxidant genes (e.g., p53 upregulated modulator of apoptosis and p53-induced gene). Thus, TP53 mutation is associated with high cellular ROS levels due to an imbalance in the antioxidant defense [119,120]. Therefore, the BRCA1 inactivation and TP53 mutation that often observed in TNBC may contribute to a significant ROS content in these cells. Hence, the effective removal of ROS in TNBC by NAC treatment can suppress ROS signaling that leads to malignant progression of this type of cancer. Notably, TP53 mutation is also prevalent in other types of cancer, including ovarian and esophageal cancer [121,122]. This suggests that survival and proliferation of these cancer cell types are ROS-dependent and that NAC treatment is of potential benefit in such cancers.
5.1.2. High ROS Production Derived from Gene Expression Changes in TNBC
The enzymatic action of NOXs also leads to ROS production (Section 2), and gene expression changes that induce high NOX-related ROS generation have also been observed in TNBC [123,124]. These NOX-derived ROS have been shown to contribute to cell proliferation, survival, and invasive behavior in TNBC. For instance, the leukotriene B4 receptor 2 (BLT2) is highly expressed in breast cancer tissues at both the mRNA and protein levels [123]. Activation of BLT2 triggers signaling pathways that promote the survival, metastasis, and adhesion of cells, and its high expression is associated with a worsened prognosis in TNBC patients [125]. Moreover, the scavenging of NOX1-related BLT2-mediated ROS by NAC treatment (5 mM) was shown to effectively induce cell apoptosis in the MDA-MB-468 TNBC cell line [123].
In addition, down-regulation of the ROS scavenger methionine sulfoxide reductase A (MsrA), has been associated with advanced cancer in human breast cancer tissues, and the same study indicated that increased ROS production by MsrA knockout in a TNBC cell line (MDA-MB-231) led to a more aggressive phenotype in both 3D-culture and tumor xenograft models [126]. Both the mitochondria and the NOX system were involved in ROS hyperproduction due to loss of MsrA, which led to decreased PTEN expression, activation of the phosphoinositide-3-kinase pathway, and increased vascular endothelial growth factor (VEGF) production [126].
High NOX4 expression has also been shown to increase mitochondria-associated ROS levels, thus resulting in the stabilization of matrix metalloproteinase-9 (MMP-9) mRNA [124]. This high MMP-9 expression significantly contributed to cancer metastasis and was linked with invasive properties of MDA-MB-231 cells [124]. In addition, cell adhesion and migration are largely dependent on the binding of integrin to extracellular matrices [127]. High β1 integrin expression has been correlated with low survival rates and advanced metastatic status in TNBC [128]. Studies have implicated the involvement of NOX complexes in integrin-mediated ROS production [127,129]. For instance, the binding of integrin α2β1 to type IV collagen in a human adenocarcinoma cell line was shown to result in the activation of NOX1 [130]. Integrin engagement activates ROS production via NOX1, which causes cancer cells including MDA-MD-231 cells to escape anoikis and survive [131]. Moreover, enhanced adhesion of MDA-MB-231 cells to proteins of extracellular matrices after radiation therapy has been observed and correlated with high ROS production and surface expression of active β1 integrin [132]. The same study demonstrated that treatment with NAC (10 mM) could inhibit this radiation therapy-induced cell adhesion. These results demonstrate that gene expression changes leading to high NOX-related ROS generation increase cell survival and induce invasive and metastatic phenotypes in TNBC, and that the reduction of ROS via NAC treatment can suppress the invasive and migratory behavior of TNBC cells.
5.1.3. High ROS Production by Cancer Stem-Like Cells in TNBC
Tumors contain phenotypically and functionally heterogeneous cancer cell populations. Consequently, the development of drug resistance and a metastatic phenotype in TNBC may be related to the presence of rare type of cancer cell referred to as cancer stem-like cells (CSCs) or tumor-initiating cells. These produce high amounts of ROS via mitochondrial OXPHOS and demonstrate self-renewal capability and aggressive characteristics such as high levels of metastasis, a tendency towards relapse after treatment, and resistance to chemotherapy [133,134,135].
Following chemotherapy, c-MYC and induced myeloid leukemia cell differentiation protein (MCL1) are frequently found to be co-overexpressed in chemotherapy-resistant human TNBC tissues as well as CSCs derived from TNBC cell lines (MDA-MB-436 and SUM159PT) [133]. Both c-MYC and MCL1 have been found to additively increase mitochondrial OXPHOS and to dramatically elevate ROS production, thus contributing to the maintenance of CSCs and the stabilization of hypoxia-inducible factor-1α (HIF-1α) [133] (Section 5.2.3). During chemotherapy, TNBC cells activate OXPHOS to induce hypoxia pathways associated with drug resistance; thus, scavenging ROS may decrease ROS overload and prevent the development of the hypoxic phenotype as well as drug resistance. The dependence of CSC maintenance upon ROS is further demonstrated by the effective treatment of CSCs within TNBC cell lines using a re-engineered CAT [136].
In addition, c-MYC has been shown to drive the dysregulation of fatty acid β-oxidation (FAO), and TNBC cells that over-express c-MYC have been shown to exhibit bioenergetic reliance upon FAO [137]. Functional mitochondria are crucial for the maintenance of stemness, and CSCs also rely on mitochondrial OXPHOS for their survival in other cancer types, including liver and head and neck cancer [138,139]. Furthermore, elevated FAO-catalyzed mitochondrial ROS production in CSCs has been reported to promote cancer metastasis [135]. Therefore, drug treatment can induce the acquisition of CSC properties in TNBC and, thus, contribute to metabolic changes, ROS-dependence in stemness, and drug resistance. This, in turn, suggests that NAC treatment can effectively target the stem cell subpopulation in TNBC.
5.2. The Interplay between Cancer Cells and the Tumor Microenvironment via ROS
A high ROS content in cancer cells can be transferred to the surrounding TME in a constant interaction that greatly influences cancer progression. For example, BRCA1 mutations induce oxidative stress in the TME [140], and a high ROS content can arise from high expression and/or activation of NOXs, or from a deficiency of antioxidant enzymes such as GPX, in the stromal cells [141,142]. Regardless of the source, high levels of ROS actively contribute to interaction between cancer cells and the TME, thus leading to more permissive microenvironment toward highly proliferative, metastatic, and drug-resistant properties of cancer (Figure 2).
5.2.1. Conversion to the Cancer-Associated Fibroblast Phenotype via ROS
Cancer-associated fibroblasts (CAFs) are a main component of the TME and actively contribute to cancer growth and malignancy by secreting various growth factors and chemokines [87,143,144,145]. The CAFs are known to exhibit a myofibroblast phenotype, and the abundance of stromal myofibroblasts that express α-smooth muscle actin (α-SMA) has been shown to predict poorer overall survival rates in various types of cancer, including breast cancer [146,147,148,149]. Although the origin of CAFs remains controversial, they are considered to be largely derived from the activation of resident fibroblasts [145]. In this respect, ROS play fundamental roles in the activation and conversion of fibroblasts into CAFs [141,146,150,151]. Specifically, ROS have been shown to promote the conversion of fibroblasts into myofibroblasts through the up-regulation of HIF-1α and CXCL12, an effect that was reduced by long-term NAC treatment (0.5 mM for 20 days) [151]. In addition, the conversion of fibroblasts into fibrotic myofibroblasts via NOX-4-dependent ROS production in association with transforming growth factor TGF-β1 (TGF-β1) has been suppressed by treatment with NAC (5 mM), either before or after treatment with TGF-β1 [141,146,150,152].
In addition to inducing myofibroblast transition, the large amounts of ROS produced by cancer cells induces metabolic changes in the neighboring fibroblasts [153,154]. In an in vitro co-culture model, ROS derived from cancer cells (MCF-7 cells) induced autophagy in fibroblasts and consequently reduced the expression of Caveolin-1 (Cav-1), which led to the development of myofibroblast characteristics (i.e., upregulation of α-SMA, calponin, and vimentin) [153,154,155]. Under these co-culture conditions, Cav-1 in fibroblasts was targeted to the lysosome for its autophagic degradation in response to high ROS levels, which could be prevented by treatment with NAC or with an autophagy inhibitor (chloroquine) [92,155]. The metabolic switch between cancer cells and fibroblasts also involves ROS. For instance, H2O2 secreted by cancer cells causes oxidative stress in adjacent fibroblasts, which leads to decreased mitochondrial activity and increased glucose uptake, thus creating a dependence on aerobic glycolysis in fibroblasts [153]. In contrast, an enhancement in mitochondrial activity has been observed in cancer cells (e.g., MDA-MB-231 and MCF7 cells) during co-culture [153]. Similarly, in a xenograft mouse model where MDA-MB-231 breast cancer cells were co-injected with wild-type or Cav-1-deficient fibroblasts, the Cav-1-deficient fibroblasts led to enhanced glycolytic enzyme synthesis to provide energy-rich metabolites (e.g., lactate), thus increasing cancer growth and angiogenesis [156]. Hence, Cav1-deficient CAFs, induced by high ROS levels, exhibit a shift towards aerobic glycolysis.
Notably, a loss of stromal Cav-1 expression is a strong predictor of poor clinical outcome in TNBC and basal-like breast cancers [157]. The BRCA1 mutation in breast cancer cells has been shown to significantly increase ROS levels in adjacent fibroblasts, thus leading to decreased Cav-1 expression and increased expression of monocarboxylate transporter 4 (MCT4), which is the main exporter of L-lactate from cells [140]. These changes were reversed by both wild-type BRCA1 and NAC treatment. Furthermore, Cav-1 has been shown to be a negative regulator of NOX proteins, such that reduced Cav-1 expression leads to increased NOX expression and, hence, a further increase in ROS production [158]. Taken together, the results described in this section indicate that the large quantities of ROS produced by cancer cells lead to myofibroblastic transition and a dependence on aerobic glycolysis in neighboring stromal fibroblasts, thus providing an energy-rich cancer-promoting microenvironment.
5.2.2. Conversion to Tumor-associated Macrophage Phenotype via ROS
In addition to CAFs, breast TMEs include tumor-associated macrophages (TAMs). These are a critical component of the TME and greatly influence cancer progression and the therapeutic response [159]. High ROS levels derived from cancer cells contribute to the recruitment of macrophages into the TME and to their conversion to a more permissive macrophage phenotype [91,105]. For instance, ROS were shown to increase the activation of aryl hydrocarbon receptor (AhR), thus leading to increased transcription of antioxidant enzymes and epidermal growth factor receptor (EGFR) ligand (amphiregulin) in malignant mammary cells [91]. In this study, AhR and amphiregulin were shown to regulate the production of chemokines (e.g., granulocyte colony-stimulating factor, CXCL1, CXCL2, and C-C motif chemokine ligand 5) to attract monocytes into the TME in a BRCA1-deleted mouse mammary tumor model. Notably, the expression of these chemokines and the infiltration of monocyte lineage cells were also correlated with the ROS levels in BRCA1 mutation-associated human breast cancer tissues in the same study. These results are further supported by those of another study in which continuous treatment with an ROS inhibitor (butylated hydroxyanisole, BHA) efficiently blocked the recruitment of monocytes into the TME and improved the prognostic values in various transgenic mouse models, including MMTV-PyMT-induced breast cancer models [93].
Cancer immune escape refers to the avoidance of immune control of cancer growth and spread and is considered an important strategy for cancer survival and development [160]. The programmed death ligand-1 (PD-L1) signaling pathway is an important component of cancer immune escape [160,161]. Antioxidant depletion or the generation of ROS by treatment with BSO or paclitaxel has been shown to positively regulate mRNA and protein levels of PD-L1 in both human and mouse macrophages in vitro [105]. These PD-L1-expressing macrophages have immunosuppressive and angiogenic properties due to the production of immunosuppressive cytokines (e.g., interleukin (IL)-4, IL-10, and IL-17) and VEGF, respectively [105]. In this study, the administration of paclitaxel for the treatment of the spontaneous-TNBC analogue BRCA1/p53-deleted mouse model mammary tumors induced PD-L1 expression in the TAMs, thus leading to an immunosuppressive TME. NF-κB signaling in response to ROS accumulation mediated the promotion of PD-L1 transcription and the release of immunosuppressive chemokines. In another study, NOX-mediated ROS generation was shown to play a critical role in macrophage differentiation into immunosuppressive M2 macrophages, whereas inhibition of ROS by BHA treatment specifically blocked the differentiation into M2, but not M1 macrophages [93]. Thus, ROS act as paracrine signaling molecules to recruit TAMs into the TME and thus alter the TAM phenotype to exhibit more immunosuppressive characteristics.
5.2.3. Activation of Hypoxic Responses via ROS-Mediated Tumor-Stromal Interaction
A hypoxic microenvironment surrounding cancer cells can also promote invasion, metastasis, and resistance to therapy [162,163,164]. Hypoxia-inducible factors (HIFs) are critical transcription regulators that respond to hypoxia [165]. These include the oxygen-regulated HIF-1α, HIF-2α, and HIF-3α subunits, and the constitutively expressed HIF-1β subunit [164]. The HIFs are subject to prolyl hydroxylation, ubiquitination, and proteasomal degradation, all of which are inhibited under hypoxic conditions; hence, the latter conditions facilitate the stabilization and accumulation of HIF-α protein [164,165]. High expression of HIFs or HIF-regulated genes is particularly common in basal-like TNBCs, thus suggesting that hypoxia may play a relevant role in promoting aggressive behaviors of these tumors [166,167]. In fact, the activation of HIF has been suggested as a mechanism by which TNBCs can acquire invasiveness and metastatic propensity [168,169,170]. Likewise, HIFs are required for the enrichment of chemotherapy-resistant CSCs in TNBC [163] (Section 5.1.3).
Hypoxic conditions also mediate the tumor-stromal interactions that lead to the elevation of cytokines, in turn, promote metastatic phenotypes in TNBC [171,172]. For instance, hypoxia has been shown to induce the recruitment of macrophages and mesenchymal stem cells (MSCs) by the production of macrophage colony-stimulating factor 1 via the interaction between TNBC cells and MSCs [171]. Hypoxia is also involved in the interaction between TNBC cells and breast CAFs via interleukin-1β (IL-1β) and interleukin receptor 1 type 1 to induce metastatic gene expression and invasive properties in TNBC cells [172]. In particular, Il-1β is highly induced in hypoxia [172] and is one of major highly-elevated proteins in TNBCs [173]. Hence, tumor-stromal inflammation induced by hypoxia stimulate the pro-metastatic phenotypes in TNBC [173].
Notably, ROS are increased in response to hypoxia and inhibit the ability of prolyl hydroxylases to hydroxylate HIFs [174]. Therefore, ROS production involving the mitochondria is required for the stabilization of HIF-1α and the subsequent transduction of the hypoxia-mediated signaling [165,175]. Hypoxia has been shown to induce ROS production in MDA-MB-468 cells, and treatment with NAC (10 mM) was shown to reverse hypoxia-induced up-regulation of N-cadherin and plasminogen activator inhibitor-1, the activation of EGFR, and cell motility [104]. In addition, exposure of MDA-MB-231 cells to hypoxia led to increased levels of ROS, the activation of two main HIF-1α transduction regulators (extracellular signal-regulated kinases and AKT), and upregulation of c-Fos protein, whereas treatment with NAC (0.3 mM) prevented these changes induced under the hypoxic conditions [172]. Furthermore, treatment with drugs that differ in their mechanisms (e.g., paclitaxel and gemcitabine) has been shown to induce hypoxic responses and CSC enrichment in TNBC cell lines (MBA-MB-231, SUM-149, and SUM-159), and this was mediated by increased ROS levels [176]. Therefore, ROS mediated hypoxic response in TNBC, and NAC could effectively inhibit the transduction of hypoxia-mediated signaling.
As noted above, ROS are actively involved in the interplay between cancer cells and the TME for cancer progression in TNBC, and NAC can suppress this process by interfering with the ROS-mediated cancer cell-TME interaction. A pilot study has demonstrated that short-term treatment (14-27 days) with NAC significantly reduces the level of stromal MCT4 expression and proliferation in breast cancer patients [124]. Therefore, NAC has been shown to be effective in the metabolic modulation of breast TME at the clinical level.
Traditional or standard chemotherapy is currently the main systemic treatment option for TNBC. Although optimal regimens still need to be established, chemotherapy regimens based on anthracyclines (e.g., doxorubicin) and taxanes (e.g., paclitaxel) represent the mainstay of TNBC treatment [177]. However, although TNBC is initially sensitive to chemotherapy, progressive resistance is a common problem that leads to disease recurrence and a poor outcome, thus limiting the benefits of standard chemotherapy [178]. The inclusion of NAC in standard chemotherapy can potentially provide additional benefits for TNBC patients, although it should be ensured that the NAC treatment does not interfere with the effectiveness of the chemotherapy. Currently, the combinational use of NAC is more focused on its effects in terms of preventing or relieving the adverse side effects of standard chemotherapy, such as peripheral neuropathy due to taxane chemotherapy [179] or cardiotoxicity caused by anthracycline chemotherapy [180,181]. Although some preclinical studies on the combination of NAC and standard chemotherapy have been published, the reported effects are inconsistent. For instance, pretreatment with NAC has been reported to potentiate the doxorubicin-induced phosphorylation of p53 and ATM, thus increasing their inhibitory effects upon cell proliferation and migration in ovarian cancer cells [182]. In addition, NAC has been shown to prevent NF-κB activation by gemcitabine and to improve the efficacy of gemcitabine in pancreatic cancer cells that were implanted in athymic nude mice [183]. By contrast, NAC has been shown to negatively alter the chemotherapeutic effectiveness of paclitaxel in a lung cancer cell line by decreasing the ROS levels, thus preventing paclitaxel-induced apoptosis [184]. However, it should be noted that studies using cell culture models may be misrepresented due to the absence of in vivo-like TMEs, which are an important component of the benefits of NAC. The potential interference of NAC with the effectiveness of standard chemotherapy critically depends on whether the specific chemotherapy depends on ROS production. For example, the anticancer efficacy of radiotherapy does not depend on ROS and, hence, NAC treatment reduces the levels of radiotherapy-induced ROS but does not interfere with radiotherapy-induced cell death [185]. In fact, high ROS is often associated with resistance to standard chemotherapy probably due to their involvement in CSC enrichment and permissive TME formation [91,163,186] (Section 5.1.3 and Section 5.2). Moreover, NAC treatment can be particularly effective in the inhibition of the tumor-stromal interaction and the reduction in CSC population, thereby decreasing chemotherapy-induced resistance and aggressiveness in TNBC.
6. Conclusions
NAC is a membrane-permeable antioxidant and has been safely used as a mucolytic and antidote for acetaminophen. However, its use in cancer treatment has resulted in varying outcomes ranging from beneficial to detrimental largely because ROS can mediate both cancer-promoting and cancer-suppressing signaling. In this respect, particularly high ROS levels are generated by TNBC cells due to gene mutation/inactivation (e.g., BRCA1 and TP53), gene expression changes, and CSC enrichment. This ROS accumulation mediates the cellular signaling necessary to maintain the survival and promote their metastatic capacity and drug resistance. Furthermore, high ROS levels derived from TNBC cells can mediate interaction between cancer cell and the TME to induce the formation of permissive TME and hypoxic responses that linked to their aggressive behavior. However, treatment with NAC effectively suppresses the ROS production and ROS-mediated signaling that contribute to cell survival, metastasis, and drug resistance, thereby inhibiting cancer progression. Meanwhile, TNBCs exhibit aggressive phenotypes, and standard chemotherapy remains the mainstay of TNBC treatment that often generate resistance and relapse. Therefore, the additional use of NAC can be an effective strategy for treating those TNBCs. However, further studies are required in order to define better molecular signatures for the identification of specific subset of TNBC patients who can benefit the most from NAC therapy and to monitor its efficacy. Moreover, for the use of NAC to be clinically relevant, further studies need to examine the potential interference of NAC with the effectiveness of standard chemotherapy, the most suitable time for NAC administration relative to the standard chemotherapy, and the appropriate treatment duration.
Funding
The present study was supported by a grant from the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (NRF-2020R1A2C1005730).
Conflicts of Interest
The author declares no conflict of interest.
References
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.O.; Shay, K.P.; Kelley, A.R.; Butler, J.A.; Hagen, T.M. Glutathione maintenance mitigates age-related susceptibility to redox cycling agents. Redox Biol. 2016, 10, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, L.; Kartha, R.; Terpstra, M.; Oz, G.; Hovde, L.; Cloyd, J.; Tuite, P. Effect of High-Dose Oral NAC on Systemic Measures of Oxidative Stress in Individuals with Parkinson Disease and Healthy Elderly Subjects. Mov. Disord. 2016, 31, E13. [Google Scholar]
- Šalamon, Š.; Kramar, B.; Marolt, T.P.; Poljsak, B.; Milisav, I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants 2019, 8, 111. [Google Scholar] [CrossRef] [Green Version]
- Altomare, A.; Baron, G.; Brioschi, M.; Longoni, M.; Butti, R.; Valvassori, E.; Tremoli, E.; Carini, M.; Agostoni, P.; Vistoli, G.; et al. N-Acetyl-Cysteine Regenerates Albumin Cys34 by a Thiol-Disulfide Breaking Mechanism: An Explanation of Its Extracellular Antioxidant Activity. Antioxidants 2020, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.; Sotgia, F.; Whitaker-Menezes, D.; Tuluc, M.; Birbe, R.; Berger, A.; Lazar, M.; Cotzia, P.; Draganova-Tacheva, R.; Lin, Z.; et al. Pilot study demonstrating metabolic and anti-proliferative effects of in vivo anti-oxidant supplementation with N-Acetylcysteine in Breast Cancer. Semin. Oncol. 2017, 44, 226–232. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.R.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; de Nicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y. Food-derived polyphenols inhibit the growth of ovarian cancer cells irrespective of their ability to induce antioxidant responses. Heliyon 2018, 4, e00753. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef]
- Dedon, P.C.; Tannenbaum, S.R. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004, 423, 12–22. [Google Scholar] [CrossRef]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; de Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxid. Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [Green Version]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wert, K.J.; Velez, G.; Cross, M.R.; Wagner, B.A.; Teoh-Fitzgerald, M.L.; Buettner, G.R.; McAnany, J.J.; Olivier, A.; Tsang, S.H.; Harper, M.M.; et al. Extracellular superoxide dismutase (SOD3) regulates oxidative stress at the vitreoretinal interface. Free Radic. Biol. Med. 2018, 124, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, A.B.; Tephly, L.A.; Venkataraman, S.; Oberley, L.W.; Zhang, Y.; Buettner, G.R.; Spitz, D.R.; Hunninghake, G.W. High Levels of Catalase and Glutathione Peroxidase Activity Dampen H2O2Signaling in Human Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2004, 31, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, C.; Lillig, C.H.; Flohã, L. Redox regulation by glutathione needs enzymes. Front. Pharmacol. 2014, 5, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 1–17. [Google Scholar] [CrossRef]
- Kawasaki, T.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Aulak, K.S.; Koeck, T.; Crabb, J.W.; Stuehr, D.J. Dynamics of protein nitration in cells and mitochondria. Am. J. Physiol. Circ. Physiol. 2004, 286, H30–H38. [Google Scholar] [CrossRef] [Green Version]
- Barrera, G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol. 2012, 2012, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titorenko, V.I.; Terlecky, S.R. Peroxisome Metabolism and Cellular Aging. Traffic 2010, 12, 252–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes Are Oxidative Organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; van Veldhoven, P.P.; Brees, C.; Rubio, N.; Nordgren, M.; Apanasets, O.I.E.; Kunze, M.; Baes, M.; Agostinis, P.; Fransen, M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 2013, 65, 882–894. [Google Scholar] [CrossRef] [Green Version]
- di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1–44. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Ogasawara, M.; Zhang, H. Redox Regulation and Its Emerging Roles in Stem Cells and Stem-Like Cancer Cells. Antioxid. Redox Signal. 2009, 11, 1107–1122. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Gross, E.; Kastner, D.B.; A Kaiser, C.; Fass, D. Structure of Ero1p, Source of Disulfide Bonds for Oxidative Protein Folding in the Cell. Cell 2004, 117, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative Innate Immune Defenses by Nox/Duox Family NADPH Oxidases. Infect. Inflamm. Impacts Oncog. 2008, 15, 164–187. [Google Scholar] [CrossRef] [Green Version]
- Zana, M.; Péterfi, Z.; Kovács, H.A.; Tóth, Z.E.; Enyedi, B.; Morel, F.; Paclet, M.-H.; Morand, S.; Leto, T.L. Interaction between p22phox and Nox4 in the endoplasmic reticulum suggests a unique mechanism of NADPH oxidase complex formation. Free Radic. Biol. Med. 2018, 116, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2009, 5, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.M.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Benov, L.T. How superoxide radical damages the cell. Protoplasma 2001, 217, 33–36. [Google Scholar] [CrossRef]
- Pani, G.; Bedogni, B.; Colavitti, R.; Anzevino, R.; Borrello, S.; Galeotti, T. Cell Compartmentalization in Redox Signaling. IUBMB Life 2001, 52, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lismont, C.; Nordgren, M.; Brees, C.; Knoops, B.; van Veldhoven, P.P.; Fransen, M. Peroxisomes as Modulators of Cellular Protein Thiol Oxidation: A New Model System. Antioxid. Redox Signal. 2019, 30, 22–39. [Google Scholar] [CrossRef]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Barygina, V.; Becatti, M.; Prignano, F.; Lotti, T.; Taddei, N.; Fiorillo, C. Fibroblasts to Keratinocytes Redox Signaling: The Possible Role of ROS in Psoriatic Plaque Formation. Antioxidants 2019, 8, 566. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.J.; Madesh, M.; Kirkpatrick, C.J.; Fisher, A.B. Superoxide Flux in Endothelial Cells via the Chloride Channel-3 Mediates Intracellular Signaling. Mol. Biol. Cell 2007, 18, 2002–2012. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Nishino, H.; Tokuda, H.; Satomi, Y.; Masuda, M.; Osaka, Y.; Yogosawa, S.; Wada, S.; Mou, X.Y.; Takayasu, J.; Murakoshi, M.; et al. Cancer prevention by antioxidants. BioFactors 2004, 22, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Wada, S. Cancer preventive effects of vitamin E. Curr. Pharm. Biotechnol. 2012, 13, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Collins, A. Antioxidant intervention as a route to cancer prevention. Eur. J. Cancer 2005, 41, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Kim, Y.-W.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol. 2009, 21, 166–179. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.; Tangen, C.M.; Lucia, M.S.; Goodman, P.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; Karp, D.D.; et al. Vitamin E and the risk of prostate cancer: Updated results of the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J. Clin. Oncol. 2012, 30, 7. [Google Scholar] [CrossRef]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.-C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nat. Cell Biol. 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; de Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perillo, B.; di Donato, M.; Pezone, A.; di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Courtney, M.J.; Laukkanen, M.O. Redox-Activated Signal Transduction Pathways Mediating Cellular Functions in Inflammation, Differentiation, Degeneration, Transformation, and Death. Oxid. Med. Cell. Longev. 2016, 2016, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [Green Version]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [Green Version]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2010, 24, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2009, 220, 263–280. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.J.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Ellis, P.; Tutt, A.N. Molecular heterogeneity of triple-negative breast cancer and its clinical implications. Curr. Opin. Oncol. 2011, 23, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Guideline Update. Arch. Pathol. Lab. Med. 2020, 144, 545–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puglisi, F.; Fontanella, C.; Amoroso, V.; Bianchi, G.V.; Bisagni, G.; Falci, C.; Fontana, A.; Generali, D.; Gianni, L.; Grassadonia, A.; et al. Current challenges in HER2-positive breast cancer. Crit. Rev. Oncol. 2016, 98, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid. Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Artacho-Cordón, A.; Artacho-Cordon, F.; Ríos-Arrabal, S.; Calvente, I.; Núñez, M.I. Tumor microenvironment and breast cancer progression. Cancer Biol. Ther. 2012, 13, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Smith, B.D.; Zhou, Y.; Kaufman, M.D.; Godwin, A.K. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene 2013, 34, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Godwin, A.K. Regulation of HGF and c-MET Interaction in Normal Ovary and Ovarian Cancer. Reprod. Sci. 2016, 24, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Chang, W.; Fang, B.; Qin, J.; Qu, X.; Cheng, F. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci. Rep. 2016, 6, 39541. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.; Balliet, R.M.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Hereditary ovarian cancer and two-compartment tumor metabolism: Epithelial loss of BRCA1 induces hydrogen peroxide production, driving oxidative stress and NFkappaB activation in the tumor stroma. Cell Cycle 2012, 11, 4152–4166. [Google Scholar] [CrossRef] [Green Version]
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Göbl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution. Cell Cycle 2010, 9, 3276–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.L.; Linnoila, I.; Liu, Z.-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Berardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef]
- le Bleu, V.S.; O’Connell, J.T.; Herrera, K.N.G.; Wikman-Kocher, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Chinen, L.T.D.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Hubalek, M.; Czech, T.; Müller, H. Biological Subtypes of Triple-Negative Breast Cancer. Breast Care 2017, 12, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Santonja, A.; Sánchez-Muñoz, A.; Lluch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacón, J.I.; Antolín, S.; Jerez, J.M.; de la Haba, J.; de Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406–26416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, C.; Jafari, S.M.; Shinde, R.; Duncan, G.; Cescon, D.W.; Silvester, J.; Chu, M.F.; Hodgson, K.; Berger, T.; Wakeham, A.; et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 2019, 116, 4326–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmiento-Salinas, F.L.; Delgado-Magallón, A.; Montes-Alvarado, J.B.; Ramírez-Ramírez, D.; Flores-Alonso, J.C.; Cortés-Hernández, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast Cancer Subtypes Present a Differential Production of Reactive Oxygen Species (ROS) and Susceptibility to Antioxidant Treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24, S36–S40. [Google Scholar] [CrossRef]
- Boukerroucha, M.; Josse, C.; El-Guendi, S.; Boujemla, B.; Frères, P.; Marée, R.; Wenric, S.; Segers, K.; Collignon, J.; Jerusalem, G.; et al. Evaluation of BRCA1-related molecular features and microRNAs as prognostic factors for triple negative breast cancers. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Wei, M.; Grushko, T.A.; Dignam, J.; Hagos, F.; Nanda, R.; Sveen, L.; Xu, J.; Fackenthal, J.; Tretiakova, M.; Das, S.; et al. BRCA1 Promoter Methylation in Sporadic Breast Cancer Is Associated with Reduced BRCA1 Copy Number and Chromosome 17 Aneusomy. Cancer Res. 2005, 65, 10692–10699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Rih, J.K.; Rosen, E.M. BRCA1 down-regulates cellular levels of reactive oxygen species. FEBS Lett. 2009, 583, 1535–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Yu, X. Tissue-Specific Carcinogens as Soil to Seed BRCA1/2-Mutant Hereditary Cancers. Trends Cancer 2020, 6, 559–568. [Google Scholar] [CrossRef]
- Li, M.; Chen, Q.; Yu, X. Chemopreventive Effects of ROS Targeting in a Murine Model of BRCA1-Deficient Breast Cancer. Cancer Res. 2017, 77, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced Up-Regulation of MnSOD and GPx but not Catalase Increases Oxidative Stress and Apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef] [Green Version]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.-P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef]
- Dhar, S.K.; Xu, Y.; Chen, Y.; Clair, D.K.S. Specificity Protein 1-dependent p53-mediated Suppression of Human Manganese Superoxide Dismutase Gene Expression. J. Biol. Chem. 2006, 281, 21698–21709. [Google Scholar] [CrossRef] [Green Version]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 3010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-A.; Lee, J.-W.; Kim, H.; Kim, E.-Y.; Seo, J.-M.; Ko, J.; Kim, J.-H. Pro-survival of estrogen receptor-negative breast cancer cells is regulated by a BLT2–reactive oxygen species-linked signaling pathway. Carcinogenesis 2009, 31, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Uchida, T.; Yoshie, T.; Mizote, Y.; Ishikawa, F.; Katsuyama, M.; Shibanuma, M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS -activated cancer cells. FEBS J. 2019, 286, 459–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Jang, J.-H.; Park, G.-S.; Chung, Y.; You, H.J.; Kim, J.H. BLT2, a leukotriene B4 receptor 2, as a novel prognostic biomarker of triple-negative breast cancer. BMB Rep. 2018, 51, 373–377. [Google Scholar] [CrossRef] [Green Version]
- de Luca, A.; Sanna, F.; Sallese, M.; Ruggiero, C.; Grossi, M.; Sacchetta, P.; Rossi, C.; de Laurenzi, V.; di Ilio, C.; Favaloro, B. Methionine sulfoxide reductase A down-regulation in human breast cancer cells results in a more aggressive phenotype. Proc. Natl. Acad. Sci. USA 2010, 107, 18628–18633. [Google Scholar] [CrossRef] [Green Version]
- Svineng, G.; Ravuri, C.; Rikardsen, O.; Huseby, N.-E.; Winberg, J.-O. The Role of Reactive Oxygen Species in Integrin and Matrix Metalloproteinase Expression and Function. Connect. Tissue Res. 2008, 49, 197–202. [Google Scholar] [CrossRef]
- Yin, H.-L.; Wu, C.-C.; Lin, C.-H.; Chai, C.-Y.; Hou, M.-F.; Chang, S.-J.; Tsai, H.-P.; Hung, W.-C.; Pan, M.-R.; Luo, C.-W. β1 Integrin as a Prognostic and Predictive Marker in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1432. [Google Scholar] [CrossRef]
- Valdivia, C.D.A.; Duran, C.; Martin, A.S. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Curr. Pharm. Des. 2015, 21, 6009–6022. [Google Scholar] [CrossRef] [Green Version]
- Honoré, S.; Kovacic, H.; Pichard, V.; Briand, C.; Rognoni, J.-B. α2β1-integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): Implication of NADPH oxidase-dependent production of ROS. Exp. Cell Res. 2003, 285, 59–71. [Google Scholar] [CrossRef]
- Zhu, P.; Tan, M.J.; Huang, R.-L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.I.; Boukamp, P.; Pan, J.Y.; Tan, S.H.; et al. Angiopoietin-like 4 Protein Elevates the Prosurvival Intracellular O2−:H2O2 Ratio and Confers Anoikis Resistance to Tumors. Cancer Cell 2011, 19, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Lee, S.H.; Wu, S. Effects of N-acetyl-L-cysteine on adhesive strength between breast cancer cell and extracellular matrix proteins after ionizing radiation. Life Sci. 2013, 93, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Qiao, J.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Mitrea, C.; Wijesinghe, P.; Marchetti, L.; Girsch, E.; Farr, R.L.; Boerner, J.L.; Mohammad, R.; Dyson, G.; Terlecky, S.R.; et al. Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep. 2017, 7, 44125. [Google Scholar] [CrossRef] [Green Version]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Liu, G.; Luo, Q.; Li, H.; Liu, Q.; Ju, Y.; Song, G. Increased Oxidative Phosphorylation is Required for Stemness Maintenance in Liver Cancer Stem Cells from Hepatocellular Carcinoma Cell Line HCCLM3 Cells. Int. J. Mol. Sci. 2020, 21, 5276. [Google Scholar] [CrossRef]
- Qian, X.; Nie, X.; Yao, W.; Klinghammer, K.; Sudhoff, H.; Kaufmann, A.M.; Albers, A.E. Reactive oxygen species in cancer stem cells of head and neck squamous cancer. Semin. Cancer Biol. 2018, 53, 248–257. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Balliet, R.; Lin, Z.; Whitaker-Menezes, D.; Birbe, R.C.; Bombonati, A.; Pavlides, S.; Lamb, R.; Sneddon, S.; Howell, A.; et al. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: Implications for breast cancer prevention with antioxidant therapies. Cell Cycle 2012, 11, 4402–4413. [Google Scholar] [CrossRef] [Green Version]
- Alili, L.; Sack, M.; Puschmann, K.; Brenneisen, P. Fibroblast-to-myofibroblast switch is mediated by NAD(P)H oxidase generated reactive oxygen species. Biosci. Rep. 2014, 34, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; Li, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2018, 8, e2562. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Park, G.-Y.; Pathak, H.B.; Godwin, A.K.; Kwon, Y. Epithelial-stromal communication via CXCL1-CXCR2 interaction stimulates growth of ovarian cancer cells through p38 activation. Cell. Oncol. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Hanley, C.J.; Mellone, M.; Ford, K.; Thirdborough, S.M.; Mellows, T.; Frampton, S.J.; Smith, D.M.; Harden, E.; Szyndralewiez, C.; Bullock, M.; et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J. Natl. Cancer Inst. 2018, 110, 109–120. [Google Scholar] [CrossRef]
- Liu, L.; Liu, L.; Yao, H.H.; Zhu, Z.Q.; Ning, Z.L.; Huang, Q. Stromal Myofibroblasts Are Associated with Poor Prognosis in Solid Cancers: A Meta-Analysis of Published Studies. PLoS ONE 2016, 11, e0159947. [Google Scholar] [CrossRef]
- Tsujino, T.; Seshimo, I.; Yamamoto, H.; Ngan, C.Y.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; Monden, M. Stromal Myofibroblasts Predict Disease Recurrence for Colorectal Cancer. Clin. Cancer Res. 2007, 13, 2082–2090. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2010, 19, 170–176. [Google Scholar] [CrossRef]
- Tobar, N.; Toyos, M.; Urra, C.; Méndez, N.; Arancibia, R.; Smith, P.C.; Martínez, J. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer 2014, 14, 640. [Google Scholar] [CrossRef] [Green Version]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Dürr, P.; Berger, P.B. ROS Signaling by NOX4 Drives Fibroblast-to-Myofibroblast Differentiation in the Diseased Prostatic Stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Bonuccelli, G.; Whitaker-Menezes, D.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Witkiewicz, A.K.; Heiden, M.G.V.; Migneco, G.; Chiavarina, B.; et al. The reverse Warburg Effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle 2010, 9, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Dasgupta, A.; Sammons, S.; Er, O.; Potoczek, M.B.; Guiles, F.; Sotgia, F.; Brody, J.R.; Mitchell, E.P.; Lisanti, M.P. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol. Ther. 2010, 10, 135–143. [Google Scholar] [CrossRef]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Dou, H.; Bagi, Z.; Han, W.; Su, Y.; Fulton, D.J.R. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Neto, V.M. MicroRNAs, Hypoxia and the Stem-Like State as Contributors to Cancer Aggressiveness. Front. Genet. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia—A mecha-nism of O-2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Tan, E.Y.; Yan, M.; Campo, L.; Han, C.; Takano, E.; Turley, H.; Candiloro, I.; Pezzella, F.; Gatter, K.C.; Millar, E.K.A.; et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br. J. Cancer 2009, 100, 405–411. [Google Scholar] [CrossRef]
- Bernardi, R.; Gianni, L. Hallmarks of triple negative breast cancer emerging at last? Cell Res. 2014, 24, 904–905. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nat. Cell Biol. 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nat. Cell Biol. 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Talia, M.; Cirillo, F.; Rigiracciolo, D.C.; Scordamaglia, D.; Guzzi, R.; Miglietta, A.M.; de Francesco, E.M.; Belfiore, A.; Sims, A.H.; et al. The IL1β-IL1R signaling is involved in the stimulatory effects triggered by hypoxia in breast cancer cells and cancer-associated fibroblasts (CAFs). J. Exp. Clin. Cancer Res. 2020, 39, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Körner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.S.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Semenza, G.L. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016, 76, 6458–6462. [Google Scholar] [CrossRef] [Green Version]
- Lebert, J.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Khalefa, H.G.; Shawki, M.A.; Aboelhassan, R.; el Wakeel, L.M. Evaluation of the effect of N-acetylcysteine on the prevention and amelioration of paclitaxel-induced peripheral neuropathy in breast cancer patients: A randomized controlled study. Breast Cancer Res. Treat. 2020, 183, 117–125. [Google Scholar] [CrossRef]
- Arıca, V.; Demir, I.; Tutanc, M.; Basarslan, F.; Arıca, S.; Karcıoglu, M.; Öztürk, H.; Nacar, A. N-Acetylcysteine prevents doxorubucine-induced cardiotoxicity in rats. Hum. Exp. Toxicol. 2012, 32, 655–661. [Google Scholar] [CrossRef]
- Farshid, A.A.; Tamaddonfard, E.; Simaee, N.; Mansouri, S.; Najafi, S.; Asri-Rezaee, S.; Alavi, H. Effects of Histidine and N-Acetylcysteine on Doxorubicin-Induced Cardiomyopathy in Rats. Cardiovasc. Toxicol. 2014, 14, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Brum, G.; Carbone, T.; Still, E.; Correia, V.; Szulak, K.; Calianese, D.; Best, C.; Cammarata, G.; Higgins, K.; Ji, F.; et al. N-acetylcysteine potentiates doxorubicin-induced ATM and p53 activation in ovarian cancer cells. Int. J. Oncol. 2012, 42, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qanungo, S.; Uys, J.D.; Manevich, Y.; Distler, A.M.; Shaner, B.; Hill, E.G.; Mieyal, J.J.; Lemasters, J.J.; Townsend, D.M.; Nieminen, A.-L. N-acetyl-l-cysteine sensitizes pancreatic cancers to gemcitabine by targeting the NFκB pathway. Biomed. Pharmacother. 2014, 68, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, P.A.; Mitsopoulos, P.; Suntres, Z.E. N-Acetylcysteine Modulates the Cytotoxic Effects of Paclitaxel. Chemotherapy 2011, 57, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X. ROS Reduction Does Not Decrease the Anticancer Efficacy of X-Ray in Two Breast Cancer Cell Lines. Oxid. Med. Cell. Longev. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The dependence of triple-negative breast cancers (TNBCs) on reactive oxygen species (ROS) signaling for their survival and malignant progression. Basal-like TNBC is related to the BRCA1 mutation/inactivation and TP53 mutation. This oncogenic dysregulation induces metabolic changes and oxidant-antioxidant imbalances that lead to high ROS production, which may be necessary for the survival and proliferation of TNBC cells. In addition, TNBC cells may further undergo gene expression changes (i.e., MsrA loss and BLT2 amplification) that increase ROS production via NOX activity, which then modulates signaling that promote cell survival and invasion. High ROS levels also stabilize gene expression (e.g., MMP-9) related to the metastatic phenotype. Moreover, the high drug-resistant and metastatic properties of TNBC are often related to an increase in the cancer stem-like cell (CSC) fraction (i.e., overexpression of c-MYC and MCL1) that produces high amounts of ROS via high oxidative phosphorylation (OXPHOS). This ROS-mediated signaling leads to TNBC progression, but is effectively attenuated by N-acetylcysteine (NAC) treatment, thereby reducing the survival and metastasis of TNBC cells.
Figure 1.
The dependence of triple-negative breast cancers (TNBCs) on reactive oxygen species (ROS) signaling for their survival and malignant progression. Basal-like TNBC is related to the BRCA1 mutation/inactivation and TP53 mutation. This oncogenic dysregulation induces metabolic changes and oxidant-antioxidant imbalances that lead to high ROS production, which may be necessary for the survival and proliferation of TNBC cells. In addition, TNBC cells may further undergo gene expression changes (i.e., MsrA loss and BLT2 amplification) that increase ROS production via NOX activity, which then modulates signaling that promote cell survival and invasion. High ROS levels also stabilize gene expression (e.g., MMP-9) related to the metastatic phenotype. Moreover, the high drug-resistant and metastatic properties of TNBC are often related to an increase in the cancer stem-like cell (CSC) fraction (i.e., overexpression of c-MYC and MCL1) that produces high amounts of ROS via high oxidative phosphorylation (OXPHOS). This ROS-mediated signaling leads to TNBC progression, but is effectively attenuated by N-acetylcysteine (NAC) treatment, thereby reducing the survival and metastasis of TNBC cells.

Figure 2.
The involvement of reactive oxygen species (ROS) in the interplay between cancer cells and the tumor microenvironment (TME). The ROS-triggered signaling actively involves in interaction between cancer cell and the TME in TNBC. These ROS-mediated interactions induce the recruitment of immune cells, conversion of stromal cells into cancer-associated phenotypes (e.g., myofibroblastic transition, metabolic changes, and immunosuppression), and hypoxic responses (e.g., metastases, inflammation, and cancer stem-like cell (CSC) enrichment), thereby creating permissive TMEs and promoting malignant progression of cancer. N-acetylcysteine (NAC) treatment can effectively interfere with cancer cell-TME interactions by suppressing the ROS signaling that mediates the invasive, drug-resistant, metastatic properties of TNBC cells.
Figure 2.
The involvement of reactive oxygen species (ROS) in the interplay between cancer cells and the tumor microenvironment (TME). The ROS-triggered signaling actively involves in interaction between cancer cell and the TME in TNBC. These ROS-mediated interactions induce the recruitment of immune cells, conversion of stromal cells into cancer-associated phenotypes (e.g., myofibroblastic transition, metabolic changes, and immunosuppression), and hypoxic responses (e.g., metastases, inflammation, and cancer stem-like cell (CSC) enrichment), thereby creating permissive TMEs and promoting malignant progression of cancer. N-acetylcysteine (NAC) treatment can effectively interfere with cancer cell-TME interactions by suppressing the ROS signaling that mediates the invasive, drug-resistant, metastatic properties of TNBC cells.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kwon, Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants 2021, 10, 169. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020169
AMA Style
Kwon Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants. 2021; 10(2):169. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020169
Chicago/Turabian StyleKwon, Youngjoo. 2021. "Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer" Antioxidants 10, no. 2: 169. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020169
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.