
Fine-Tuning the Biological Profile of Multitarget Mitochondriotropic Antioxidants for Neurodegenerative Diseases

 , , , , , , , , ,
, , , , , , , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
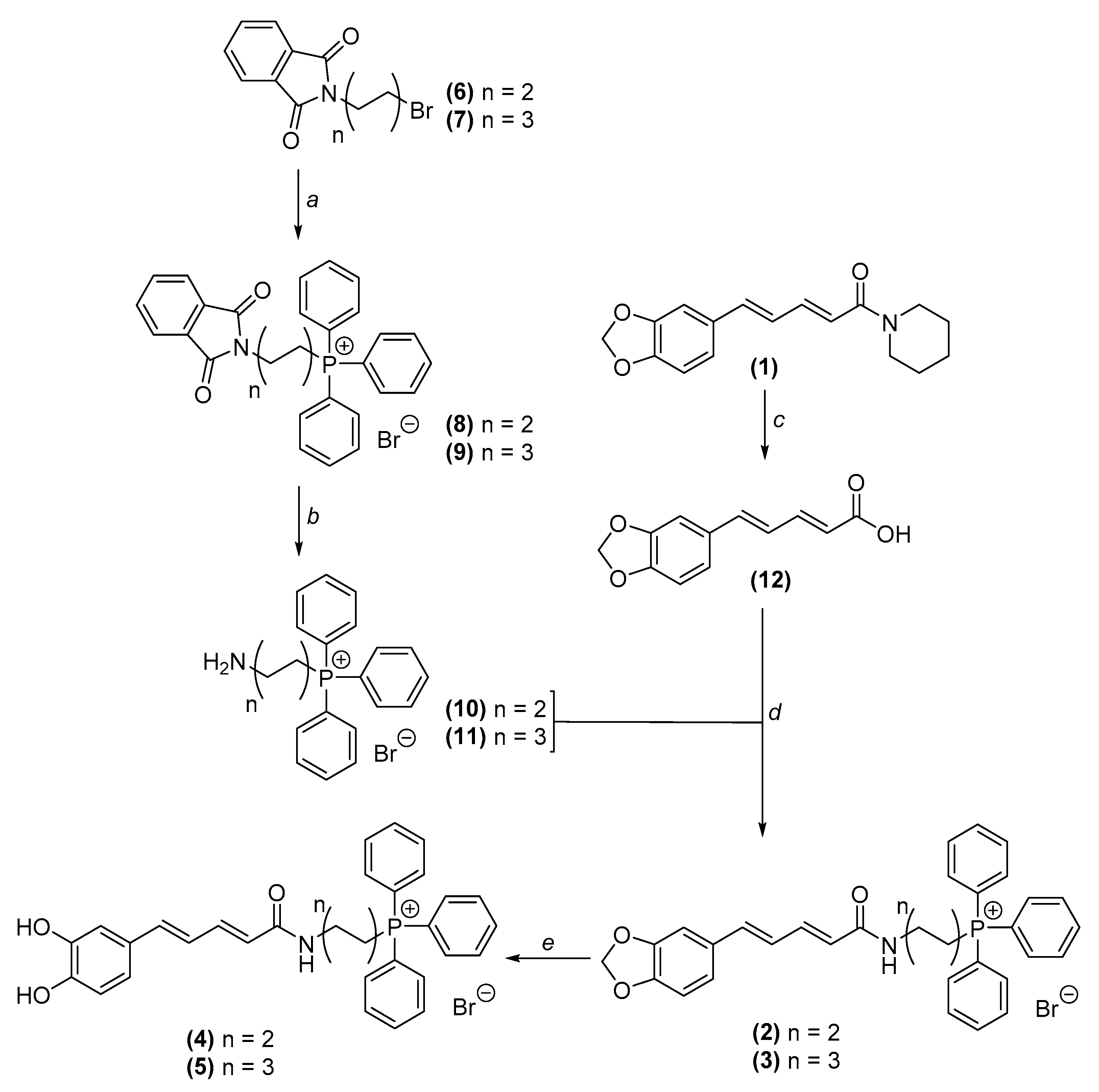
2.1. Chemistry
2.1.1. Synthesis of AntiOXCIN2 and AntiOXCIN3
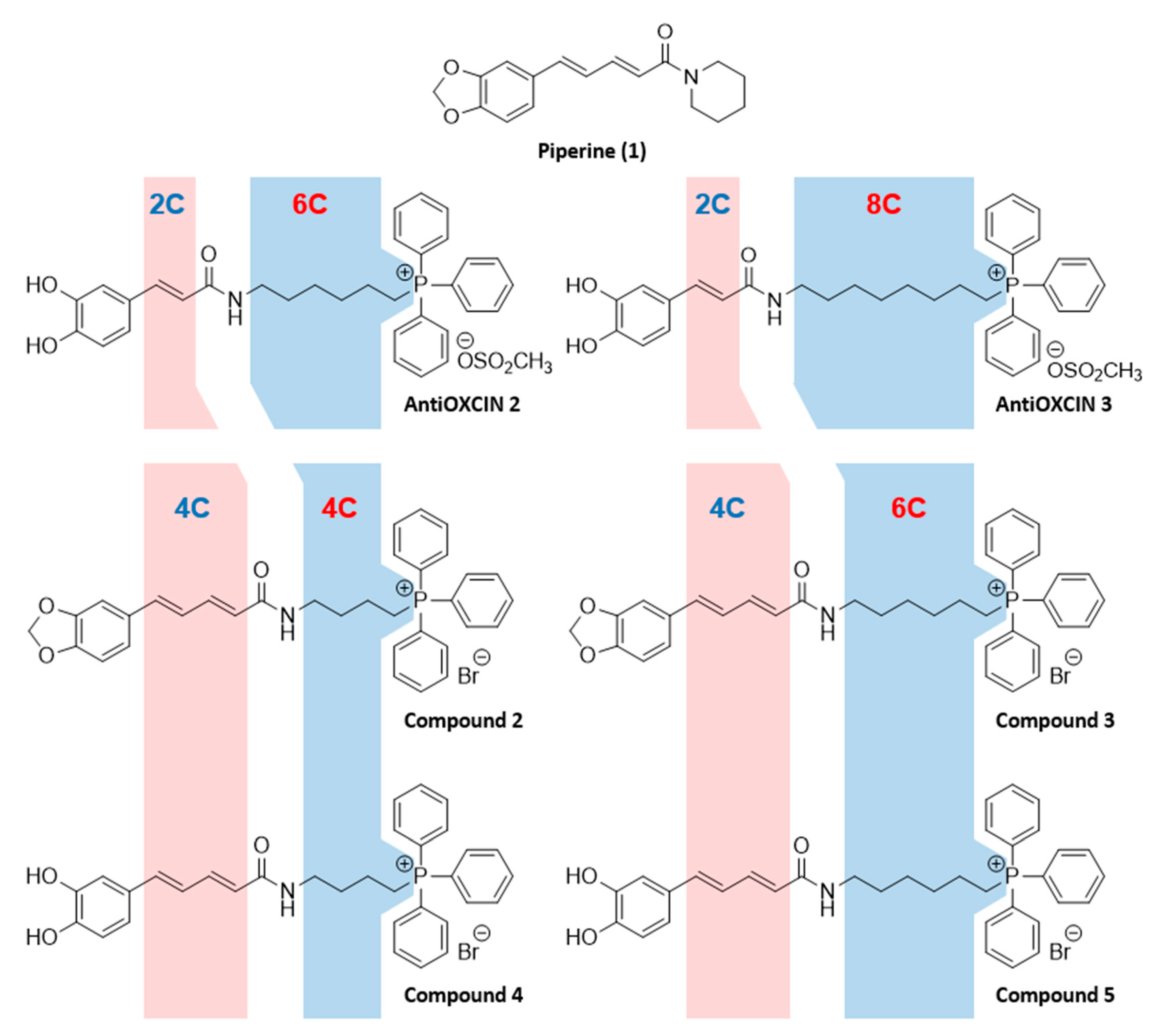
2.1.2. Synthesis of Mitochondria-Targeted Agents Inspired on Piperine
General Procedure for the Obtention of Phtalimidylalkylltriphenylphosphonium Salts
General Procedure for the Obtention of Aminoalkylltriphenylphosphonium Salts
Synthesis of (2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)Penta-2,4-Dienoic Acid (12)
Piperic Acid Amidation
Demethylenation Reaction
2.2. Enzymatic Assays
2.2.1. Acetylcholinesterase and Butyrylcholinesterase
Evaluation of Eel Acetylcholinesterase and Equine Butyrylcholinesterase Inhibitory Activity
Evaluation of Human Acetylcholinesterase and Human Butyrylcholinesterase Inhibitory Activities
Crystallization, X-ray Data Collection and Processing
2.2.2. Evaluation of Human Monoamine Oxidase Inhibitory Activity
2.3. Oxygen Radical Absorbance Capacity (ORAC-FL) Assay
2.4. Electrochemical Measurements
2.5. In Vitro Toxicology
2.5.1. Materials
2.5.2. Cell Lines and Culture Conditions
2.5.3. Cytotoxicity
2.5.4. Statistical Analysis
2.6. Evaluation of the Chromatographic Hydrophobicity Index
2.7. Estimation of Drug-Like Properties
3. Results and Discussion
3.1. Chemistry
3.2. Cholinesterase Inhibition Studies
3.2.1. Evaluation of Electric Eel Acetylcholinesterase and Equine Butyrylcholinesterase Inhibitory Activities
3.2.2. Evaluation of Human Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities
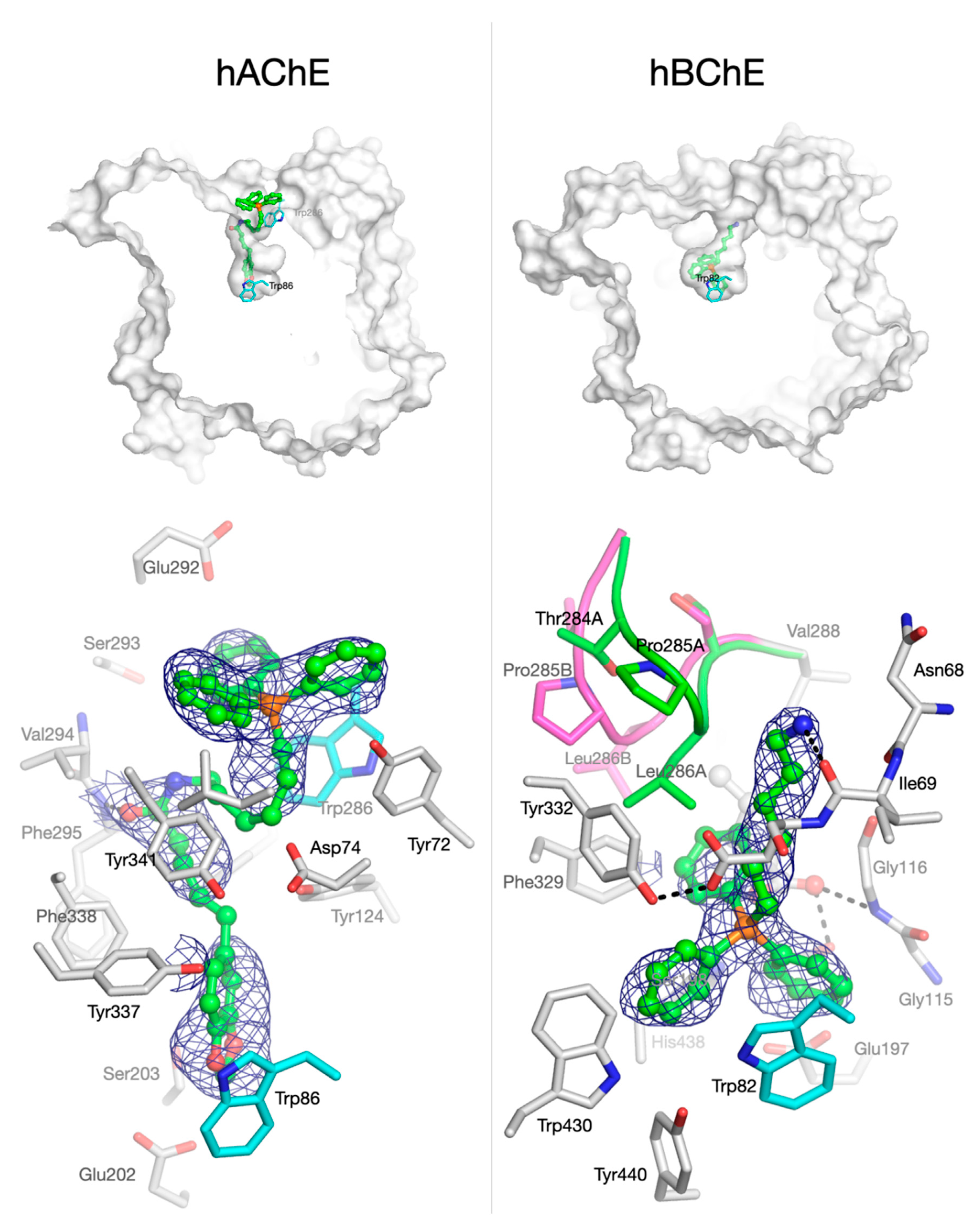
3.2.3. Crystallographic Studies with Human Cholinesterases
3.3. Monoamine Oxidase Inhibition Studies
3.4. Antioxidant Activity
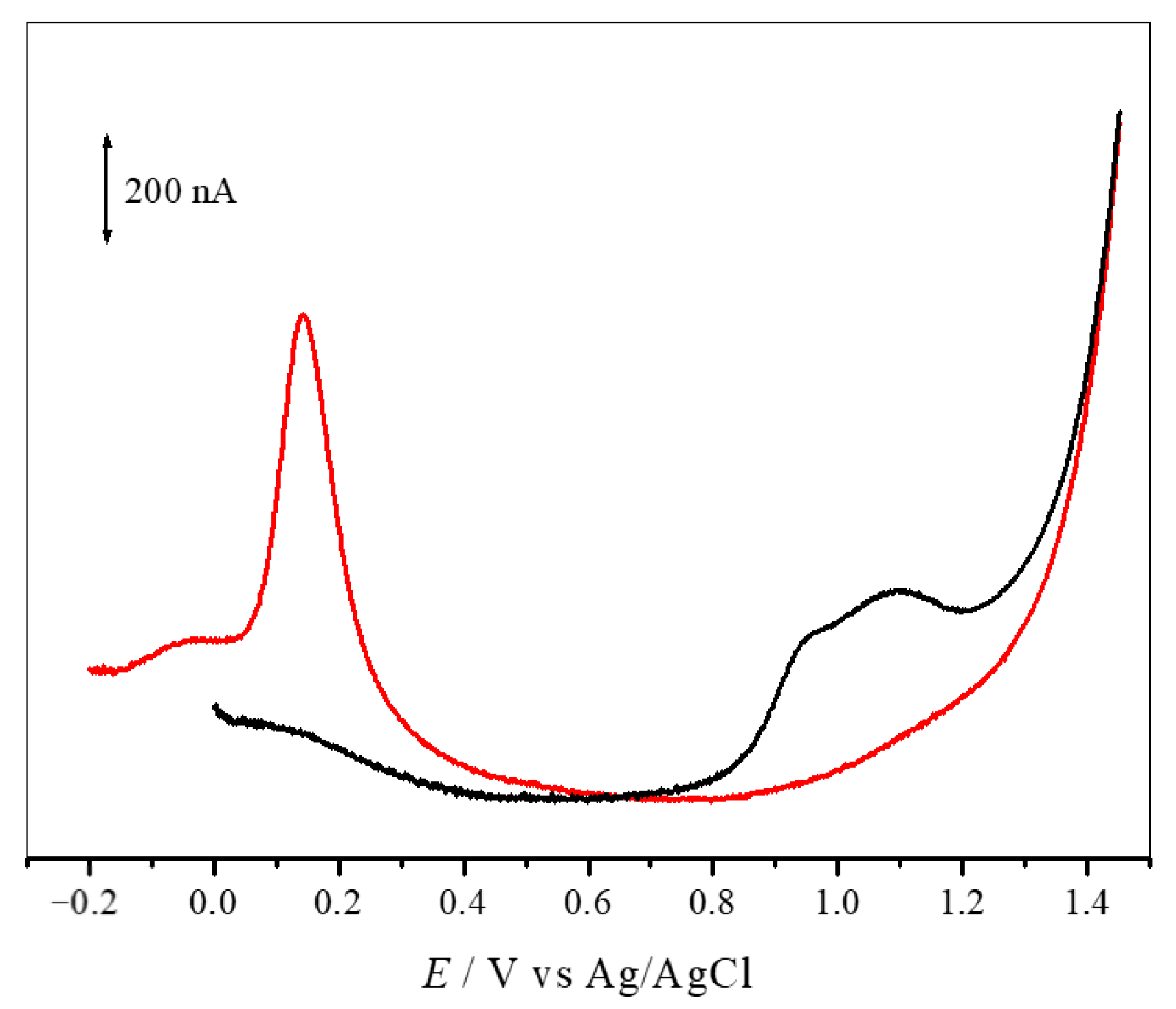
3.5. Electrochemical Studies
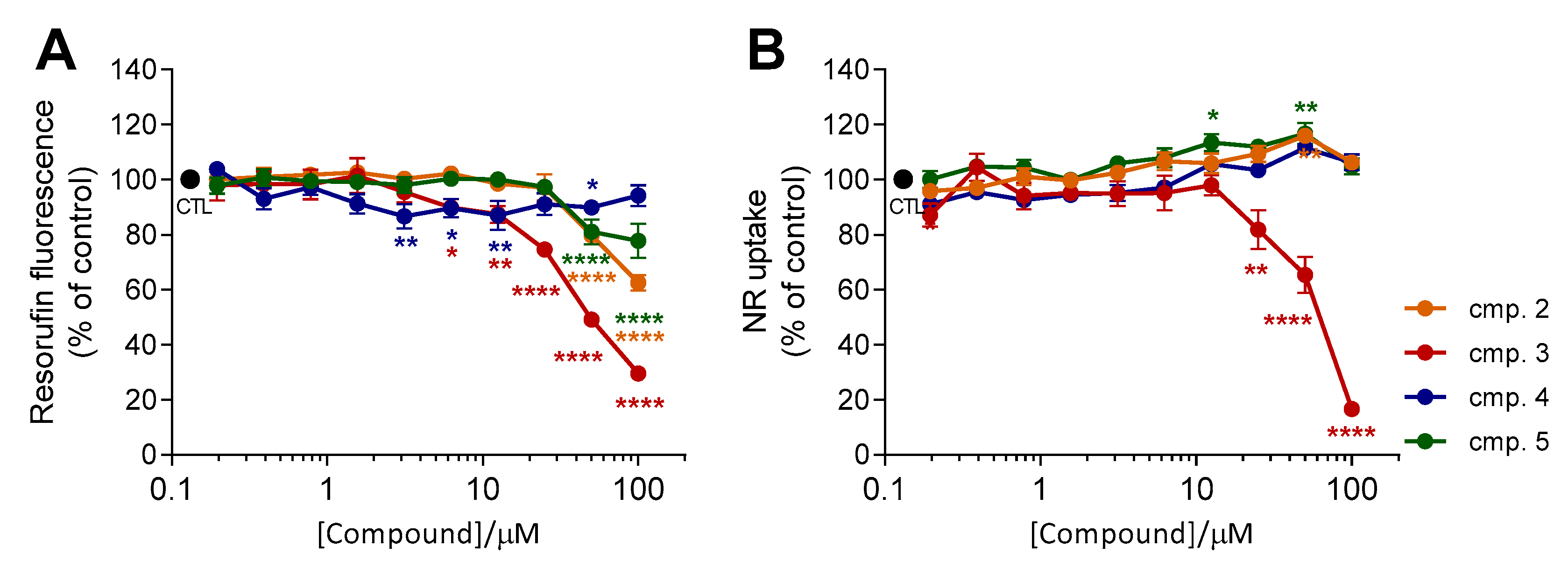
3.6. Evaluation of Cytotoxicity Profile
3.7. Evaluation of Drug-Like Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramsay, R.R.; Majekova, M.; Medina, M.; Valoti, M. Key Targets for Multi-Target Ligands Designed to Combat Neurodegeneration. Front. Neurosci. 2016, 10, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- Elmabruk, A.; Das, B.; Yedlapudi, D.; Xu, L.; Antonio, T.; Reith, M.E.A.; Dutta, A.K. Design, Synthesis, and Pharmacological Characterization of Carbazole Based Dopamine Agonists as Potential Symptomatic and Neuroprotective Therapeutic Agents for Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 396–411. [Google Scholar] [CrossRef]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef]
- Fetisova, E.K.; Avetisyan, A.V.; Izyumov, D.S.; Korotetskaya, M.V.; Chernyak, B.V.; Skulachev, V.P. Mitochondria-targeted antioxidant SkQR1 selectively protects MDR (Pgp 170)-negative cells against oxidative stress. FEBS Lett. 2010, 584, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Facecchia, K.; Fochesato, L.A.; Ray, S.D.; Stohs, S.J.; Pandey, S. Oxidative toxicity in neurodegenerative diseases: Role of mitochondrial dysfunction and therapeutic strategies. J. Toxicol. 2011, 2011, 683728. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Biasutto, L.; Mattarei, A.; La Spina, M.; Azzolini, M.; Parrasia, S.; Szabò, I.; Zoratti, M. Strategies to target bioactive molecules to subcellular compartments. Focus on natural compounds. Eur. J. Med. Chem. 2019, 181, 11557. [Google Scholar] [CrossRef] [PubMed]
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release 2015, 207, 40–58. [Google Scholar] [CrossRef]
- Guzman-Villanueva, D.; Weissig, V. Mitochondria-Targeted Agents: Mitochondriotropics, Mitochondriotoxics, and Mitocans. Handb. Exp. Pharmacol. 2017, 240, 423–438. [Google Scholar] [CrossRef]
- He, H.; Li, D.W.; Yang, L.Y.; Fu, L.; Zhu, X.J.; Wong, W.K.; Jiang, F.L.; Liu, Y. A novel bifunctional mitochondria-targeted anticancer agent with high selectivity for cancer cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Porteous, C.M.; Logan, A.; Evans, C.; Ledgerwood, E.C.; Menon, D.K.; Aigbirhio, F.; Smith, R.A.; Murphy, M.P. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: Implications for mitochondria-specific therapies and probes. Biochim. Biophys. Acta 2010, 1800, 1009–1017. [Google Scholar] [CrossRef]
- Oliveira, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Sequeira, L.; Mesiti, F.; Silva, T.; Garrido, J.; Remiao, F.; Vilar, S.; et al. Hydroxybenzoic Acid Derivatives as Dual-Target Ligands: Mitochondriotropic Antioxidants and Cholinesterase Inhibitors. Front. Chem. 2018, 6, 126. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.; Oliveira, C.; Amorim, R.; Cagide, F.; Garrido, J.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; Oliveira, P.J.; et al. Development of hydroxybenzoic-based platforms as a solution to deliver dietary antioxidants to mitochondria. Sci. Rep. 2017, 7, 6842. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Cagide, F.; Benfeito, S.; Soares, P.; Garrido, J.; Baldeiras, I.; Ribeiro, J.A.; Pereira, C.M.; Silva, A.F.; Andrade, P.B.; et al. Development of a Mitochondriotropic Antioxidant Based on Caffeic Acid: Proof of Concept on Cellular and Mitochondrial Oxidative Stress Models. J. Med. Chem. 2017, 60, 7084–7098. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Soares, P.; Benfeito, S.; Gaspar, A.; Garrido, J.; Murphy, M.P.; Borges, F. Rational discovery and development of a mitochondria-targeted antioxidant based on cinnamic acid scaffold. Free Radic. Res. 2012, 46, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Oliveira, C.; Fernandes, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Garrido, J.; Martins, C.; Sarmento, B.; Silva, R.; et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur. J. Med. Chem. 2019, 167, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Benfeito, S.; Fernandes, C.; Vilar, S.; Remião, F.; Uriarte, E.; Borges, F. Exploring the multi-target performance of mitochondriotropic antioxidants against the pivotal Alzheimer’s disease pathophysiological hallmarks. Molecules 2020, 25, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Chen, Y.H.; Liu, H.; Qu, H.D. Neuroprotective effects of piperine on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease mouse model. Int. J. Mol. Med. 2015, 36, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattanathorn, J.; Chonpathompikunlert, P.; Muchimapura, S.; Priprem, A.; Tankamnerdthai, O. Piperine, the potential functional food for mood and cognitive disorders. Food Chem. Toxicol. 2008, 46, 3106–3110. [Google Scholar] [CrossRef]
- Chonpathompikunlert, P.; Wattanathorn, J.; Muchimapura, S. Piperine, the main alkaloid of Thai black pepper, protects against neurodegeneration and cognitive impairment in animal model of cognitive deficit like condition of Alzheimer’s disease. Food Chem. Toxicol. 2010, 48, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, M.; Wang, X.; Wang, Y.; Duan, C.; Gao, G.; Lu, L.; Wu, X.; Wang, X.; Yang, H. Piperine induces autophagy by enhancing protein phosphotase 2A activity in a rotenone-induced Parkinson’s disease model. Oncotarget 2016, 7, 60823–60843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Q.Q.; Huang, Z.; Ip, S.P.; Xian, Y.F.; Che, C.T. Protective effects of piperine against corticosterone-induced neurotoxicity in PC12 cells. Cell. Mol. Neurobiol. 2012, 32, 531–537. [Google Scholar] [CrossRef]
- Lee, S.A.; Hong, S.S.; Han, X.H.; Hwang, J.S.; Oh, G.J.; Lee, K.S.; Lee, M.K.; Hwang, B.Y.; Ro, J.S. Piperine from the fruits of Piper longum with inhibitory effect on monoamine oxidase and antidepressant-like activity. Chem. Pharm. Bull. 2005, 53, 832–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavarria, D.; Cagide, F.; Pinto, M.; Gomes, L.R.; Low, J.N.; Borges, F. Development of piperic acid-based monoamine oxidase inhibitors: Synthesis, structural characterization and biological evaluation. J. Mol. Struct. 2019, 1182, 298–307. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williard, P.G.; Fryhle, C.B. Boron trihalide-methyl sulfide complexes as convenient reagents for dealkylation of aryl ethers. Tetrahedron Lett. 1980, 21, 3731–3734. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Borloz, A.; Urbain, A.; Marston, A.; Hostettmann, K.; Carrupt, P.A.; Reist, M. In vitro screening assays to identify natural or synthetic acetylcholinesterase inhibitors: Thin layer chromatography versus microplate methods. Eur. J. Pharm. Sci. 2008, 33, 109–119. [Google Scholar] [CrossRef]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Wandhammer, M.; Ronco, C.; Trovaslet, M.; Jean, L.; Lockridge, O.; Renard, P.Y.; Nachon, F. Human butyrylcholinesterase produced in insect cells: Huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallog. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Hagenow, S.; Stasiak, A.; Ramsay, R.R.; Stark, H. Ciproxifan, a histamine H3 receptor antagonist, reversibly inhibits monoamine oxidase A and B. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Mura, F.; Silva, T.; Castro, C.; Borges, F.; Zuniga, M.C.; Morales, J.; Olea-Azar, C. New insights into the antioxidant activity of hydroxycinnamic and hydroxybenzoic systems: Spectroscopic, electrochemistry, and cellular studies. Free Radic. Res. 2014, 48, 1473–1484. [Google Scholar] [CrossRef]
- Garrido, J.; Gaspar, A.; Garrido, E.M.; Miri, R.; Tavakkoli, M.; Pourali, S.; Saso, L.; Borges, F.; Firuzi, O. Alkyl esters of hydroxycinnamic acids with improved antioxidant activity and lipophilicity protect PC12 cells against oxidative stress. Biochimie 2012, 94, 961–967. [Google Scholar] [CrossRef]
- Fernandes, C.; Pinto, M.; Martins, C.; Gomes, M.J.; Sarmento, B.; Oliveira, P.J.; Remião, F.; Borges, F. Development of a PEGylated-Based Platform for Efficient Delivery of Dietary Antioxidants Across the Blood-Brain Barrier. Bioconjug. Chem. 2018, 29, 1677–1689. [Google Scholar] [CrossRef]
- Camurri, G.; Zaramella, A. High-throughput liquid chromatography/mass spectrometry method for the determination of the chromatographic hydrophobicity index. Anal. Chem. 2001, 73, 3716–3722. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch. Pharm. Res. 2013, 36, 375–399. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, B. Donepezil: A review. Expert Opin. Drug Metab. Toxicol. 2005, 1, 527–536. [Google Scholar] [CrossRef]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, R.; Caria, P.; Matos, M.J.; Vina, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, D.; Fallarero, A.; Brunhofer, G.; Mayer, C.; Prakash, O.; Mohan, C.G.; Vuorela, P.; Erker, T. The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 2012, 47, 190–205. [Google Scholar] [CrossRef]
- Moorad, D.R.; Luo, C.; Saxena, A.; Doctor, B.P.; Garcia, G.E. Purification and determination of the amino acid sequence of equine serum butyrylcholinesterase. Toxicol. Methods 1999, 9, 219–227. [Google Scholar] [CrossRef]
- Afonine, P.V.; Moriarty, N.W.; Mustyakimov, M.; Sobolev, O.V.; Terwilliger, T.C.; Turk, D.; Urzhumtsev, A.; Adams, P.D. FEM: Feature-enhanced map. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 646–666. [Google Scholar] [CrossRef] [Green Version]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Deng, Z.; Wu, T.; Liu, R.; Loewen, S.; Tsao, R. Microwave-assisted extraction of phenolics with maximal antioxidant activities in tomatoes. Food Chem. 2012, 130, 928–936. [Google Scholar] [CrossRef]
- Carp, O.E.; Moraru, A.; Pinteala, M.; Arvinte, A. Electrochemical behaviour of piperine. Comparison with control antioxidants. Food Chem. 2021, 339. [Google Scholar] [CrossRef]
- Silva, F.S.; Starostina, I.G.; Ivanova, V.V.; Rizvanov, A.A.; Oliveira, P.J.; Pereira, S.P. Determination of Metabolic Viability and Cell Mass Using a Tandem Resazurin/Sulforhodamine B Assay. Curr. Protoc. Toxicol. 2016, 68, 2–24. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Chavarria, D.; Fernandes, C.; Silva, V.; Silva, C.; Gil-Martins, E.; Soares, P.; Silva, T.; Silva, R.; Remiao, F.; Oliveira, P.J.; et al. Design of novel monoamine oxidase-B inhibitors based on piperine scaffold: Structure-activity-toxicity, drug-likeness and efflux transport studies. Eur. J. Med. Chem. 2020, 185, 111770. [Google Scholar] [CrossRef]
- Valko, K.; Bevan, C.; Reynolds, D. Chromatographic Hydrophobicity Index by Fast-Gradient RP-HPLC: A High-Throughput Alternative to log P/log D. Anal. Chem. 1997, 69, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, K.; Du, C.M.; Bevan, C.; Reynolds, D.P.; Abraham, M.H. Rapid method for the estimation of octanol/water partition coefficient (Log Poct) from gradient RP-HPLC retention and a hydrogen bond acidity term (∑α2H). Curr. Med. Chem. 2001, 8, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Entry Code | 6ZWE | 6ZWI |
|---|---|---|
| X-ray source | PROXIMA 2 | PROXIMA 2 |
| Wavelenght | 0.9801 | 0.9801 |
| Resolution range | 78.09–3.0 (3.107–3.0) | 41.48–1.85 (1.916–1.85) |
| Space group | P 61 | I 4 2 2 |
| Unit cell | a = 211.35, b = 211.35, c = 115.9 α = β = 90.0, γ = 120 | a = 154.3, b = 154.3, c = 127.7 α = β = γ = 90.0 |
| Total reflections | 2352044 (194838) | 3366060 (268864) |
| Unique reflections | 59080 (5885) | 65496 (6485) |
| Multiplicity | 39.8 (33.1) | 51.4 (41.5) |
| Completeness (%) | 99.87 (99.66) | 99.96 (99.92) |
| Mean I/sigma(I) | 20.81 (2.80) | 29.97 (0.81) |
| Wilson B-factor | 85.22 | 47.10 |
| R-merge | 0.1637 (1.526) | 0.1014 (6.341) |
| R-meas | 0.1658 (1.55) | 0.1025 (6.418) |
| R-pim | 0.02615 (0.2668) | 0.01425 (0.9876) |
| CC1/2 | 0.999 (0.933) | 1 (0.31) |
| CC * | 1 (0.982) | 1 (0.688) |
| Reflections used in refinement | 59025 (5879) | 65488 (6482) |
| Reflections used for R-free | 1180 (118) | 1309 (129) |
| R-work | 0.1982 (0.3401) | 0.1878 (0.3676) |
| R-free | 0.2411 (0.3895) | 0.2241 (0.3844) |
| Number of non-hydrogen atoms | 8782 | 4808 |
| macromolecules | 8363 | 4294 |
| ligands | 274 | 286 |
| solvent | 145 | 228 |
| Protein residues | 1073 | 527 |
| RMS(bonds) | 0.004 | 0.016 |
| RMS(angles) | 0.88 | 1.27 |
| Ramachandran favored (%) | 95.03 | 95.62 |
| Ramachandran allowed (%) | 4.87 | 4.19 |
| Ramachandran outliers (%) | 0.09 | 0.19 |
| Rotamer outliers (%) | 0.11 | 0.43 |
| Clashscore | 11.09 | 5.34 |
| Average B-factor | 97.42 | 56.31 |
| macromolecules | 96.06 | 53.61 |
| ligands | 143.73 | 96.23 |
| solvent | 88.44 | 57.18 |
| Compound | IC50/µM | SI (1) b | IC50/µM | SI (2) c | ||
|---|---|---|---|---|---|---|
| eeAChE | eqBChE | hAChE | hBChE | |||
| 1 | ___ a | ___a | ___ | ___ e | ___ e | ___ |
| 2 | 6.39 ± 0.28 | 0.0282 ± 0.0014 | 226 | 11 ± 1 | 0.20 ± 0.04 | 55 |
| 3 | 5.74 ± 0.32 | 0.0179 ± 0.0009 | 314 | 2.0 ± 0.2 | 3.0 ± 0.4 | 0.67 |
| 4 | 2.23 ± 0.12 | 0.0619 ± 0.0042 | 36 | 22 ± 2 | 3.0 ± 0.2 | 7.3 |
| 5 | 2.14 ± 0.06 | 0.0341 ± 0.0029 | 62 | 9.0 ± 0.5 | 23 ± 2 | 0.39 |
| AntiOXCIN2 | 6.32 ± 0.14 d | 0.124 ± 0.007 d | 51 d | ___ e | ___ e | ___ |
| AntiOXCIN3 | 5.08 ± 0.22 | 0.325 ± 0.09 | 16 | ___ e | ___ e | ___ |
| Donepezil | 0.0129 ± 0.0008 | 2.50 ± 0.09 | 0.0052 | ___ e | ___ e | ___ |
| Compound | IC50/µM | SI (3) b | |
|---|---|---|---|
| hMAO-A | hMAO-B | ||
| 1 | ___ a,d | 1.05 ± 0.08 d | >10 c,d |
| 2 | 0.888 ± 0.022 | 12.4 ± 1.9 | 0.07 |
| 3 | 1.23 ± 0.13 | 4.64 ± 0.29 | 0.26 |
| 4 | 5.17 ± 0.54 | 13.5 ± 1.1 | 0.38 |
| 5 | 2.17 ± 0.28 | 10.9 ± 0.7 | 0.20 |
| AntiOXCIN2 | ___ a | ___ a | __ |
| AntiOXCIN3 | ___ a | ___ a | __ |
| R-(−)-Deprenyl | 20.1 ± 1.9 | 0.0386 ± 0.0043 | 522 |
| Rasagiline | 3.65 ± 0.31 | 147.3 ± 249 | 24 |
| Safinamide | ___ a | 0.0231 ± 0.0026 | >433 c |
| Clorgyline | 0.00274 ± 0.00047 | 2.21 ± 0.26 | 0.00124 |
| Compound | ORAC-FL Index | Ep/mV |
|---|---|---|
| 1 | ___ | n.d. |
| 2 | ___ | 942; 1070 |
| 3 | ___ | 931; 1083 |
| 4 | 3.3 ± 0.1 | 125 |
| 5 | 3.1 ± 0.3 | 144 |
| AntiOXCIN2 | 2.8 ± 0.1 | 166 a |
| AntiOXCIN3 | 2.6 ± 0.1 | 164 a |
| Compound | CHI a | CHI LogPoct b | MW c | TPSA c | HBA c | HBD c | RB c |
|---|---|---|---|---|---|---|---|
| 1 | 73.4 | 2.35 | 285.3 | 38.77 | 3 | 0 | 4 |
| 2 | 42.8 | 1.27 | 614.5 | 61.15 | 4 | 1 | 12 |
| 3 | 45.8 | 1.41 | 642.6 | 61.15 | 4 | 1 | 14 |
| 4 | 34.4 | 1.59 | 602.5 | 83.15 | 4 | 3 | 12 |
| 5 | 37.2 | 1.73 | 630.6 | 83.15 | 4 | 3 | 14 |
| AntiOXCIN2 | 37.9 | 1.76 | 619.7 | 83.15 | 4 | 3 | 14 |
| AntiOXCIN3 | 41.4 | 1.92 | 647.8 | 83.15 | 4 | 3 | 16 |
| CNS+ drugs | ___ | ___ | <500 [60] | <90 [60] | <7 [61] | <3 [61] | <8 [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavarria, D.; Da Silva, O.; Benfeito, S.; Barreiro, S.; Garrido, J.; Cagide, F.; Soares, P.; Remião, F.; Brazzolotto, X.; Nachon, F.; et al. Fine-Tuning the Biological Profile of Multitarget Mitochondriotropic Antioxidants for Neurodegenerative Diseases. Antioxidants 2021, 10, 329. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020329
Chavarria D, Da Silva O, Benfeito S, Barreiro S, Garrido J, Cagide F, Soares P, Remião F, Brazzolotto X, Nachon F, et al. Fine-Tuning the Biological Profile of Multitarget Mitochondriotropic Antioxidants for Neurodegenerative Diseases. Antioxidants. 2021; 10(2):329. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020329
Chicago/Turabian StyleChavarria, Daniel, Ophelie Da Silva, Sofia Benfeito, Sandra Barreiro, Jorge Garrido, Fernando Cagide, Pedro Soares, Fernando Remião, Xavier Brazzolotto, Florian Nachon, and et al. 2021. "Fine-Tuning the Biological Profile of Multitarget Mitochondriotropic Antioxidants for Neurodegenerative Diseases" Antioxidants 10, no. 2: 329. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020329