
Signal Transducer and Activator of Transcription 3 (STAT3) Suppresses STAT1/Interferon Signaling Pathway and Inflammation in Senescent Preadipocytes

 , ,
, ,  , ,
, ,  and
and
Abstract
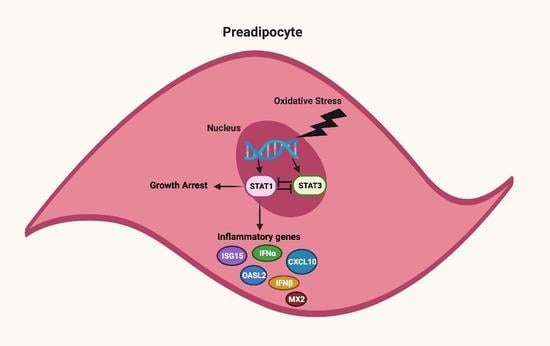
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. The 3T3-L1 Cell Culture and Senescence Induction Protocol
2.2. Mouse Experiments
2.3. Experiments on Human Adipose Tissue Samples
2.4. Senescence Associated β-Galactosidase (SA-β-Gal) Staining
2.5. Growth Curve
2.6. Western Blotting
2.7. Quantitative Polymerase Chain Reaction (qPCR)
2.8. Adipocyte Differentiation and Oil Red O Staining
2.9. Transcriptomics and Ingenuity Pathway Analysis (IPA)
2.10. Dot Blotting
2.11. Generation of Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR Associated Protein 9 (CRISPR/Cas9) Lentiviruses
2.12. Data Analysis and Statistics
3. Results
3.1. Induced Senescence in 3T3-L1 Preadipocytes with Repeated H2O2 Treatment Impairs Adipogenesis
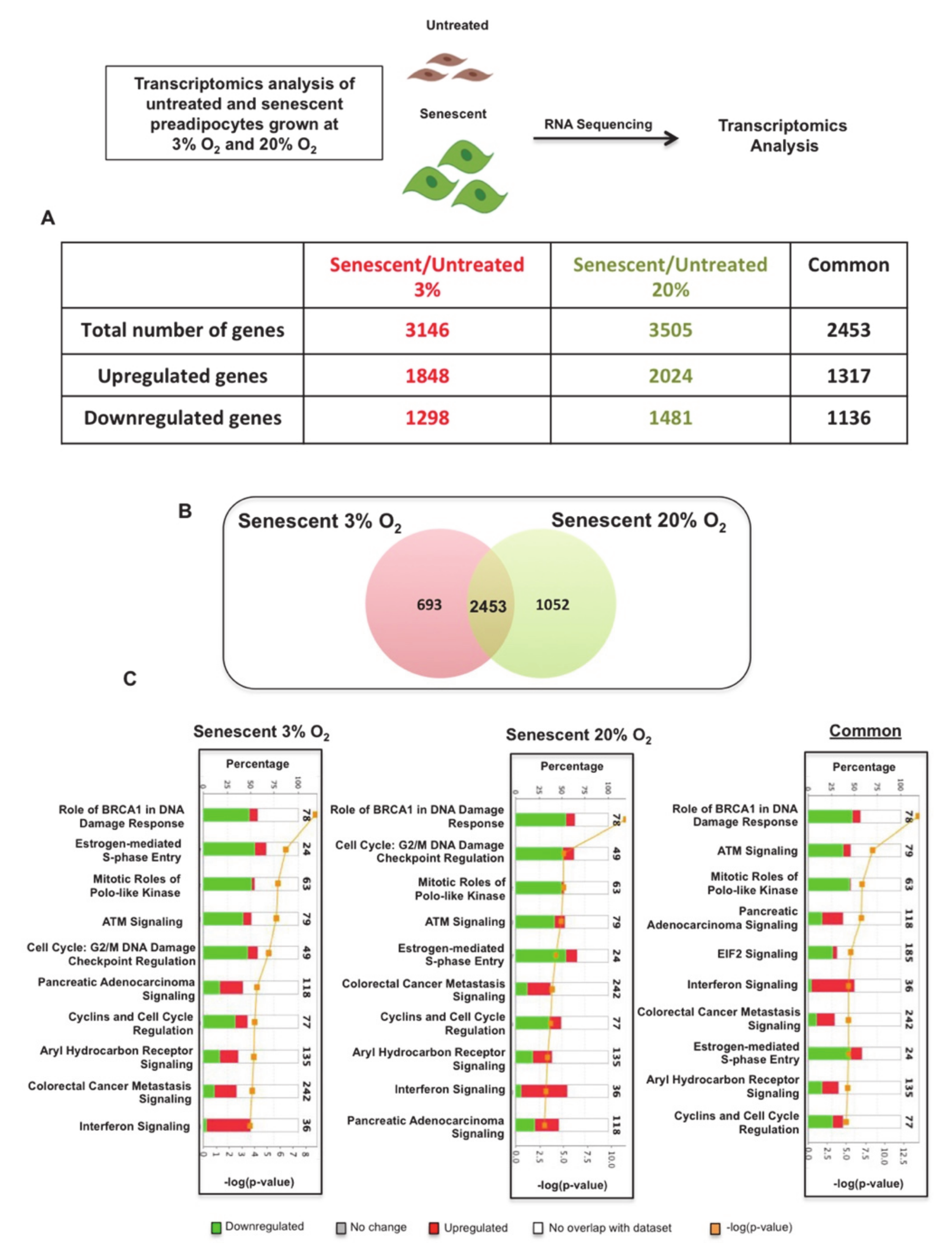
3.2. Transcriptomics and Enriched Pathway Analysis Uncover Critical Activated Pro-Inflammatory Pathways in Senescent Preadipocytes
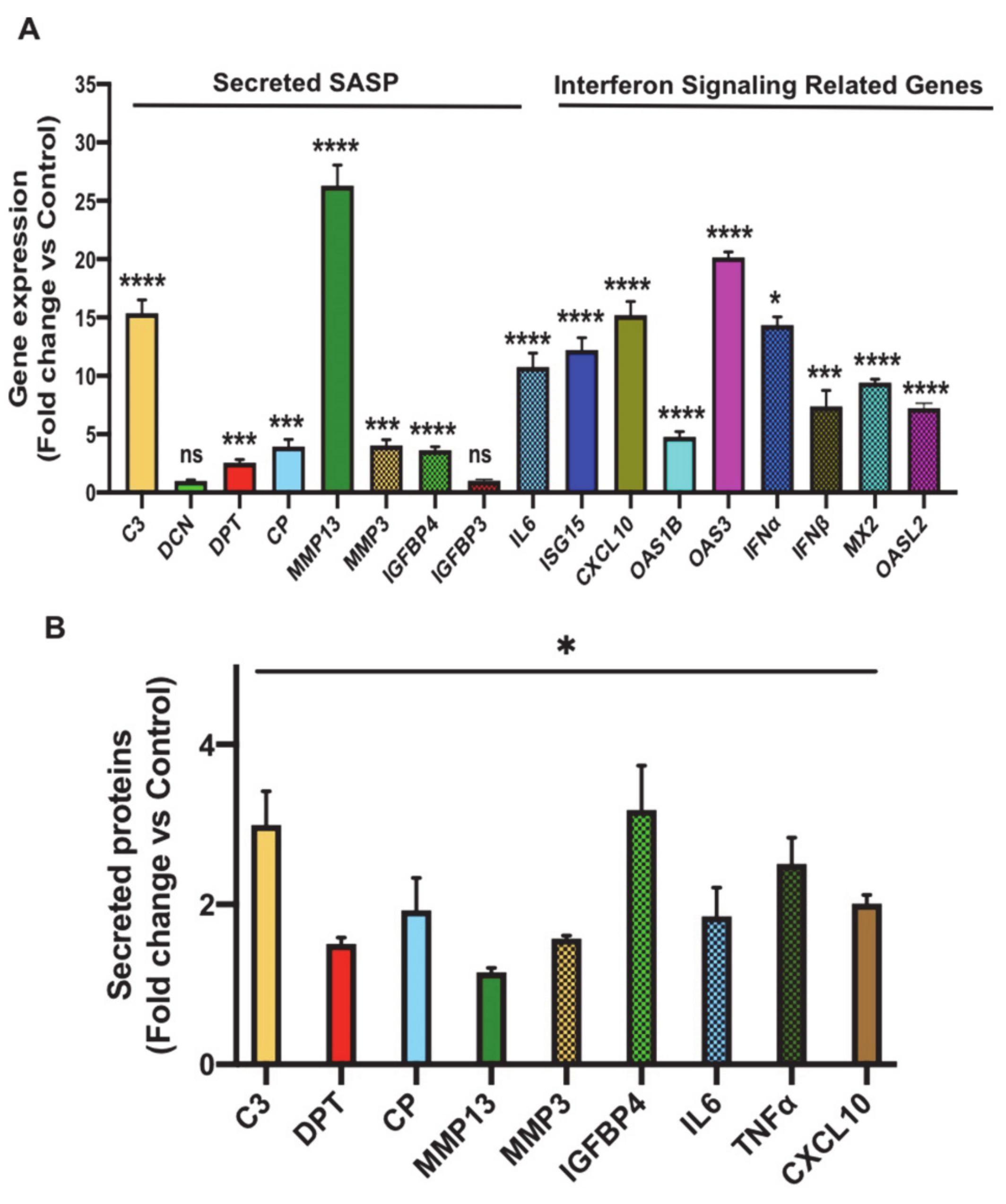
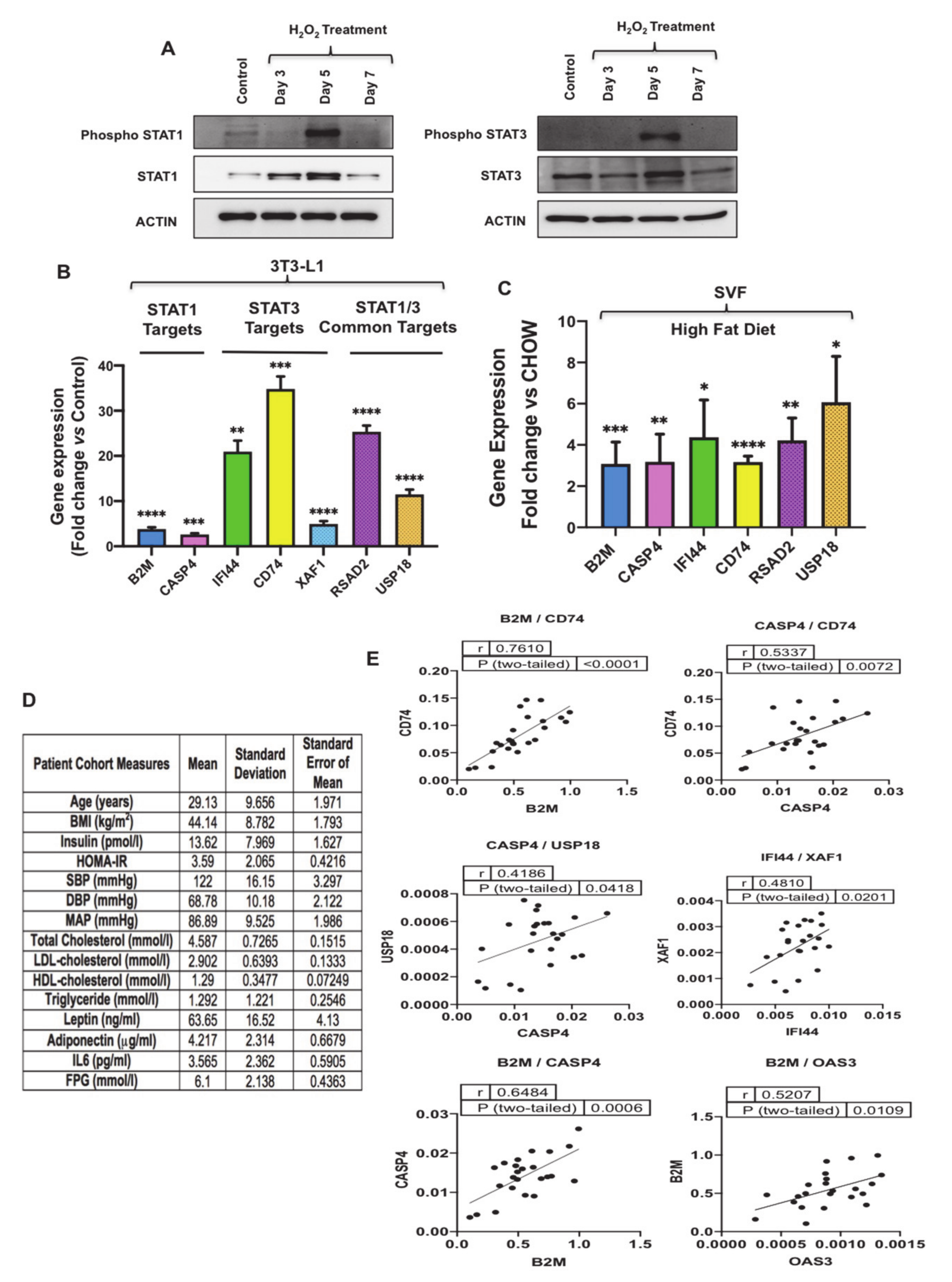
3.3. Oxidative Stress Promotes Senescence-Associated Secretory Phenotype (SASP) Secretion, Interferon Signaling and Signal Transducer and Activator of Transcription 1 (STAT1) Expression in Senescent 3T3-L1 Preadipocytes
3.4. Oxidative Stress Activates STAT1 and STAT3 and Promotes Induction of Their Targets
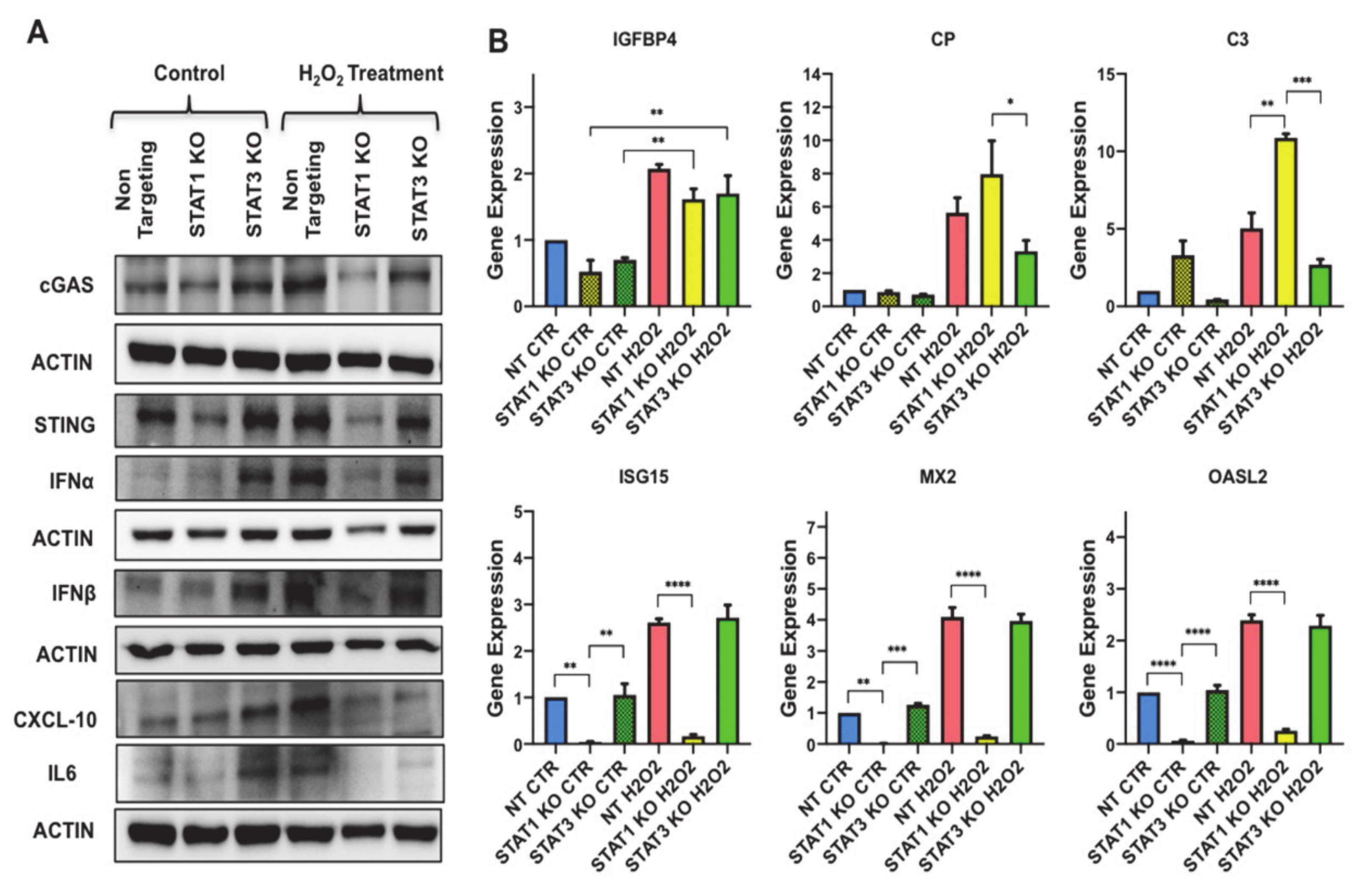
3.5. GMP-AMP Synthase-Stimulator of Interferon Genes (cGAS-STING) Pathway Regulates C-X-C Motif Chemokine Ligand 10 (CXCL10) and Type I Interferon Effector Molecules in Preadipocytes
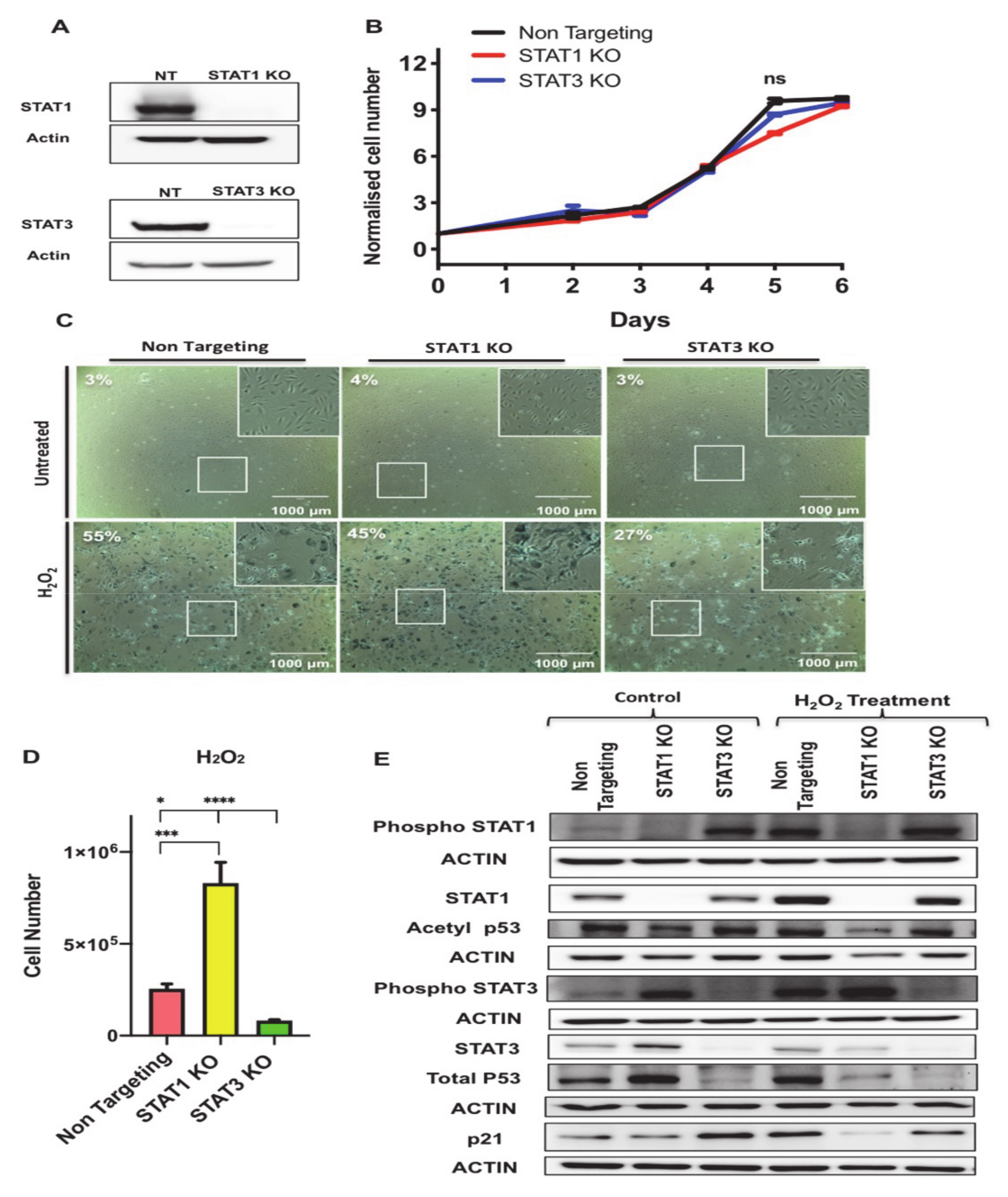
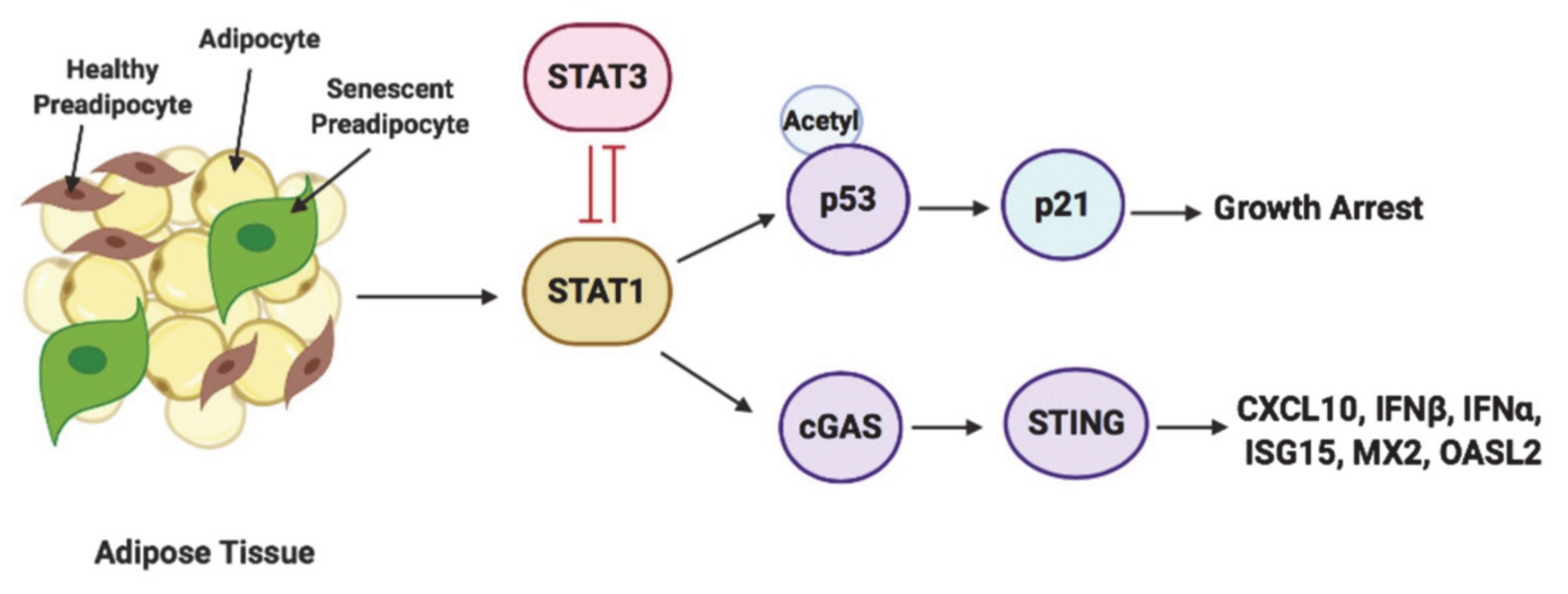
3.6. STAT1 and STAT3 Exhibit Opposing Functions in Regulating Senescence-Associated Growth Arrest Phenotype
3.7. STAT3 Aantagonizes STAT1 Regulation of STING and Downstream Molecules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, R.M.; Baker, D.J.; Van Deursen, J.M. Senescent Cells: A Novel Therapeutic Target for Aging and Age-Related Diseases. Clin. Pharmacol. Ther. 2013, 93, 105–116. [Google Scholar] [CrossRef]
- McGill, M.; Blonde, L.; Chan, J.C.; Khunti, K.; LaValle, F.J.; Bailey, C.J. The interdisciplinary team in type 2 diabetes management: Challenges and best practice solutions from real-world scenarios. J. Clin. Transl. Endocrinol. 2017, 7, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahima, R.S. Connecting obesity, aging and diabetes. Nat. Med. 2009, 15, 996–997. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, A.R.; Coppe, J.-P.; Campisi, J.; Desprez, P.-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Tagele, B.M.; Cheema, P.; Madan, A.K.; Tichansky, D.S. Release of 12 Adipokines by Adipose Tissue, Nonfat Cells, and Fat Cells From Obese Women. Obesity 2010, 18, 890–896. [Google Scholar] [CrossRef]
- Wu, D.; Ren, Z.; Pae, M.; Guo, W.; Cui, X.; Merrill, A.H.; Meydani, S.N. Aging Up-Regulates Expression of Inflammatory Mediators in Mouse Adipose Tissue. J. Immunol. 2007, 179, 4829–4839. [Google Scholar] [CrossRef] [Green Version]
- Charrière, G.; Cousin, B.; Arnaud, E.; André, M.; Bacou, F.; Pénicaud, L.; Casteilla, L. Preadipocyte Conversion to Macrophage. J. Biol. Chem. 2003, 278, 9850–9855. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; LaPoint, K.; Martinez, K.; Kennedy, A.; Sandberg, M.B.; McIntosh, M.K. Preadipocytes Mediate Lipopolysaccharide-Induced Inflammation and Insulin Resistance in Primary Cultures of Newly Differentiated Human Adipocytes. Endocrinology 2006, 147, 5340–5351. [Google Scholar] [CrossRef]
- Vitseva, O.I.; Tanriverdi, K.; Tchkonia, T.T.; Kirkland, J.L.; McDonnell, M.E.; Apovian, C.M.; Freedman, J.; Gokce, N. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity 2008, 16, 932–937. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Childs, B.G.; Khazaie, K.; Miller, J.D.; Van Deursen, J.M.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; Van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdesselem, H.; Madani, A.; Hani, A.; Alnoubi, M.N.; Goswami, N.; Ben Hamidane, H.; Billing, A.M.; Pasquier, J.; Bonkowski, M.S.; Halabi, N.M.; et al. SIRT1 Limits Adipocyte Hyperplasia through c-Myc Inhibition. J. Biol. Chem. 2016, 291, 2119–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almuraikhy, S.; Kafienah, W.; Elrayess, M.A.; Bashah, M.; Diboun, I.; Jaganjac, M.; Al-Khelaifi, F.; Abdesselem, H.; Mazloum, N.A.; Alsayrafi, M.; et al. Interleukin-6 induces impairment in human subcutaneous adipogenesis in obesity-associated insulin resistance. Diabetologia 2016, 59, 2406–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudge, J.M.; Harrow, J. Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm. Genome 2015, 26, 366–378. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, H.; Meuth, M. An established pre-adipose cell line and its differentiation in culture. Cell 1974, 3, 127–133. [Google Scholar] [CrossRef]
- Morrison, S.; McGee, S.L. 3T3-L1 adipocytes display phenotypic characteristics of multiple adipocyte lineages. Adipocyte 2015, 4, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef]
- Su, L.; David, M. Distinct mechanisms of STAT phosphorylation via the interferon-alpha/beta receptor. Selective inhibition of STAT3 and STAT5 by piceatannol. J. Biol. Chem. 2000, 275, 12661–12666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Hald, A.; Andrés, R.; Salskov-Iversen, M.; Kjellerup, R.; Iversen, L.; Johansen, C. STAT1 expression and activation is increased in lesional psoriatic skin. Br. J. Dermatol. 2013, 168, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pan, X.; Mao, L.; Pan, H.; Xu, W.; Hu, Y.; Yu, X.; Chen, Z.; Qian, S.; Ye, Y.; et al. Phospho-Tyr705 of STAT3 is a therapeutic target for sepsis through regulating inflammation and coagulation. Cell Commun. Signal. 2020, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-Y.; Chiu, C.-J.; Kuan, C.-H.; Chen, F.-H.; Shen, Y.-C.; Wu, C.-H.; Hsu, Y.-H. IL-29 promoted obesity-induced inflammation and insulin resistance. Cell. Mol. Immunol. 2019, 17, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Vergoni, B.; Cornejo, P.-J.; Gilleron, J.; Djedaini, M.; Ceppo, F.; Jacquel, A.; Bouget, G.; Ginet, C.; Gonzalez, T.; Maillet, J.; et al. DNA Damage and the Activation of the p53 Pathway Mediate Alterations in Metabolic and Secretory Functions of Adipocytes. Diabetes 2016, 65, 3062–3074. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity 2015, 23, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Lee, K.-H.; Lee, Y.-S.; Mun, E.-G.; Kwon, D.-Y.; Cha, Y.-S. Transcriptome analysis and promoter sequence studies on early adipogenesis in 3T3-L1 cells. Nutr. Res. Pract. 2007, 1, 19–28. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.E.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Karkhaneh, M.; Qorbani, M.; Mohajeri-Tehrani, M.R.; Hoseini, S. Association of serum complement C3 with metabolic syndrome components in normal weight obese women. J. Diabetes Metab. Disord. 2017, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.Y.; Shin, M.-J.; Moon, J.; Chung, J.H. Plasma ceruloplasmin as a biomarker for obesity: A proteomic approach. Clin. Biochem. 2011, 44, 351–356. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, A.J.; Stephens, J.M. The role of JAK–STAT signaling in adipose tissue function. Biochim. Biophys. Acta 2014, 1842, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, H.; Inoue, T.; Kunimoto, H.; Nakajima, K. IL-6-STAT3 signaling and premature senescence. JAK-STAT 2013, 2, e25763. [Google Scholar] [CrossRef]
- MacKenzie, K.J.; Carroll, P.; Osborn, R.T.; Wheeler, A.P.; Nowotny, M.; Gilbert, N.; Chandra, T.; Reijns, M.A.M.; Jackson, A.P.; Martin, C.-A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, C.; Mohr, I. Restriction of Human Cytomegalovirus Replication by ISG15, a Host Effector Regulated by cGAS-STING Double-Stranded-DNA Sensing. J. Virol. 2017, 91, e02483-16. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Yang, D.; Wang, B.; Wu, C.; Wu, Y.; Li, S.; Liu, X.; Lassen, K.; Dai, L.; Yang, S. Gasdermin D in macrophages restrains colitis by controlling cGAS-mediated inflammation. Sci. Adv. 2020, 6, eaaz6717. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Shao, L.; DeLuca, N.A.; Thorne, S.H.; Rathinam, V.A.; Li, P.; Sarkar, S.N.; Sampath, P.; Zhao, B.; Patel, N.V.; et al. Oligoadenylate-Synthetase-Family Protein OASL Inhibits Activity of the DNA Sensor cGAS during DNA Virus Infection to Limit Interferon Production. Immunity 2019, 50, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Motani, K.; Ito, S.; Nagata, S. DNA-Mediated Cyclic GMP-AMP Synthase-Dependent and -Independent Regulation of Innate Immune Responses. J. Immunol. 2015, 194, 4914–4923. [Google Scholar] [CrossRef] [Green Version]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFalpha-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [Green Version]
- DeMaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [Green Version]
- Dimco, G.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010, 9, 4638–4649. [Google Scholar] [CrossRef] [Green Version]
- Ishii, K.J.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A Toll-like receptor–independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2005, 7, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; You, M.; Ding, L.; Fan, H.; Liu, F.; Ren, D.; Hou, Y. STING Negatively Regulates Double-Stranded DNA-Activated JAK1-STAT1 Signaling via SHP-1/2 in B Cells. Mol. Cells 2015, 38, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, H.-C.; Hutti, J.E.; Baldwin, A.S. Cytosolic DNA Promotes Signal Transducer and Activator of Transcription 3 (STAT3) Phosphorylation by TANK-binding Kinase 1 (TBK1) to Restrain STAT3 Activity. J. Biol. Chem. 2017, 292, 5405–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.-B.; Levy, D.E.; Lee, C.-K. STAT3 Negatively Regulates Type I IFN-Mediated Antiviral Response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef]
- Yue, C.; Shen, S.; Deng, J.; Priceman, S.J.; Li, W.; Huang, A.; Yu, H. STAT3 in CD8+ T Cells Inhibits Their Tumor Accumulation by Downregulating CXCR3/CXCL10 Axis. Cancer Immunol. Res. 2015, 3, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, J.; Wang, T.C. Inflammation and Cancer: IL-6 and STAT3 Complete the Link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madani, A.Y.; Majeed, Y.; Abdesselem, H.B.; Agha, M.V.; Vakayil, M.; Sukhun, N.K.A.; Halabi, N.M.; Kumar, P.; Hayat, S.; Elrayess, M.A.; et al. Signal Transducer and Activator of Transcription 3 (STAT3) Suppresses STAT1/Interferon Signaling Pathway and Inflammation in Senescent Preadipocytes. Antioxidants 2021, 10, 334. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020334
Madani AY, Majeed Y, Abdesselem HB, Agha MV, Vakayil M, Sukhun NKA, Halabi NM, Kumar P, Hayat S, Elrayess MA, et al. Signal Transducer and Activator of Transcription 3 (STAT3) Suppresses STAT1/Interferon Signaling Pathway and Inflammation in Senescent Preadipocytes. Antioxidants. 2021; 10(2):334. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020334
Chicago/Turabian StyleMadani, Aisha Y., Yasser Majeed, Houari B. Abdesselem, Maha V. Agha, Muneera Vakayil, Nour K. Al Sukhun, Najeeb M. Halabi, Pankaj Kumar, Shahina Hayat, Mohamed A. Elrayess, and et al. 2021. "Signal Transducer and Activator of Transcription 3 (STAT3) Suppresses STAT1/Interferon Signaling Pathway and Inflammation in Senescent Preadipocytes" Antioxidants 10, no. 2: 334. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10020334