
Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development

 , ,
, ,  and
and
Abstract
:1. Introduction
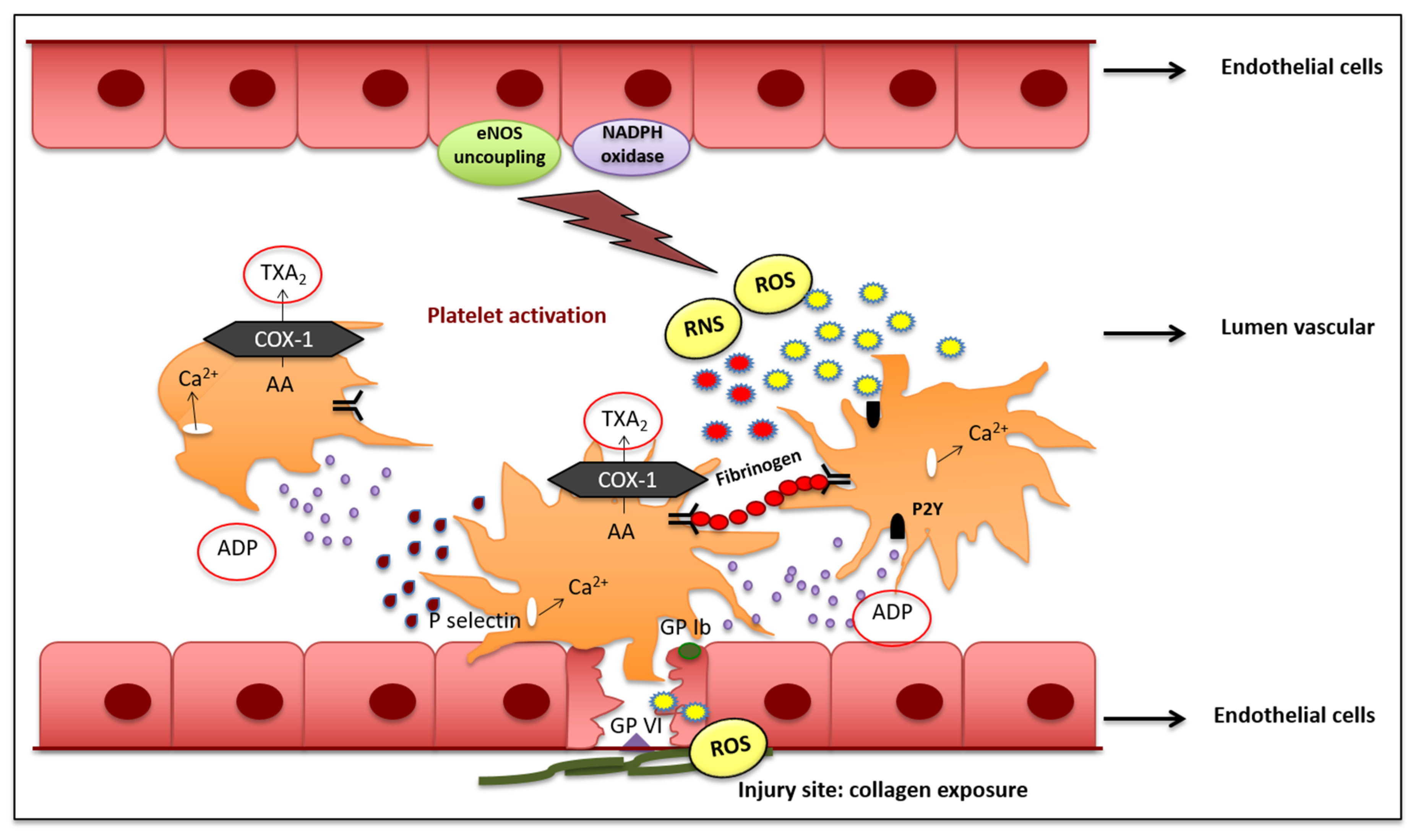
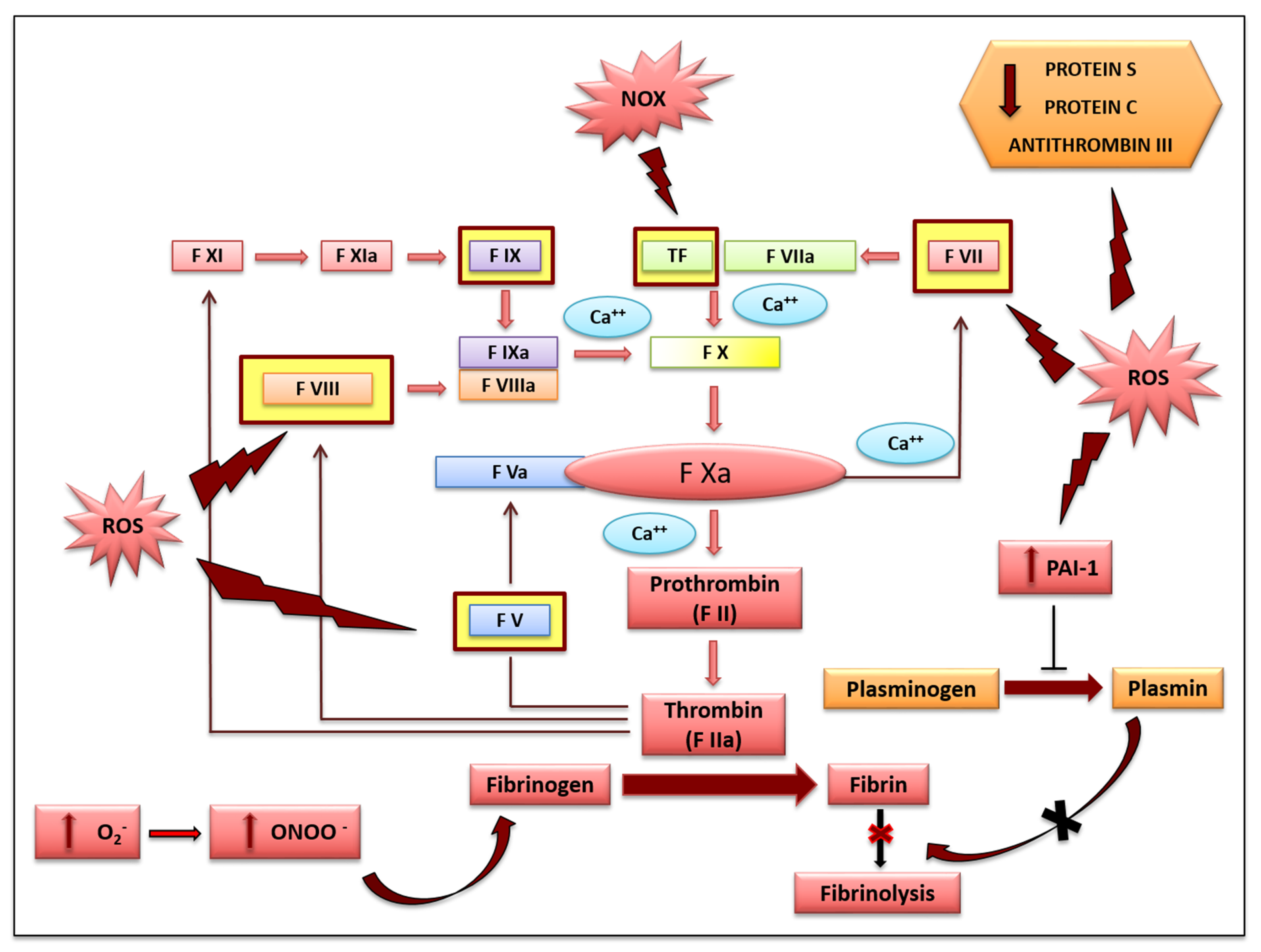
2. Role of Oxidative Stress in Vascular Haemostasis
2.1. Primary Haemostasis and Platelet Activation
2.2. Secondary Haemostasis and Clotting Formation
2.3. Fibrinolysis
3. ROS and Endothelial Dysfunction
4. Autophagy in Endothelial Cell Biology
5. Crosstalk between Oxidative Stress and Autophagy
6. The Critical Role of Autophagy in the Development of Atherosclerosis: Focus on Endothelial Cells
6.1. The Modulation of EC Autophagy in the Onset and Development of Atherothrombosis
6.2. Crosstalk between Non-Coding RNAs and Autophagy in the Atherosclerotic Process

{kind=link}
{kind=link}
{kind=link}
| In Vitro/In Vivo Model | Results | References |
|---|---|---|
| - shAtg7 HUVECs treated with 50 μg mL−1 oxLDLs - Atg7endo mice (fl/fl;VE-Cadherin Cre) mice - HFD-fed apoE−/−Atg7endo mice | ↓ LC3-II/ LC3I, ↑ oxLDLs ↑ intracellular 125I-LDL chol ↑ transcytosis intracellular 125I-LDL chol ↑ dil-oxLDLs in RPE and choroidal endothelium ↑ atherosclerotic lesion size ↑ necrotic area | [136] |
| - HUVECs treated with 50 mg/mL oxLDLs - ApoE−/− mice | ↓ mTOR, ↑ Atg13, ↓p-Atg13 ↑ apoptosis of endothelial cells ↑ atherosclerotic lesion areas ↑ instability of plague, ↑ lipid deposition, ↑SMCs ↑ macrophage cell area ↑ MMP-2 MMP-9 activity ↑ ICAM-1, VCAM-1; IL-6, IL-8 | [141] |
| - HFD-fed apoE−/−Atg5flox/flox mice - HFD-fed apoE−/−Atg5flox/flox; VE-cadherin-cre mice - Atg5flox/flox;VE-cadherin-cre - Atg7flox/flox;VE-cadherin-cre mice - wortmannin-inhibited autophagy in HUVECs exposed to high SS - sh-Atg5 lentivirus in HUVECs exposed to high SS - HFD-fed Atg5 or Atg7-deficient mice | ↑ plague formation, ↑ plague size in atheroresistant regions ↑ serum glucose levels - disturbed endothelial alignment in response to flow ↓ KLF2, ↑ ICAM-1, and MCP-1 ↑; p53, ↑ p16, ↑ SA-β-gal | [143,150] |
| - HUVECs treated with 20–40 μg/mL oxLDLs - LDLR knockout mice | ↑ Beclin-1, ↑LC3-II/ LC3I, ↑ LOX-1 ↑ mtDNA damage, ↑ TLR9, ↑ CD45 and CD68 | [147] |
| - BAECs treated with 100 μM oxLDLs | ↓ Beclin-1, LC3-II/LC3I, ↑ LOX-1 ↑ MDA, ↑apoptosis | [148] |
| - young HUVECs or HAECs transfected with adenovirus-mediated overexpression of ARG2 or inactive ARG2 (H160F) - HFD-fed apoE−/−Arg2+/+ mice - HFD-fed apoE−/−Arg2−/− mice | ↓ LC3II/LC3I, Atg12–Atg5 conjugate, ↑ p62 ↑ SA-β-gal ↓ PRKAA-T172/PRKAA, ↑ RICTOR ↑ AKT-S473/AKT, RPS6-S235/236:RPS6 ↑ TP53-S15/TP53 | [149] |
| - HUVECs treated with 100 μg/mL oxLDLs | ↓ miRNA-126, ↓ cell viability ↑ caspase-3 activity, ↑ apoptosis rate, ↑ LDH release - defective autophagic flux and degradation ↑ Beclin-1, ↑ LC3-II/LC3I, ↑ p62, ↑ p-mTOR/mTOR ↑ p-PI3K p85/ PI3K p85, ↑ p-Akt/Akt | [152] |
| - young HUVECs transfected with miRNA216a - HAECs transfected with miRNA216a - HCAECs transfected with miRNA216a - young HUVECs transfected with iRNA216a and treated with 100 μg/mL oxLDLs | ↓ Beclin-1 and Atg5 mRNA and protein levels ↓ LC3II/LC3I, ↑ oxLDL accumulation ↑ monocyte adhesion | [153] |
| - CD31+ aortic endothelial cells derived from HFD-fed apoE−/− mice - young HUVECs transfected with miRNA214-3p and treated with 100 μg/mL oxLDLs | ↓ Arg5 and Atg12, ↑ p62, ↓ LC3II ↑ oxLDL accumulation, ↑ monocyte adhesion ↓ cell viability | [154] |
| - HUVECs treated with 25, 50, or 100 μg/L oxLDLs - HUVECs transfected with si-LncRNAFA2H-2 and treated with 25, 50, or 100 μg/L oxLDLs - HFD-fed apoE−/− mice infected with si-LncRNAFA2H-2 | ↑ VCAM-1, MCP-1, TNF-α ↑ IL-6, IL-1 β, IL-18, IL-8 ↓ IL-10 - impaired autophagic degradation ↑ LC3-II/LC3I, p62, LAMP1 ↓ cathepsin D ↓ LncRNAFA2H-2, ↑ MLKL ↓ p-PI3K, p-Akt, p-mTOR, ↑ AMPK ↑ lesion areas, proliferative and disarranged intima ↑ lipid-laden foam cells ↑ cholesterol crystals | [155] |
6.3. Macrophage Autophagy in Atherosclerosis
6.4. The Link between Autophagy and Ferroptosis in Atherothrombosis
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Chegodaev, Y.S.; Wu, W.K.; Orekhov, A.N. Oxidative Stress and Antioxidants in Atherosclerosis Development and Treatment. Biology 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and Oxidative Stress: Mechanisms and Management in Elderly. Antioxid. Redox Signal. 2017, 27, 1083–1124. [Google Scholar] [CrossRef]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, D. The LDL modification hypothesis of atherogenesis: An update. J. Lipid Res. 2009, S376–S381. [Google Scholar] [CrossRef] [Green Version]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.J.; Castillo, A.I.; Zaragoza, C.; Ibáñez, B.; Andrés, V. Animal models of atherosclerosis. Prog. Mol. Biol. Transl. Sci. Elsevier 2012, 105, 1–23. [Google Scholar] [CrossRef]
- Davi, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Perrotta, I.; Aquila, S. The role of oxidative stress and autophagy in atherosclerosis. Oxid. Med. Cell. Longev. 2015, 2015, 130315. [Google Scholar] [CrossRef] [Green Version]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e41–e52. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: The central role of LDL and oxidative burst. Endocr. Metab. Immun. Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef]
- Farbstein, D.; Soloveichik, Y.Z.; Levy, N.S.; Levy, A.P. Genetics of redox systems and their relationship with cardiovascular disease. Curr. Atheroscler. Rep. 2011, 13, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS. thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective Autophagy in Parkinson’s Disease: Role of Oxidative Stress. Mol. Neurobiol. 2012, 46, 639–661. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Sachdev, U.; Lotze, M.T. Perpetual change: Autophagy, the endothelium, and response to vascular injury. J. Leukoc Biol. 2017, 102, 221–235. [Google Scholar] [CrossRef]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The Role of Autophagy in Vascular Biology Samuel C. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Kirshenbaum, L.A. Regulation of autophagy in the heart in health and disease. J. Cardiovasc. Pharmacol. 2012, 60, 109. [Google Scholar] [CrossRef]
- Schrijvers, D.M.; de Meyer, G.R.Y.; Martinet, W. Autophagy in atherosclerosis: A potential drug target for plaque stabilization. Arterioscler Thromb. Vasc Biol. 2011, 31, 2787–2791. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelialdysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Franchini, M.; Targher, G. Arterial thrombus formation in cardiovascular disease. Nat. Rev. Cardiol. 2011, 8, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, L.A.; E Albuquerque, R.P.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Clemetson, K.J. Platelets and primary haemostasis. Thromb. Res. 2012, 129, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of Platelet Activation and Coagulation andIts Role in Vascular Injury and Arterial Thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vilahur, G.; Gutiérrez, M.; Arzanauskaite, M.; Mendieta, G.; Ben-Aicha, S.; Badimon, L. Intracellular platelet signalling as a target for drug development. Vascul Pharmacol. 2018, 111, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Del Principe, D.A.; Menichelli, W.; De Matteis, S.; Di Giulio, M.; Giordani, I.; Savini, A.F. Hydrogen peroxide is an intermediate in the platelet activation cascade triggered by collagen, but not by thrombin. Agro Thromb. Res. 1991, 62, 365–375. [Google Scholar] [CrossRef]
- Senis, Y.A. Protein-tyrosine phosphatases: A new frontier in platelet signal transduction. J. Thromb. Haemost. 2013, 11, 1800–1813. [Google Scholar] [CrossRef]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Wang, S.B.; Park, S.J.; Oh, G.T.; Lee, S.H.; Ho, Y.S.; Chang, T.S. Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Franchini, M.; Lippi, G. Aging hemostasis: Changes to laboratory markers of hemostasis as we age—A narrative review. Semin. Thromb. Hemost. 2014, 40, 621–633. [Google Scholar] [CrossRef]
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancie, H. Poly-morphonuclear leukocytes modulate tissue factor production by mononuclear cells: Role of reactive oxygen species. J. Immunol. 2000, 164, 3822–3828. [Google Scholar] [CrossRef] [Green Version]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Gorlach, A. NADPH oxidase mediates tissue factor- dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier:Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, G.; Cirillo, P.; Ragni, M.; Conte, S.; Uccello, G.; Golino, P. Reactive oxygen species induce a procoagulant state in endothelial cells by inhibiting tissue factor pathway inhibitor. J. Thromb. Thrombolysis 2015, 40, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. [Google Scholar] [CrossRef] [Green Version]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015, 29, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Sandrini, L.; Ieraci, A.; Amadio, P.; Zarà, M.; Barbieri, S.S. Impact of Acute and Chronic Stress on Thrombosis in Healthy Individuals and Cardiovascular Disease Patients. Int. J. Mol. Sci. 2020, 21, 7818. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowska, M.; Szemraj, J.; Al-Nedawi, K.N.; Pawlowska, Z. Reactive oxygen species upregulate expression of PAI-1 in endothelial cells. Cell Mol. Biol. Lett. 2002, 7, 1065–1071. [Google Scholar] [PubMed]
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thromb. Res. 2009, 124, 439–446. [Google Scholar] [CrossRef]
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH oxidase in oxidized LDL-induced upregulation of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E104–E111. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. J. Mol. Cell Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol. Biol. Cell. 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Aljofan, M.; Triggle, C.R. Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells. J. Cell Physiol. 2007, 212, 682–689. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Faraci, F.M. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol. Med. 2008, 14, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, G.; Rizza, S.; Montagna, C.; Filomeni, G. Established principles and emerging concepts on the interplay between mitochondrial physiology and S-(De)nitrosylation: Implications in cancer and neurodegeneration. Int. J. Cell Biol. 2012, 2012, 361872. [Google Scholar] [CrossRef] [Green Version]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, C.; Marco Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid β peptide induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflamm. 2015, 12–18, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of nitric oxide synthases by oxyLDL: Role in vascular inflammation and atherosclerosis development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [Green Version]
- Tassone, E.J.; Perticone, M.; Sciacqua, A.; Mafrici, S.F.; Settino, C.; Malara, N.; Mollace, V.; Sesti, G.; Perticone, F. Low dose of acetylsalicylic acid and oxidative stress-mediated endothelial dysfunction in diabetes: A short-term evaluation. Acta Diabetol. 2015, 52, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Łuczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathwayin the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ding, Y.; Ramprasath, T.; Zou, M.H. Oxidative Stress, GTPCH1, and Endothelial Nitric Oxide Synthase Uncoupling in Hypertension. Antioxid. Redox Signal. 2020. [Google Scholar] [CrossRef]
- Cosentino, F.; Barker, J.E.; Brand, M.P.; Heales, S.J.; Werner, E.R.; Tippins, J.R.; West, N.; Channon, K.M.; Volpe, M.; Lüscher, T.F. Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhanam, A.V.; d’Uscio, L.V.; Smith, L.A.; Katusic, Z.S. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J. Neurochem. 2012, 122, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A Lysosome-Dependent Process with Implications in Cellular Redox Homeostasis and Human Disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Hughes, W.E.; Beyer, A.M.; Gutterman, D.D. Vascular autophagy in health and disease. Basic Res. Cardiol. 2020, 115, 41. [Google Scholar] [CrossRef] [PubMed]
- King, J.S.; Veltman, D.M.; Insall, R.H. The induction of autophagy by mechanical stress. Autophagy 2011, 7, 1490–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2016, 219, 382–408. [Google Scholar] [CrossRef]
- Bharath, L.P.; Mueller, R.; Li, Y.; Ruan, T.; Kunz, D.; Goodrich, R.; Mills, T.; Deeter, L.; Sargsyan, A.; Anandh Babu, P.V.; et al. Impairment of autophagy in endothelial cells prevents shear-stress-induced increases in nitric oxide bioavailability. Can J. Physiol. Pharmacol. 2014, 92, 605–612. [Google Scholar] [CrossRef]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef]
- Tian, J.; Popal, M.S.; Huang, R.; Zhang, M.; Zhao, X.; Zhang, M.; Song, X. Caveolin as a Novel Potential Therapeutic Target in Cardiac and Vascular Diseases: A Mini Review. Aging Dis. 2020, 11, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Kalamida, D.; Karagounis, I.V.; Giatromanolaki, A.; Koukourakis, M.I. Important Role of Autophagy in Endothelial Cell Response to Ionizing Radiation. PLoS ONE 2014, 9, e102408. [Google Scholar] [CrossRef]
- Chen, F.; Chen, B.; Xiao, F.Q.; Wu, Y.T.; Wang, R.H.; Sun, Z.W.; Fu, G.S.; Mou, Y.; Tao, W.; Hu, X.S.; et al. Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high- glucose-induced endothelial cells. Cell Physiol. Biochem. 2014, 33, 1058–1074. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Flint, N.; Feng, B.; Bretón-Romero, R.; Linder, E.A.; Berk, B.D.; Duess, M.A.; Farb, M.G.; Gokce, N.; et al. Restoration of Autophagy in Endothelial Cells from Patients with Diabetes Mellitus Improves Nitric Oxide Signaling. Atherosclerosis 2016, 247, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezabakhsh, A.; Ahmadi, M.; Khaksar, M.; Montaseri, A.; Malekinejad, H.; Rahbarghazi, R.; Garjani, A. Rapamycin inhibits oxidative/nitrosative stress and enhances angiogenesis in high glucose-treated human umbilical vein endothelial cells: Role of autophagy. Biomed. Pharmacother. 2017, 93, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; He, Y.; Hao, S.; Wang, B.; Xu, M.; Qi, H.; Liu, S.; Du, Y.; Yu, X. Vps15 is critical to mediate autophagy in AngII treated HUVECs probably by PDK1/PKC signaling pathway. Life Sci. 2019, 233, 116701. [Google Scholar] [CrossRef] [PubMed]
- Rezabakhsh, A.; Rahbarghazi, R.; Malekinejad, H.; Fathi, F.; Montaseri, A.; Garjani, A. Quercetin alleviates high glucose-induced damage on human umbilical vein endothelial cells by promoting autophagy. Phytomedicine 2019, 56, 183–193. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, J.; Zhou, M.; Zhang, Y.; Liu, Y.; Hou, P.; Zeng, X.; Yi, L.; Mi, M. Resveratrol attenuates endothelial oxidative injury by inducing autophagy via the activation of transcription factor EB. Nutr. Metab. (Lond) 2019, 16, 42. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef] [Green Version]
- Neill, T.; Buraschi, S.; Kapoor, A.; Iozzo, R.V. Proteoglycan-driven Autophagy: A Nutrient-independent Mechanism to Control Intracellular Catabolism. J. Histochem. Cytochem. 2020, 68, 733–746. [Google Scholar] [CrossRef]
- Du, J.; Teng, R.J.; Guan, T.; Eis, A.; Kaul, S.; Konduri, G.G.; Shi, Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C383–C391. [Google Scholar] [CrossRef] [Green Version]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell. 2017, 16, 17–26. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; Agostinis, P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ. 2019, 26, 665–679. [Google Scholar] [CrossRef] [Green Version]
- Torisu, T.; Torisu, K.; Lee, I.H.; Liu, J.; Malide, D.; Combs, C.A.; Wu, X.S.; Rovira, I.I.; Fergusson, M.M.; Weigert, R.; et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 2013, 19, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Singh, K.K.; Hou, Y.; Lei, X.; Ramadan, A.; Quan, A.; Teoh, H.; Kuebler, W.M.; Al-Omran, M.; Yanagawa, B.; et al. Endothelial-specific deletion of autophagy-related 7 (ATG7) attenuates arterial thrombosis in mice. J. Thorac. Cardiovasc Surg. 2017, 154, 978–988.e1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sequeira, R.C.; Zabalawi, M.; Madenspacher, J.; Boudyguina, E.; Ou, T.; Nelson, J.M.; Nie, Y.; Zhao, Q.; Fessler, M.B.; et al. Myeloid atg5 deletion impairs n-3 PUFA-mediated atheroprotection. Atherosclerosis 2020, 295, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Q.; Jeon, S.H.; Bae, W.J.; Choi, S.W.; Jeong, H.C.; Kim, K.S.; Kim, S.J.; Cho, H.J.; Ha, U.; Hong, S.H. Efficient promotion of autophagy and angiogenesis using mesenchymal stem cell therapy enhanced by the low-energy shock waves in the treatment of erectile dysfunction. Stem Cells Int. 2018, 2018, 1302672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, J.; Wang, K.; Zhelyabovska, O.; Saad, Y.; Kolattukudy, P.E. MCP-1-induced protein promotes endothelial-like and angiogenic properties in human bone marrow monocytic cells. J. Pharmacol. Exp. Ther. 2013, 347, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, Z.S.; Cao, X.N.; Wen, Q.; Mo, X.D.; Zhao, H.Y.; Chen, Y.H.; Wang, Y.; Chang, Y.J.; Xu, L.P.; Zhang, X.H.; et al. Autophagy in endothelial cells regulates their haematopoiesis-supporting ability. EBioMedicine 2020, 53, 102677. [Google Scholar] [CrossRef]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [Green Version]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, L.; Wang, X.B.; Wang, J.S.; Geng, Y.D.; Yang, C.S.; Kong, L.y. Calyxin Y induces hydrogen peroxide-dependent autophagy and apoptosis via JNK activation in human nonsmall cell lung cancer NCI-H460 cells. Cancer Lett. 2013, 340, 51–62. [Google Scholar] [CrossRef]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71C, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Portal-Núñez, S.; Esbrit, P.; Alcaraz, M.J.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2016, 108, 1–10. [Google Scholar] [CrossRef]
- Shiomi, M.; Miyamae, M.; Takemura, G.; Kaneda, K.; Inamura, Y.; Onishi, A.; Koshinuma, S.; Momota, Y.; Minami, T.; Figueredo, V.M. Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J. Anesth. 2014, 28, 593–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2019, 26, 1749–1760. [Google Scholar] [CrossRef]
- Scherz-shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. P38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Liu, G.Y.; Jiang, X.X.; Zhu, X.; He, W.Y.; Kuang, Y.L.; Ren, K.; Lin, Y.; Gou, X. ROS activates JNK-mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta Pharmacol. Sin. 2015, 36, 1473–1479. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotesapoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. P38 MAPK links oxidative stress to autophagyrelated gene expression in cachectic musclewasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oude Ophuis, R.J.; Wijers, M.; Bennink, M.B.; van de Loo, F.A.; Fransen, J.A.; Wieringa, B.; Wansink, D.G. A tail-anchored myotonic dystrophy protein kinase isoform induces perinuclear clustering of mitochondria, autophagy, and apoptosis. PLoS ONE 2009, 4, e8024. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/ Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Johansen, T. p62/SQSTM1: A missing link between protein aggregates and the autophagy machinery. Autophagy 2006, 2, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redoxdependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Giles, N.M.; Gutowski, N.J.; Giles, G.I.; Jacob, C. Redox catalysts as sensitisers towards oxidative stress. FEBS Lett. 2003, 535, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Rivera, E.; Jayaraman, P.; Parikh, F.; Davies, M.A.; Ekmekcioglu, S.; Izadmehr, S.; Milton, D.R.; Chipuk, J.E.; Grimm, E.A.; Estrada, Y.; et al. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Res. 2014, 74, 1067–1078. [Google Scholar] [CrossRef] [Green Version]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. (Lond) 2019, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in Vascular Disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Terman, A.; Brunk, U.T. Autophagy, ageing and apoptosis: The role of oxidative stress and lysosomal iron. Arch. Biochem. Biophys. 2007, 462, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Gliozzi, M.; Carresi, C.; Musolino, V.; Palma, E.; Muscoli, C.; Vitale, C.; Gratteri, S.; Muscianisi, G.; Janda, E.; Muscoli, S.; et al. The Effect of Bergamot-Derived Polyphenolic Fraction on LDL Small Dense articles and Non Alcoholic Fatty Liver Disease in Patients with Metabolic Syndrome. Adv. Biol. Chemestry 2014, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Tian, C.; Chen, H.; Yang, Y.; Zhu, D.; Gao, P.; Jiankang, L. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic. Biol. Med. 2013, 55, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016, 15, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.; Zabirnyk, O.; Duerrschmidt, N.; Borlak, J.; Spanel-Borowski, K. No upregulation of lectin-like oxidized low-density lipoprotein receptor-1 in serum-deprived EA.hy926 endothelial cells under oxLDL exposure, but increase in autophagy. Eur. J. Cell Biol. 2007, 86, 605–616. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metabolism. 2012, 15, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metabolism. 2012, 15, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Le Guezennec, X.; Brichkina, A.; Huang, Y.F.; Kostromina, E.; Han, W.; Bulavin, D.V. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metabolism. 2012, 16, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E−/− mice. Sci. Rep. 2014, 4, 5519. [Google Scholar] [CrossRef] [Green Version]
- Martinet, W.; De Meyer, G.R. Autophagy in AS: A cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009, 104, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, 1077. [Google Scholar] [CrossRef] [Green Version]
- Mollace, V.; Gliozzi, M.; Musolino, V.; Carresi, C.; Muscoli, S.; Mollace, R.; Tavernese, A.; Gratteri, S.; Palma, E.; Morabito, C.; et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int. J. Cardiol. 2015, 184, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yepuri, G.; Forbiteh, M.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signalling in advanced atherosclerosis. Autophagy 2014, 10, 2223–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheloufi, M.; Vion, A.C.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Endothelial autophagic flux hampers atherosclerotic lesion development. Autophagy 2018, 14, 173–175. [Google Scholar] [CrossRef]
- Natarelli, L.; Schober, A. MicroRNAs and the response to injury in atherosclerosis. Hamostaseologie 2015, 35, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Yang, T.L. MicroRNA-126 alleviates endothelial cells injury in atherosclerosis by restoring autophagic flux via inhibiting of PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1482–1489. [Google Scholar] [CrossRef]
- Menghini, R.; Casagrande, V.; Marino, A.; Marchetti, V.; Cardellini, M.; Stoehr, R.; Rizza, S.; Martelli, E.; Greco, S.; Mauriello, A.; et al. MiR-216a: A link between endothelial dysfunction and autophagy. Cell Death Dis. 2014, 5, e1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.N.; Xu, S.B.; Wu, H.; Dai, B.; Jian, D.D.; Yang, M.; Wu, Y.T.; Feng, Q.; Zhu, J.H.; et al. MicroRNA-214-3p: A link between autophagy and endothelial cell dysfunction in atherosclerosis. Acta Physiol. (Oxf.) 2018, 222, 3. [Google Scholar] [CrossRef]
- Guo, F.X.; Wu, Q.; Li, P.; Zheng, L.; Ye, S.; Dai, X.Y.; Kang, C.M.; Lu, J.B.; Xu, B.M.; Xu, Y.J.; et al. The role of the LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ. 2019, 26, 26–1670. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, P.M.; Heinecke, J.W. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr. Opin. Lipidol. 2015, 26, 388–393. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Grassia, G.; Platt, A.M.; Carnuccio, R.; Ialenti, A.; Maffia, P. Macrophage autophagy in atherosclerosis. Mediat. Inflamm. 2013, 2013, 584715. [Google Scholar] [CrossRef]
- Zhu, Y.N.; Fan, W.J.; Zhang, C.; Guo, F.; Li, W.; Wang, Y.F.; Jiang, Z.S.; Qu, S.L. Role of autophagy in advanced atherosclerosis. Mol. Med. Rep. 2017, 15, 2903–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Kouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Peng, X.; Zhang, M.; Jia, Y.; Yu, B.; Tian, J. Revisiting Tumors and the cardiovascular System: Mechanistic Intersections and Divergences in Ferroptosis. Oxid. Med. Cell Longev. 2020, 2020, 9738143. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, S.; Li, J.; Li, Z.; Quan, J.; Liu, X.; Tang, Y.; Liu, B. The Latest View on the Mechanism of Ferroptosis and Its Research Progress in Spinal Cord Injury. Oxid. Med. Cell Longev. 2020, 2020, 6375938. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Tang, D. Autophagy and ferroptosis–what’s the connection? Curr. Pathobiol. Rep. 2017, 5, 153–159. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2019, 66, 89–100. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [Green Version]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Y.; Yuan, S.; Zhang, P.; Zhang, J.; Li, H.; Li, X.; Shen, H.; Wang, Z.; Chen, G. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral haemorrhage. Brain Res. 2018, 1701, 112–125. [Google Scholar] [CrossRef]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y.; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay between Lipid Metabolism and Autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2018, 508, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bai, Y.; Jia, Y.; Zhao, Y.; Kang, R.; Tang, D.; Dai, E. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 2018, 503, 1550–1556. [Google Scholar] [CrossRef]
- Guo, Z.; Ran, Q.; Roberts, L.J.; Zhou, L.; Richardson, A.; Sharan, C.; Wu, D.; Yang, H. Suppression of atherogenesis by overexpression of glutathione peroxidase- 4 in apolipoprotein E-deficient mice. Free Radic. Biol. Med. 2008, 44, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.J.; Zhang, D.; Wu, Y.; Jia, Q.H.; Zhang, L.; Li, Y.X.; Yang, Y.F.; Wang, H.; Wu, C.T.; Wang, L.S. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 2019, 515, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Bio. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Lavandero, S.; Chiong, M.; Rothermel, B.A.; Hill, J.A. Autophagy in cardiovascular biology. J. Clin. Investig. 2015, 12, 55–64. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carresi, C.; Mollace, R.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Coppoletta, A.R.; Guarnieri, L.; Ruga, S.; Zito, M.C.; et al. Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development. Antioxidants 2021, 10, 387. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030387
Carresi C, Mollace R, Macrì R, Scicchitano M, Bosco F, Scarano F, Coppoletta AR, Guarnieri L, Ruga S, Zito MC, et al. Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development. Antioxidants. 2021; 10(3):387. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030387
Chicago/Turabian StyleCarresi, Cristina, Rocco Mollace, Roberta Macrì, Miriam Scicchitano, Francesca Bosco, Federica Scarano, Anna Rita Coppoletta, Lorenza Guarnieri, Stefano Ruga, Maria Caterina Zito, and et al. 2021. "Oxidative Stress Triggers Defective Autophagy in Endothelial Cells: Role in Atherothrombosis Development" Antioxidants 10, no. 3: 387. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10030387