
Total Analysis of the Major Secoiridoids in Extra Virgin Olive Oil: Validation of an UHPLC-ESI-MS/MS Method

 , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. SEC Extraction for UHPLC-ESI-MS/MS Analysis
2.3. UHPLC-ESI-MS/MS Analysis
2.4. Validation of the UHPLC-ESI-MS/MS Method
2.5. Matrix Effect and Recovery
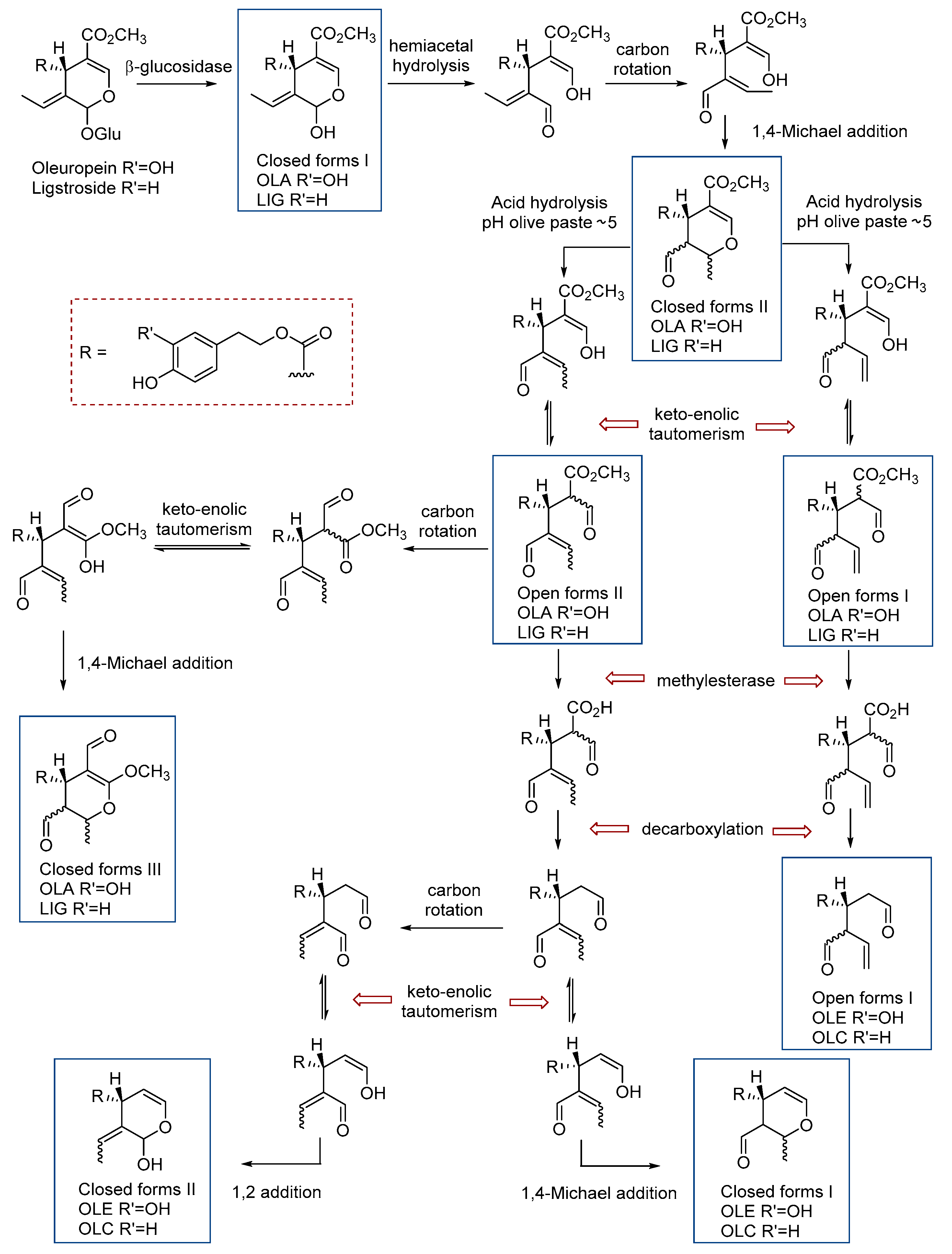
3. Results and Discussion
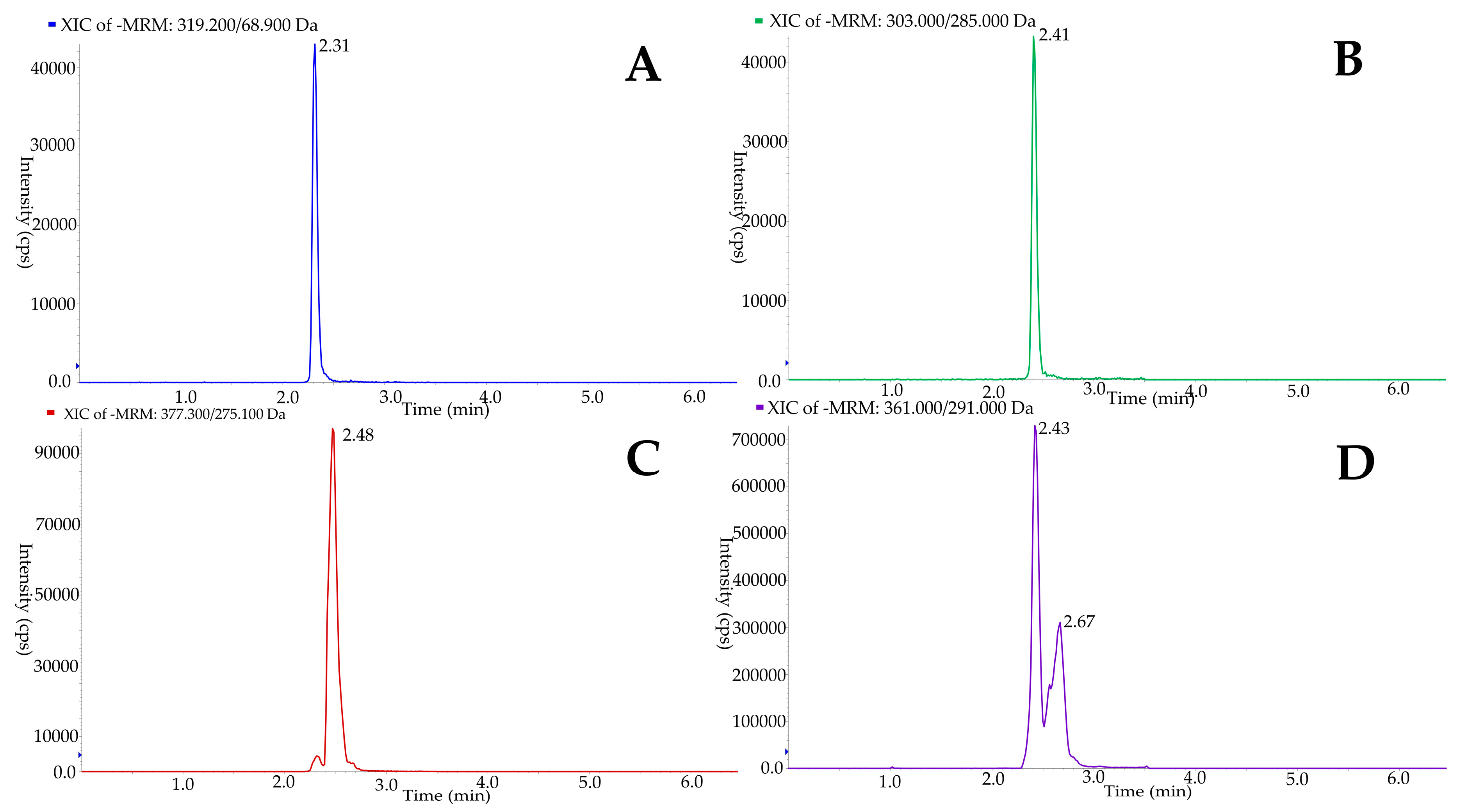
3.1. Analytical Method Development
3.2. Analytical Method Validation
3.3. Matrix Effect and Recovery
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gotsis, E.; Anagnostis, P.; Mariolis, A.; Vlachou, A.; Katsiki, N.; Karagiannis, A. Health benefits of the mediterranean diet: An update of research over the last 5 years. Angiology 2015, 66, 304–318. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Corella, D.; Salas-Salvadó, J.; Estruch, R.; Ros, E. Prevention of Diabetes With Mediterranean Diets. Ann. Intern. Med. 2014, 161, 157. [Google Scholar] [CrossRef]
- Martínez-Lapiscina, E.H.; Clavero, P.; Toledo, E.; Estruch, R.; Salas-Salvadó, J.; San Julián, B.; Sanchez-Tainta, A.; Ros, E.; Valls-Pedret, C.; Martinez-Gonzalez, M.Á. Mediterranean diet improves cognition: The PREDIMED-NAVARRA randomised trial. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1318–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guasch-Ferré, M.; Hu, F.B.; Martínez-González, M.A.; Fitó, M.; Bulló, M.; Estruch, R.; Ros, E.; Corella, D.; Recondo, J.; Gómez-Gracia, E.; et al. Olive oil intake and risk of cardiovascular disease and mortality in the PREDIMED Study. BMC Med. 2014, 12, 12–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yubero-serrano, E.M.; Lopez-moreno, J.; Gomez-delgado, F.; Lopez-miranda, J. Extra virgin olive oil: More than a healthy fat. Eur. J. Clin. Nutr. 2019, 72, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prata, R.; Pereira, J.A.; Rodrigues, N.; Dias, L.G.; Veloso, A.C.A.; Casal, S.; Peres, A.M. Olive Oil Total Phenolic Contents and Sensory Sensations Trends during Oven and Microwave Heating Processes and Their Discrimination Using an Electronic Tongue. J. Food Qual. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [Green Version]
- López-Yerena, A.; Lozano-Castellón, J.; Olmo-Cunillera, A.; Tresserra-Rimbau, A.; Quifer-Rada, P.; Jiménez, B.; Pérez, M.; Vallverdú-Queralt, A. Effects of organic and conventional growing systems on the phenolic profile of extra-virgin olive oil. Molecules 2019, 24, 1986. [Google Scholar] [CrossRef] [Green Version]
- Kanakis, P.; Termentzi, A.; Michel, T.; Gikas, E.; Halabalaki, M.; Skaltsounis, A.L. From olive drupes to olive OilAn HPLC-orbitrap-based qualitative and quantitative exploration of olive key metabolites. Planta Med. 2013, 79, 1576–1587. [Google Scholar]
- Jerman Klen, T.; Golc Wondra, A.; Vrhovšek, U.; Mozetič Vodopivec, B. Phenolic Profiling of Olives and Olive Oil Process-Derived Matrices Using UPLC-DAD-ESI-QTOF-HRMS Analysis. J. Agric. Food Chem. 2015, 63, 3859–3872. [Google Scholar] [CrossRef]
- Abbattista, R.; Losito, I.; De Ceglie, C.; Basile, G.; Calvano, C.D.; Palmisano, F.; Cataldi, T.R.I. Structural characterization of the ligstroside aglycone isoforms in virgin olive oils by liquid chromatography–high-resolution Fourier-transform mass spectrometry and H/Dexchange. J. Mass Spectrom. 2019, 54, 843–855. [Google Scholar] [CrossRef]
- Pérez-Trujillo, M.; Gómez-Caravaca, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Parella, T. Separation and identification of phenolic compounds of extra virgin Olive Oil from Olea europaea L. by HPLC-DAD-SPE-NMR/MS. Identification of a new diastereoisomer of the aldehydic form of oleuropein aglycone. J. Agric. Food Chem. 2010, 58, 9129–9136. [Google Scholar] [CrossRef]
- Lozano-Castellón, J.; López-Yerena, A.; Rinaldi de Alvarenga, J.F.; Romero del Castillo-Alba, J.; Vallverdú-Queralt, A.; Escribano-Ferrer, E.; Lamuela-Raventós, R.M. Health-promoting properties of oleocanthal and oleacein: Two secoiridoids from extra-virgin olive oil. Crit. Rev. Food Sci. Nutr. 2020, 60, 2532–2548. [Google Scholar] [CrossRef]
- Beauchamp, G.K.; Keast, R.S.J.; Morel, D.; Lin, J.; Pika, J.; Han, Q.; Lee, C.H.; Smith, A.B.; Breslin, P.A.S. Ibuprofen-like activity in extra-virgin olive oil. Nature 2005, 437, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, K.; Melliou, E.; Li, X.; Pedersen, T.L.; Wang, S.C.; Magiatis, P.; Newman, J.W.; Holt, R.R. Oleocanthal-rich extra virgin olive oil demonstrates acute anti-platelet effects in healthy men in a randomized trial. J. Funct. Foods 2017, 36, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Mete, M.; Aydemir, I.; Unsal, U.U.; Collu, F.; Vatandas, G.; Gurcu, B.; Duransoy, Y.K.; Taneli, F.; Tuglu, M.I.; Selcuki, M. Neuroprotective effects of oleocanthal, a compound in virgin olive oil, in a rat model of traumatic brain injury. Turk. Neurosurg. 2018, 28, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Abuznait, A.H.; Qosa, H.; Busnena, B.A.; El Sayed, K.A.; Kaddoumi, A. Olive-oil-derived oleocanthal enhances β-amyloid clearance as a potential neuroprotective mechanism against Alzheimer’s disease: In vitro and in vivo studies. ACS Chem. Neurosci. 2013, 4, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Khanfar, M.A.; Bardaweel, S.K.; Akl, M.R.; El Sayed, K.A. Olive Oil-derived Oleocanthal as Potent Inhibitor of Mammalian Target of Rapamycin: Biological Evaluation and Molecular Modeling Studies. Phyther. Res. 2015, 14, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal Enhances Amyloid-β Clearance from the Brains of TgSwDI Mice and in Vitro across a Human Blood-Brain Barrier Model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef] [Green Version]
- Sindona, G.; Caruso, A.; Cozza, A.; Fiorentini, S.; Lorusso, B.; Marini, E.; Nardi, M.; Procopio, A.; Zicari, S. Anti-Inflammatory Effect of 3,4-DHPEA-EDA [2-(3,4 -Hydroxyphenyl) ethyl (3S, 4E)-4-Formyl-3-(2-Oxoethyl)Hex-4-Enoate] on Primary Human Vascular Endothelial Cells. Curr. Med. Chem. 2012, 19, 4006–4013. [Google Scholar] [CrossRef]
- Czerwińska, M.E.; Kiss, A.K.; Naruszewicz, M. Inhibition of human neutrophils NEP activity, CD11b/CD18 expression and elastase release by 3,4-dihydroxyphenylethanol-elenolic acid dialdehyde, oleacein. Food Chem. 2014, 153, 1–8. [Google Scholar] [CrossRef]
- Fabiani, R.; De Bartolomeo, A.; Rosignoli, P.; Servili, M.; Selvaggini, R.; Montedoro, G.F.; Di Saverio, C.; Morozzi, G. Virgin olive oil phenols inhibit proliferation of human promyelocytic leukemia cells (HL60) by inducing apoptosis and differentiation. J. Nutr. 2006, 136, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Polini, B.; Digiacomo, M.; Carpi, S.; Bertini, S.; Gado, F.; Saccomanni, G.; Macchia, M.; Nieri, P.; Manera, C.; Fogli, S. Oleocanthal and oleacein contribute to the in vitro therapeutic potential of extra virgin oil-derived extracts in non-melanoma skin cancer. Toxicol. In Vitro 2018, 52, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Vazquez-Martin, A.; Garcia-Villalba, R.; Carrasco-Pancorbo, A.; Oliveras-Ferraros, C.; Fernandez-Gutierrez, A.; Segura-Carretero, A. TabAnti-HER2 (erbB-2) oncogene effects of phenolic compounds directly isolated from commercial Extra-Virgin Olive Oil (EVOO). BMC Cancer 2008, 8, 377. [Google Scholar] [CrossRef] [Green Version]
- Busnena, B.A.; Foudah, A.I.; Melancon, T.; El Sayed, K.A. Olive secoiridoids and semisynthetic bioisostere analogues for the control of metastatic breast cancer. Bioorg. Med. Chem. 2013, 21, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Jemai, H.; Bouaziz, M.; Fki, I.; El Feki, A.; Sayadi, S. Hypolipidimic and antioxidant activities of oleuropein and its hydrolysis derivative-rich extracts from Chemlali olive leaves. Chem. Biol. Interact. 2008, 176, 88–98. [Google Scholar] [CrossRef]
- Ceci, L.N.; Mattar, S.B.; Carelli, A.A. Chemical quality and oxidative stability of extra virgin olive oils from San Juan province (Argentina). Food Res. Int. 2017, 100, 764–770. [Google Scholar] [CrossRef]
- López-Huertas, E.; Lozano-Sánchez, J.; Segura-Carretero, A. Olive oil varieties and ripening stages containing the antioxidants hydroxytyrosol and derivatives in compliance with EFSA health claim. Food Chem. 2021, 342, 128291. [Google Scholar] [CrossRef]
- López-Yerena, A.; Ninot, A.; Lozano-Castellón, J.; Escribano-Ferrer, E.; Romero-Aroca, A.J.; Belaj, A.; Vallverdú-Queralt, A.; Lamuela-Raventós, R.M. Conservation of Native Wild Ivory-White Olives from the MEDES Islands Natural Reserve to Maintain Virgin Olive Oil Diversity. Antioxidants 2020, 9, 1009. [Google Scholar] [CrossRef]
- Taticchi, A.; Esposto, S.; Veneziani, G.; Urbani, S.; Selvaggini, R.; Servili, M. The influence of the malaxation temperature on the activity of polyphenoloxidase and peroxidase and on the phenolic composition of virgin olive oil. Food Chem. 2013, 136, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Miho, H.; Moral, J.; López-González, M.A.; Díez, C.M.; Priego-Capote, F. The phenolic profile of virgin olive oil is influenced by malaxation conditions and determines the oxidative stability. Food Chem. 2020, 314, 126183. [Google Scholar] [CrossRef] [PubMed]
- Polari, J.J.; Wang, S.C. Hammer Mill Sieve Design Impacts Olive Oil Minor Component Composition. Eur. J. Lipid Sci. Technol. 2019, 121, 1900168. [Google Scholar] [CrossRef]
- Reboredo-Rodríguez, P.; Valli, E.; Bendini, A.; Di Lecce, G.; Simal-Gándara, J.; Gallina Toschi, T. A widely used spectrophotometric assay to quantify olive oil biophenols according to the health claim (EU Reg. 432/2012). Eur. J. Lipid Sci. Technol. 2016, 118, 1593–1599. [Google Scholar] [CrossRef]
- Olmo-Cunillera, A.; López-Yerena, A.; Lozano-Castellón, J.; Tresserra-Rimbau, A.; Vallverdú-Queralt, A.; Pérez, M. NMR spectroscopy: A powerful tool for the analysis of polyphenols in extra virgin olive oil. J. Sci. Food Agric. 2020, 100, 1842–1851. [Google Scholar] [CrossRef]
- Olmo-García, L.; Fernández-Fernández, C.; Hidalgo, A.; Vílchez, P.; Fernández-Gutiérrez, A.; Marchal, R.; Carrasco-Pancorbo, A. Evaluating the reliability of specific and global methods to assess the phenolic content of virgin olive oil: Do they drive to equivalent results? J. Chromatogr. A 2019, 1585, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, S.; Ieri, F.; Romani, A. Minor polar compounds in extra virgin olive oil: Correlation between HPLC-DAD-MS and the Folin-Ciocalteu spectrophotometric method. J. Agric. Food Chem. 2014, 62, 826–835. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Santangelo, E.; Cuyàs, E.; Micol, V.; Joven, J.; Ariza, X.; Segura-Carretero, A.; García, J.; Menendez, J.A. Computer-aided discovery of biological activity spectra for anti-aging and anti-cancer olive oil oleuropeins. Aging (Albany NY) 2014, 6, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Diamantakos, P.; Velkou, A.; Killday, K.B.; Gimisis, T.; Melliou, E.; Magiatis, P. Oleokoronal and oleomissional: New major phenolic ingredients of extra virgin olive oil. Olivae 2015, 122, 22–33. [Google Scholar]
- Sánchez de Medina, V.; Miho, H.; Melliou, E.; Magiatis, P.; Priego-Capote, F.; Luque de Castro, M.D. Quantitative method for determination of oleocanthal and oleacein in virgin olive oils by liquid chromatography–tandem mass spectrometry. Talanta 2017, 162, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pancorbo, A.; Neusüß, C.; Pelzing, M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. CE- and HPLC-TOF-MS for the characterization of phenolic compounds in olive oil. Electrophoresis 2007, 28, 806–821. [Google Scholar] [CrossRef]
- Suárez, M.; Macià, A.; Romero, M.P.; Motilva, M.J. Improved liquid chromatography tandem mass spectrometry method for the determination of phenolic compounds in virgin olive oil. J. Chromatogr. A 2008, 1214, 90–99. [Google Scholar] [CrossRef]
- Di Donna, L.; Benabdelkamel, H.; Mazzotti, F.; Napoli, A.; Nardi, M.; Sindona, G. High-throughput assay of oleopentanedialdheydes in extra virgin olive oil by the UHPLC-ESI-MS/MS and isotope dilution methods. Anal. Chem. 2011, 83, 1990–1995. [Google Scholar] [CrossRef]
- Karkoula, E.; Skantzari, A.; Melliou, E.; Magiatis, P. Direct measurement of oleocanthal and oleacein levels in olive oil by quantitative 1H NMR. Establishment of a new index for the characterization of extra virgin olive oils. J. Agric. Food Chem. 2012, 60, 11696–11703. [Google Scholar] [CrossRef]
- Karkoula, E.; Skantzari, A.; Melliou, E.; Magiatis, P. Quantitative measurement of major secoiridoid derivatives in olive oil using qNMR. Proof of the artificial formation of aldehydic oleuropein and ligstroside aglycon isomers. J. Agric. Food Chem. 2014, 62, 600–607. [Google Scholar] [CrossRef]
- Diamantakos, P.; Giannara, T.; Skarkou, M.; Melliou, E.; Magiatis, P. Influence of Harvest Time and Malaxation Conditions on the Concentration of Individual Phenols in Extra Virgin Olive Oil Related to Its Healthy Properties. Molecules 2020, 25, 2449. [Google Scholar] [CrossRef] [PubMed]
- Miho, H.; Díez, C.M.; Mena-Bravo, A.; Sánchez de Medina, V.; Moral, J.; Melliou, E.; Magiatis, P.; Rallo, L.; Barranco, D.; Priego-Capote, F. Cultivar influence on variability in olive oil phenolic profiles determined through an extensive germplasm survey. Food Chem. 2018, 266, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Celano, R.; Piccinelli, A.L.; Pugliese, A.; Carabetta, S.; Di Sanzo, R.; Rastrelli, L.; Russo, M. Insights into the Analysis of Phenolic Secoiridoids in Extra Virgin Olive Oil. J. Agric. Food Chem. 2018, 66, 6053–6063. [Google Scholar] [CrossRef] [PubMed]
- Luque-Muñoz, A.; Tapia, R.; Haidour, A.; Justicia, J.; Cuerva, J.M. Direct determination of phenolic secoiridoids in olive oil by ultra-high performance liquid chromatography-triple quadruple mass spectrometry analysis. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, A.L.; Cavaliere, C.; Crescenzi, C.; Foglia, P.; Nescatelli, R.; Samperi, R.; Laganà, A. Comparison of extraction methods for the identification and quantification of polyphenols in virgin olive oil by ultra-HPLC-QToF mass spectrometry. Food Chem. 2014, 158, 392–400. [Google Scholar] [CrossRef] [PubMed]
- AOAC International. Appendix K: Guidelines for Dietary Supplements and Botanicals, Part 1 AOAC Guidelines for Single-Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals. 2013. Available online: https://members.aoac.org/AOAC_Docs/StandardsDevelopment/eoma_appendix_k.pdf (accessed on 10 May 2019).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 10 May 2019).
- Kiser, M.M.; Dolan, J.W. Selecting the best curve fit. LC-GC Eur. 2004, 17, 138–143. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Emwas, A.H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar] [PubMed]
- Montedoro, G.; Servili, M.; Baldioli, M.; Miniati, E. Simple and hydrolyzable phenolic compounds in virgin olive oil. 2. Initial characterization of the hydrolyzable fraction. J. Agric. Food Chem. 1992, 40, 1577–1580. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| OLE | OLC | OLA | LIG | |
|---|---|---|---|---|
| MRM Transition | 361/291 | 303/285 | 377/275 | 319/69 |
| Declustering Potential | −40 | −40 | −45 | −30 |
| Focusing Potential | −170 | −170 | −140 | −170 |
| Entrance Potential | −5 | −5 | −5 | −5 |
| Collision Energy | −10 | −10 | −15 | −30 |
| Dwell Time (ms) | 150 | 50 | 150 | 50 |
| Retention time (min) | 2.7 | 2.4 | 2.5 | 2.3 |
| OLE | OLC | OLA | ||
|---|---|---|---|---|
| Calibration curve | Slope | 27,987 | 33,800 | 82,737 |
| Interception | −19,765 | −7875 | −74,897 | |
| R2 | 0.9991 | 0.9873 | 0.9907 | |
| Weighting | none | 1/x2 | 1/x | |
| Limits | LOD (mg·kg−1) | 0.0343 | 0.0186 | 0.0334 |
| LOQ (mg·kg−1) | 0.114 | 0.0621 | 0.111 | |
| Accuracies | 1 mg·kg−1 | 93% | 107% | 87% |
| 2 mg·kg−1 | 100% | 89% | 101% | |
| 5 mg·kg−1 | 103% | 97% | 113% | |
| 8 mg·kg−1 | 96% | 95% | 98% | |
| 10 mg·kg−1 | 102% | 100% | 110% | |
| 20 mg·kg−1 | 99% | 114% | 94% | |
| Repeatability Relative Standard Deviation | Intraday 1 mg·kg−1 | 2.3% | 2.5% | 5.8% |
| Intraday 5 mg·kg−1 | 1.9% | 2.7% | 1.8% | |
| Intraday10 mg·kg−1 | 1.1% | 5.7% | 3.7% | |
| Interday 1 mg·kg−1 | 2.4% | 3.3% | 7.5% | |
| Interday 5 mg·kg−1 | 5.7% | 4.1% | 7.0% | |
| Interday 10 mg·kg−1 | 1.2% | 6.0% | 3.1% |
| LOD (mg·kg−1) | LOQ (mg·kg−1) | R2 | RSD (%) a | EVOO (g) | Instrument | Time of Analysis (min) | Study | |
|---|---|---|---|---|---|---|---|---|
| OLE | 0.0343 | 0.114 | 0.999 | 1.8 | 0.5 | UHPLC-ESI-MS/MS | 6.5 | Current study |
| 0.128 | 0.428 | - | 3.1 | 45 | UPLC-ESI-MS/MS | 40 | [40] | |
| 0.002 | 0.005 | 0.999 | 10 | 1 | HPLC-ESI-MS/MS | 10 | [38] | |
| 0.001 | 0.0033 | 0.995 | 1.9 | 1 | UHPLC-ESI-MS/MS b | 7.5 | [47] | |
| 1 | 10 | 0.994 | 4.3 | 5 | NMR | - | [42] | |
| OLC | 0.0186 | 0.0621 | 0.987 | 3.6 | 0.5 | UHPLC-ESI-MS/MS | 6.5 | Current study |
| 0.072 | 0.244 | - | 3 | 45 | UPLC-ESI-MS/MS | 40 | [40] | |
| 0.004 | 0.01 | 0.992 | 11 | 1 | HPLC-ESI-MS/MS | 10 | [38] | |
| 0.004 | 0.012 | 0.994 | 4.4 | 1 | UHPLC-ESI-MS/MS b | 7.5 | [47] | |
| 1 | 10 | 0.999 | 4.3 | 5 | NMR | - | [42] | |
| OLA | 0.0334 | 0.111 | 0.991 | 3.8 | 0.5 | UPLC-ESI-MS/MS | 6.5 | Current study |
| 0.0003 | 0.0009 | 0.996 | 2.5 | 1 | UHPLC-ESI-MS/MS b | 7.5 | [47] | |
| 1 | 10 | 0.999 | 4.1 | 5 | NMR | - | [43] |
| OLE | OLC | OLA | ||
|---|---|---|---|---|
| Matrix effect (%) | 1 mg·kg−1 | 56 | 93 | 20 |
| 2 mg·kg−1 | 50 | 76 | 27 | |
| 5 mg·kg−1 | 86 | 129 | 57 | |
| 8 mg·kg−1 | 79 | 123 | 51 | |
| 10 mg·kg−1 | 82 | 134 | 56 | |
| 20 mg·kg−1 | 93 | 152 | 69 | |
| Recoveries (%) | 1 mg·kg−1 | 55 | 59 | 66 |
| 2 mg·kg−1 | 98 | 96 | 88 | |
| 5 mg·kg−1 | 80 | 61 | 74 | |
| 8 mg·kg−1 | 96 | 62 | 106 | |
| 10 mg·kg−1 | 89 | 61 | 88 | |
| 20 mg·kg−1 | 94 | 60 | 93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano-Castellón, J.; López-Yerena, A.; Olmo-Cunillera, A.; Jáuregui, O.; Pérez, M.; Lamuela-Raventós, R.M.; Vallverdú-Queralt, A. Total Analysis of the Major Secoiridoids in Extra Virgin Olive Oil: Validation of an UHPLC-ESI-MS/MS Method. Antioxidants 2021, 10, 540. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040540
Lozano-Castellón J, López-Yerena A, Olmo-Cunillera A, Jáuregui O, Pérez M, Lamuela-Raventós RM, Vallverdú-Queralt A. Total Analysis of the Major Secoiridoids in Extra Virgin Olive Oil: Validation of an UHPLC-ESI-MS/MS Method. Antioxidants. 2021; 10(4):540. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040540
Chicago/Turabian StyleLozano-Castellón, Julián, Anallely López-Yerena, Alexandra Olmo-Cunillera, Olga Jáuregui, Maria Pérez, Rosa Mª Lamuela-Raventós, and Anna Vallverdú-Queralt. 2021. "Total Analysis of the Major Secoiridoids in Extra Virgin Olive Oil: Validation of an UHPLC-ESI-MS/MS Method" Antioxidants 10, no. 4: 540. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10040540