Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress
by
 , and
, and
Aishwarya R. Vaidya
1, 
Nina Wolska
1,
Dina Vara
2,
Reiner K. Mailer
1,
Katrin Schröder
3 and
Giordano Pula
1,* 1
Institute of Clinical Chemistry and Laboratory Medicine, University Medical Center Hamburg-Eppendorf, D-20246 Hamburg, Germany
2
Biotherapeutics Division, National Institute for Biological Standards and Control (NIBSC), Medicines and Healthcare Products Regulatory Agency (MHRA), London EN6 3QG, UK
3
Institute of Cardiovascular Physiology, Goethe-University, D-60596 Frankfurt, Germany
*
Author to whom correspondence should be addressed.
Antioxidants 2021, 10(5), 706; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050706
Submission received: 30 March 2021
/
Revised: 19 April 2021
/
Accepted: 27 April 2021
/
Published: 29 April 2021
(This article belongs to the Special Issue Redox-Dependent Regulation of Haemostasis in Health and Disease)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Diabetes mellitus is the fifth most common cause of death worldwide. Due to its chronic nature, diabetes is a debilitating disease for the patient and a relevant cost for the national health system. Type 2 diabetes mellitus is the most common form of diabetes mellitus (90% of cases) and is characteristically multifactorial, with both genetic and environmental causes. Diabetes patients display a significant increase in the risk of developing cardiovascular disease compared to the rest of the population. This is associated with increased blood clotting, which results in circulatory complications and vascular damage. Platelets are circulating cells within the vascular system that contribute to hemostasis. Their increased tendency to activate and form thrombi has been observed in diabetes mellitus patients (i.e., platelet hyperactivity). The oxidative damage of platelets and the function of pro-oxidant enzymes such as the NADPH oxidases appear central to diabetes-dependent platelet hyperactivity. In addition to platelet hyperactivity, endothelial cell damage and alterations of the coagulation response also participate in the vascular damage associated with diabetes. Here, we present an updated interpretation of the molecular mechanisms underlying vascular damage in diabetes, including current therapeutic options for its control.
1. Introduction
Diabetes mellitus (DM) is a heterogeneous metabolic disorder characterized by persistent hyperglycemia (HG), which can be diagnosed by quantifying glycated hemoglobin (HbA1c) in peripheral blood. The HbA1c test provides information about glycemic levels in the 3 months preceding blood collection and allows one to distinguish pre-diabetes (HbA1c between 5.7% and 6.5%) from diabetes (HbA1c > 6.5%). Insufficient release of insulin by pancreatic β cells or loss of responsiveness of cells to insulin are the cause of HG in DM. Two main forms of DM are described: type 1 (or T1DM), which is caused by genetic deficiency in insulin release, and type 2 (or T2DM), which is caused by insulin resistance and has mixed genetic and lifestyle determinants. Poor glycemic control in DM patients is accompanied by altered hematological parameters, such as hypercholesterolemia and dyslipidemia, and by a range of serious health complications, including cardiovascular diseases, nerve damage (neuropathy), kidney damage (nephropathy), eye damage (retinopathy), and limb and other peripheral tissue conditions.
Population studies suggest an increase in DM diagnosis from a current count of over 415 million patients globally to over 600 million in 2042 [1]. Six percent of the world mortality rate (i.e., over 3 million deaths annually) is attributed to complications of diabetes. DM patients display a two- to fourfold increase in mortality caused by cardiovascular diseases as compared to the normal population [2]. The risk of developing microvascular complications is particularly increased with HbA1C > 7.9% (>53 mmol/mol) [3]. Disturbed glycemic control increases the propensity for microvascular complications (e.g., retinopathy, neuropathy, and nephropathy) and contributes significantly to the risk factors for macrovascular events, such as age, physical activity, lifestyle, and ethnicity, leading to cardiovascular disease [4].
Longitudinal studies suggest that women with diabetes (35–59 years) have a higher mortality rate due to vascular occlusion than healthy women. However, men with diabetes have increased risk of developing cardiovascular diseases later in life (59–70 years) [5]. Overall, sexual dimorphism in the cardiovascular risk in diabetes is attributable to a combination of diabetes symptoms like dyslipidemia and hormonal control of vascular balance [6].
The vascular damage associated with DM appears to have multiple and simultaneous causes. Platelet hyperactivity, blood hypercoagulability, reduced thrombolysis, and endothelial damage have all been reported and appear to contribute to vascular frailty in DM patients.
2. Platelet Hyperactivity in Diabetes
Platelets are anucleated circulating cells derived from megakaryocytes and function to prevent blood loss resulting from injury (i.e., hemostasis). They patrol the vasculature in healthy individuals and are activated in the presence of vascular insults like endothelial denudation, leading to sub-endothelial collagen exposure, or in response to other injury-dependent stimuli such as thrombin and von Willebrand factor (vWF). Platelets also play a pivotal role in thrombotic and inflammatory pathologies [7]. Increased responsiveness of platelets (hyperactivity) has been suggested as a critical driver for cardiovascular complications of diabetes [8,9]. Platelet hyperactivity is suggested by the detection of increased levels of thromboxane B2 in the urine of T2DM patients [10,11]. Thromboxane B2 is a stable degradation product of thromboxane A2, a secondary agonist released by activated platelets.
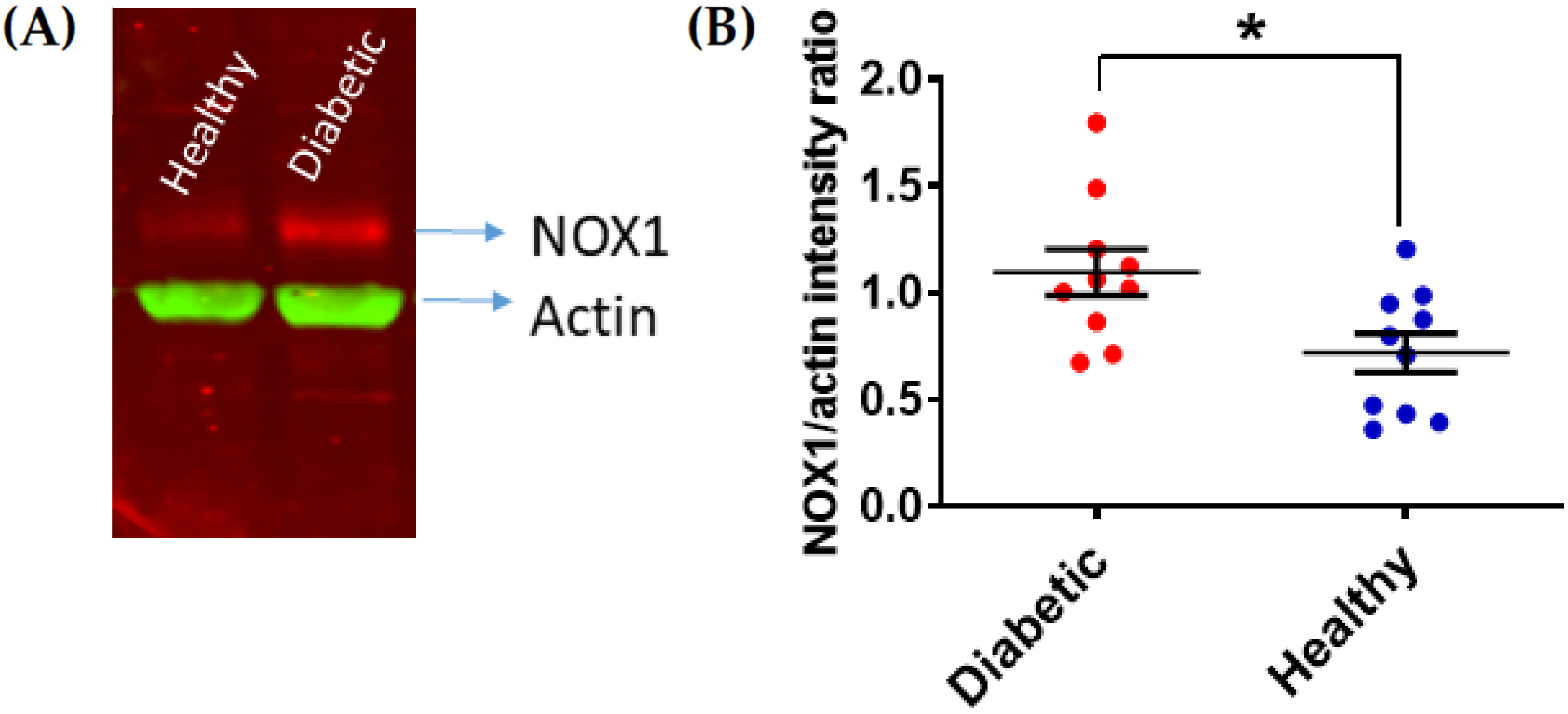
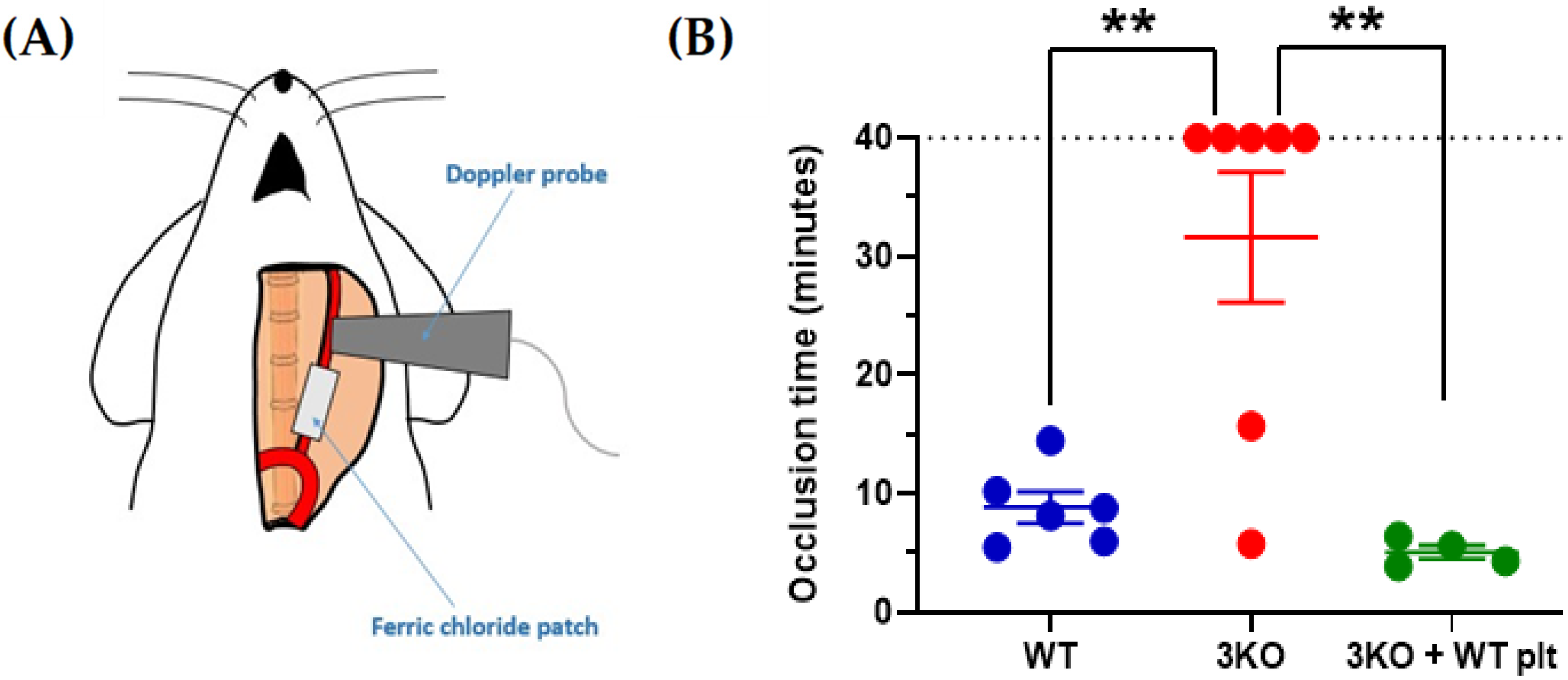
DM-associated HG affects the expression of key platelet enzymes and receptors at the megakaryocyte stage. The expression levels of the receptor of the negative platelet regulator prostacyclin are decreased in T2DM, which in turn enhances platelet responsiveness [12]. In parallel, P2Y12, a key receptor for the secondary agonist adenosine diphosphate (ADP), has been reported to be significantly upregulated in T2DM platelets [13]. The upregulation of P2Y12 expression is supported by the activation of oxidative stress-dependent transcription factor nuclear factor-κB (NF-κB) in megakaryocytes. Insulin-like growth factor 1 receptor (IGF1R) is another important receptor upregulated in T2DM patients, which makes platelets from these patients more responsive to IGF1 [14]. Since IGF1 has been described as a positive regulator of platelet signaling and responses [15], the upregulation of IGF1R is likely to contribute to platelet hyperactivity in T2DM. Changes in the membrane expression levels have also been proposed for integrin β3; however, the modulation of the surface expression of this receptor occurs in platelets by microparticle shedding [16]. Work in our laboratory showed that platelets from DM patients with poor glycemic control express significantly higher levels of the pro-oxidant enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (NOX1) (Figure 1). NOXs have been described as important positive regulators of platelet activity [17,18,19,20]. In view of the pro-thrombotic role of platelet NOXs both in vitro and in vivo (Figure 2), the upregulation of NOX1 in DM patients contributes significantly to platelet hyperresponsiveness. Further studies in this direction are required to elucidate the mechanism of this interaction.
In addition to changes in the proteome of platelets caused by alteration of gene expression, transcription, or protein turnover, DM also regulates platelet function via modulation of different signaling pathways. Markers of platelet activation, such as P-selectin and CD40L, are increased in T1DM and T2DM patients, which suggests raised levels of platelet activation in these patients [21,22]. HG has been shown to directly correlate with the levels of CD40L release (sCD40L) in vitro [22].
High plasma glucose results in increased levels of advanced glycation end products (AGEs) in plasma [24]. AGEs have been shown to activate platelets via activation of the receptor for AGEs (RAGE) [25]. Alternatively, the scavenger receptor CD36 also recognizes AGEs and stimulates platelet activation [26]. Increased pro-coagulant activity of platelets has also been described for T2DM platelets, which was integrin αIIbβ3 dependent [27].
One of the first mechanistic explanations of the hyperactivity of platelets in diabetes suggested a negative regulatory role of insulin in the ADP receptor P2Y12 and platelet function. Therefore, insulin resistance and ultimately loss of insulin secretion results in the dysregulation of platelet activation [28]. The insulin-dependent activation of the protein kinase PKB and the modulation of the inhibitory intracellular messenger cAMP support the negative regulatory activity of insulin. Another factor driving platelet hyperactivity can be dyslipidemia, which is often present alongside diabetes. Increased plasma levels of lipids and cholesterol enhance platelet reactivity. Although the evidence was initially only observational [29], recent studies have highlighted the molecular mechanisms linking plasma lipids (low-density lipoprotein, or LDL, in particular) to platelet responsiveness. Typically, dyslipidemia associated with T2DM is accompanied by increased levels of LDL oxidation (ox-LDL) [30]. Ox-LDL has been shown to activate the scavenger receptor CD36 in different cell types, including platelets [31]. The signaling pathway activated by CD36 includes tyrosine kinase- and protein kinase C-dependent activation of NOX2 and generation of reactive oxygen species (ROS), ultimately counteracting the negative regulatory function of the cyclic nucleotides cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP). Recent studies from our laboratory highlighted the involvement of both NOX1 and NOX2 in the signaling of ox-LDL [17] and confirmed the negative modulation of the cyclic nucleotide pathways by NOXs [18]. In addition to enzymatic ROS sources, HG causes metabolic overload in platelet mitochondria, which results in the leakage of electrons from the respiration chain and the release of ROS [32]. As a result, protein tyrosine phosphatases are inhibited and the protein kinase signaling pathways are potentiated, which ultimately leads to the potentiation of platelets responses. ROS-dependent inhibition of the protein tyrosine phosphatase Src homology 2 (SH2) domain-containing phosphatase 2 (SHP2) has been shown to lead to increased activity of the protein kinase spleen tyrosine kinase (Syk) and the potentiation of collagen-induced platelet responses [33,34,35].
The role of platelets in vascular health and disease has recently been widened by the discovery of their involvement in the formation of neutrophil extracellular traps (NETs) [36]. NETs have been shown to contribute significantly to thrombotic diseases [37]. Diabetes has been shown to increase NET formation [38,39]. Further studies are required to ascertain whether platelets are a cause for increased NET formation or whether NET formation contributes to DM-dependent thrombosis by inducing platelet activation and vascular occlusion.
3. Coagulation and Fibrinolysis
Increased plasma levels for different coagulation factors have been reported for DM patients [40]. Fibrinogen (factor I), pro-thrombin (factor II), pre-kallikrein, factor V, factor VII, factor VIII, factor X, and factor XI have been detected at higher than normal concentration in the plasma of T1DM and T2DM patients [41]. Some coagulation-related proteins are only elevated in T2DM, such as kininogen, factor IX, and factor XIII [42,43]. Interestingly, activated factor XII is downregulated in T1DM, while it is upregulated in T2DM [44]. Tissue factor (TF) is elevated in both T1DM and T2DM [45], although the plasma concentration of TF in response to experimental HG and hyperinsulinemia (HI) is increased in T2DM but not in T1DM. In addition, several anticoagulant proteins have a reduced plasma concentration in both types of diabetes, including anti-thrombin, protein C, and protein S [46]. Overall, the alteration of plasma levels of coagulation factors promotes the hypercoagulative state of DM patients [47]. Oxidative post-translational modification of plasma proteins in T2DM (especially coagulation factor carbonylation) has been proposed as a novel and important factor promoting the procoagulant state of this disease [48].
In parallel, the dissolution of clots (i.e., fibrinolysis) is decreased in DM. Plasminogen activation to form plasmin is the central biochemical reaction of fibrinolysis. Plasmin is a serine protease that acts to dissolve fibrin blood clots. The activation of plasminogen to form plasmin via tissue plasminogen activator (t-PA) and urokinase plasminogen activator (u-PA) promotes fibrinolysis. Increased cross-fibrin cross-linking caused by HG has been suggested to increase clot strength [49] and reduce the fibrinolytic rate in T2DM patients [50]. In addition, HG in T1DM has been suggested to mediate glycation and other post-translational modifications of plasminogen that prevent its activation and limit the formation of plasmin, which ultimately impairs fibrinolysis [51]. Disease duration appears to have a stronger influence on clot strength and fibrinolysis than glycemia control [52]. The implications of this observation remain to be fully understood.
In T2DM, an increased concentration of plasminogen activator inhibitor-1 (PAI-1) reduces fibrinolysis [53,54]. In addition, the concentration of other inhibitors of fibrinolysis, such as thrombin-activatable fibrinolysis inhibitor (TAFI) [55] and α2-macroglobulin [56], are increased in both T1DM and T2DM. In addition, the concentration of α2-antiplasmin is elevated in T2DM [57], while the literature is inconclusive about α2-antiplasmin in T1DM. Taken together, the above studies suggest that reduced fibrinolysis is a likely source of increased thrombotic risk for DM patients. As the consensus is that hypercoagulation is particularly relevant for venous thrombosis, while platelet hyperactivity participates in the onset and progression of arterial thrombosis, the imbalance between coagulation and fibrinolysis may explain the increased risk of both venous and arterial thrombosis in DM patients.
4. Endothelial Cell Dysfunction
The endothelium is a cellular monolayer that lines the whole of the vasculature. One of the key physiological roles of the endothelium is the release of antithrombotic signals, such as nitric oxide (NO) and prostacyclin. Endothelial damage induces thrombotic complications by reducing the bioavailability of the abovementioned antithrombotic substances and exposing subendothelial substances that stimulate blood platelets and coagulation (via exposure of platelet activators such as collagen). Vascular inflammation and endothelial cell damage have also been shown to increase the release of vWF [54] and plasminogen activator inhibitor-1 (PAI-1) [58] by endothelial cells. Therefore, as vWF promotes platelet adhesion [59] and PAI-1 inhibits fibrinolysis [60], the overall effect of DM-dependent endothelial cell damage is the increase in platelet adhesion and clot formation. Ultimately, the endothelial cell-dependent modulation of platelets and fibrinolysis participates in the increase in thrombotic risk for DM patients [9]. Raised systemic levels of vWF are associated with vascular comorbidities, making them cardiovascular risk predictors and diagnostic markers in T2D patients [61].
High plasma glucose results in increased levels of advanced glycation end products (AGEs) in plasma [24] and vascular endothelial cells [62]. Endothelial damage also contributes to thickening of the vascular wall, impairs vasodilation, and results in vessel stiffening [63]. The presence of AGEs promotes the activity of the endothelial receptor for advanced glycation end products (RAGE), which contributes to ROS production [64]. Endothelial oxidative stress in DM also derives from Ras-related C3 botulinum toxin substrate 1 (RAC1) and T cell lymphoma invasion and metastasis (TIAM1)-dependent activation of NOX2 [65] and diacylglycerol (DAG)/protein kinase C (PKC)-dependent phosphorylation and stimulation of the NOX activator p47phox [66]. Importantly, endothelial oxidative stress is associated with the formation of superoxide anion, which directly quenches NO by forming peroxynitrite ions and in turn inhibits endothelial nitric oxide synthase (eNOS), a phenomenon that involves the generation of the endogenous intermediate asymmetric dimethylarginine (ADMA), which is an eNOS inhibitor [67].
In addition to the negative regulation of eNOS activity, endothelial oxidative stress also inhibits prostacyclin synthase [68] and activates the pro-inflammatory transcription factor NF-κB [69]. The upregulation of endothelin-1, thrombomodulin, the adhesion molecules ICAM, VCAM, and E-selectin [70], and the increase in the circulating levels of cytokines like IL-1β and TNF-α and chemoattractants like MCP-1 [71] are amongst the most important pro-inflammatory effects of NF-κB activation.
5. Therapeutic Intervention
Antiplatelet drugs are commonly prescribed for DM patients. The cyclooxygenase-1 (COX-1) inhibitor acetylsalicylic acid (ASA), the P2Y12 receptor antagonists clopidogrel, prasugrel, cangrelor, and ticagrelor, and the GPIIb/IIIa inhibitors abciximab (ReoPro), eptifibatide (Integrilin), and tirofiban (Aggrastat) are the most utilized antiplatelet therapies in clinical practice. The combination of ASA with another antiplatelet drug is known as dual antiplatelet therapy (DAPT), and ADP receptor inhibitors such as clopidogrel, ticagrelor, and prasugrel are usually combined with ASA [72].
Recent studies have highlighted the poor efficacy of ASA in DM patients. The ASCEND trial revealed almost no difference between the number of serious vascular events between the ASA and placebo participants in high-risk DM patients within a 7-year follow-up period [73]. A recent meta-analysis of 12 randomized studies showed that aspirin for primary prevention only reduces major adverse cardiovascular events by 11% [74]. There is evidence for cardiovascular primary prevention in DM patients by DAPT [75]. Nonetheless, a DAPT regime of 75–100 mg ASA/day and 75 mg clopidogrel/day is only advised for secondary prevention in patients with diabetes [76] or for primary prevention in T1DM and T2DM patients with high cardiovascular risk, at age ≥ 50 years and no risk of bleeding [77]. High risk is defined as a family history of at least one condition amongst atherosclerotic cardiovascular disease, hypertension, dyslipidemia, or albuminuria. The increase in bleeding risk for DM patients limits the adoption of DAPT for preventive purposes and highlights the necessity of alternative therapeutic options [76]. Another recent and important study focusing on DAPT concluded that DM patients have less of a positive response to continued pharmacological treatment compared to non-DM patients [78]. More encouraging results are coming from a yet-to-be-completed study exploring the use of DAPT combining ticagrelor and ASA, which significantly reduces major adverse cardiovascular or cerebrovascular events (MACCEs) compared to treatment with ASA alone [79].
Alternatives to ASA may represent an improvement over existing therapeutic options. The thromboxane synthase inhibitor picotamide did not prove more effective in preventing mortality than aspirin, although it reduced unwanted bleeding side effects [80]. The thrombin inhibitor bivalirudin gave some positive results in the ACUITY study with regard to composite ischemia and major bleeding complications [81]. P2Y12 receptor antagonists have also been tested on their own in DM patients, highlighting a complex picture in which, depending on comorbidities, clopidogrel, prasugrel, or ticagrelor are the preferred therapeutic options [82]. Overall, cardiovascular prophylaxis in DM remains an unresolved challenge. A better understanding of the molecular mechanisms increasing the cardiovascular risk for DM patients may help to design novel and more efficacious treatments. For example, the participation of oxidative stress in the vascular dysfunction in both T1DM and T2DM may offer some interventional opportunities. NOX inhibitors have been proven to be effective for vascular protection in DM animal models [83,84,85,86,87,88] and clinical studies in humans are ongoing [89]. Dietary antioxidants have also shown some promise to prevent or slow down vascular degradation in DM and protect against cardiovascular complications of this disease. Polyphenols from mushrooms, tea, coffee, and dark chocolate have shown some degree of effectiveness in controlling platelet hyperactivity, mitochondrial stress, and superoxide formation [90] and decreasing the level of plasma oxidized LDL [91]. Food supplements may therefore be designed that help to protect vascular health in DM patients.
In addition to antiplatelet drugs, anticoagulants have been investigated for the treatment of vascular conditions in DM patients. The factor X inhibitor anticoagulant rivaroxaban has been shown to increase the effectiveness of DAPT (ASA + clopidogrel) against MACCEs in acute coronary syndrome patients [92]. As anticoagulants are generally prescribed for atrial fibrillation [93], recent studies have focused on the comparison between traditional vitamin K inhibitor anticoagulants (e.g., warfarin) and novel oral anticoagulants (NOACs, e.g., apixaban, dabigatran, rivaroxaban, and edoxaban). NOACs display better efficacy against thromboembolism events and major adverse limb events than warfarin in DM patients [94]. In addition, NOACs appear to be safer than warfarin as they are associated with a lower risk of major bleeding [95,96]. NOACs should therefore be considered in the diabetic AF population with a high atherosclerotic burden, although a negative interaction with insulin treatment has been noted, which limits their therapeutic efficacy in DM [89].
Although endothelial cell damage is an established pathological mechanism in diabetes, and despite ongoing clinical studies on the use of NADPH oxidase inhibitors [97], there are no effective drugs for endothelial cell protection that have been adopted in this disease.
6. Conclusions
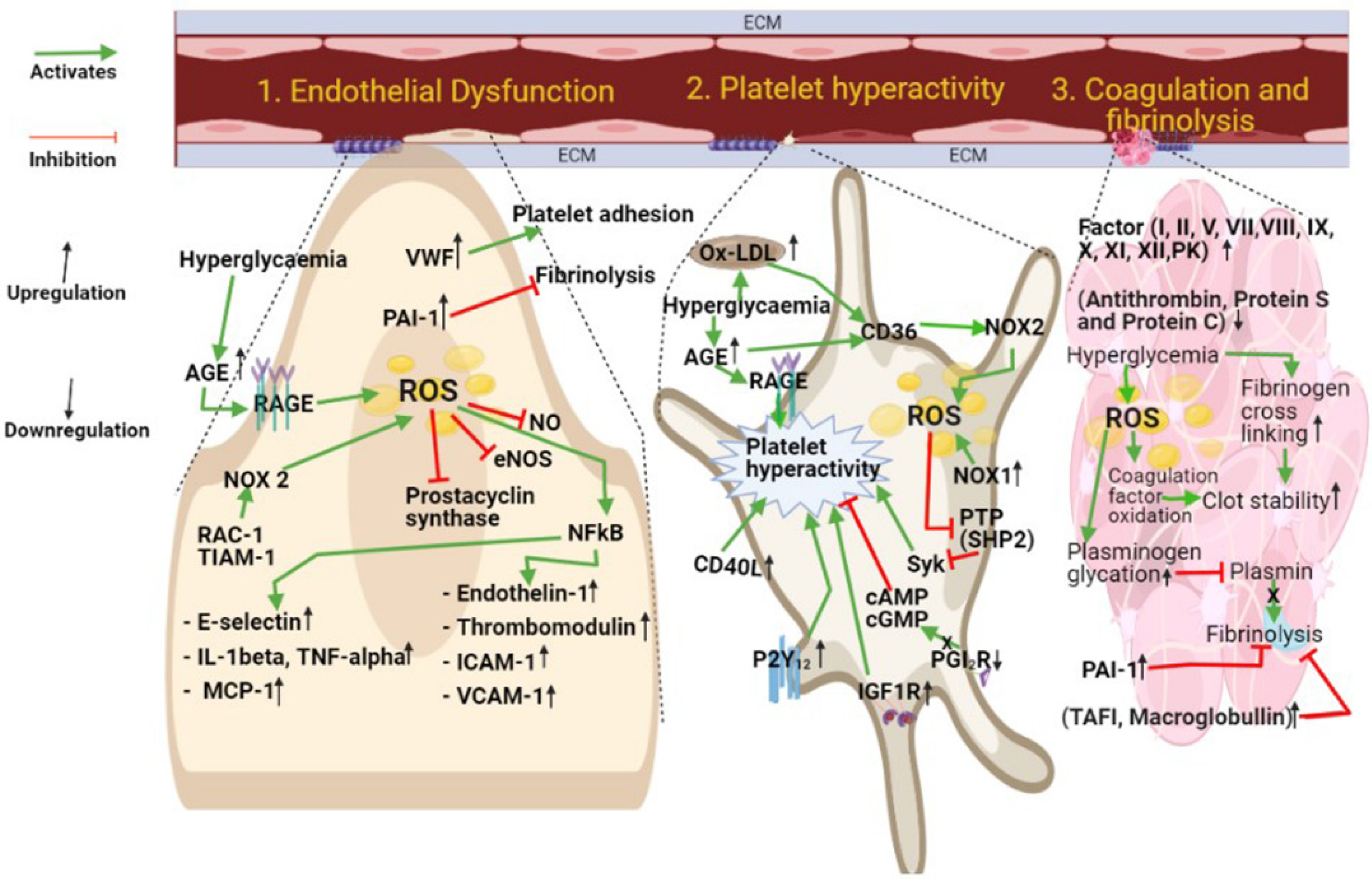
Taken together, the correlation between vascular frailty and DM has been described, which underlies the increased incidence of cardiovascular diseases in both T1DM and T2DM. Multiple mechanisms have been suggested affecting platelets, the coagulation cascade, or the physiology of the vascular endothelium (Figure 3). Although a significant amount of research in recent years has led to important advances in our understanding of the causes for increased cardiovascular risk in DM, several questions remain unanswered in this area of medical research. In particular, there is an urgent need to improve the therapeutic options available for the clinical management of cardiovascular diseases in DM. An accelerated translation of preclinical discoveries into effective clinical tools via targeted clinical studies appears to be the only viable direction that the biomedical research community has, to resolve this unmet clinical challenge.
Author Contributions
Funding
This work was funded by British Heart Foundation (PG/15/40/31522), Alzheimer’s Research UK (ARUK-PG2017A-3), and Werner Otto Foundation (#3/97) grants to Giordano Pula, and DFG grants (CRC815 TPA1 and SFB834 TPA2) to Katrin Schröder. Nina Wolska was supported by an ETIUDA7 doctoral scholarship funded by the National Science Centre, Poland (2019/32/T/NZ3/00333).
Institutional Review Board Statement
All experiments performed with human blood conformed to the principles outlined in the Declaration of Helsinki and were in accordance with local ethics (according to the existing ethics approval, Ethik-Kommission der Ärztekammer Hamburg n. 2322). All experiments on animals performed were in accordance with German animal protection law and were carried out after approval by the local authorities (Behörde für Gesundheit und Verbraucherschutz Freie und Hansestadt Hamburg, approval number 76/16).
Informed Consent Statement
According to approval n. 2322 (see above), the usage of anonymized blood samples from the diagnostic units of the Institute of Clinical Chemistry and Laboratory Medicine for research purposes is authorized without informed consent by the patients.
Data Availability Statement
Data in this article will be shared on request to the corresponding author by email.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zinman, B. The International Diabetes Federation World Diabetes Congress 2015. Eur. Endocrinol. 2015, 11, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannel, W.B.; D’Agostino, R.B.; Wilson, P.W.; Belanger, A.J.; Gagnon, D.R. Diabetes, fibrinogen, and risk of cardiovascular disease: The Framingham experience. Am. Heart J. 1990, 120, 672–676. [Google Scholar] [CrossRef]
- Laiteerapong, N.; Ham, S.A.; Gao, Y.; Moffet, H.H.; Liu, J.Y.; Huang, E.S.; Karter, A.J. The Legacy Effect in Type 2 Diabetes: Impact of Early Glycemic Control on Future Complications (The Diabetes & Aging Study). Diabetes Care 2019, 42, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahkoska, A.R.; Geybels, M.S.; Klein, K.R.; Kreiner, F.F.; Marx, N.; Nauck, M.A.; Pratley, R.E.; Wolthers, B.O.; Buse, J.B. Validation of distinct type 2 diabetes clusters and their association with diabetes complications in the DEVOTE, LEADER and SUSTAIN -6 cardiovascular outcomes trials. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Prospective Studies Collaboration; Asia Pacific Cohort Studies Colaboration. Sex-specific relevance of diabetes to occlusive vascular and other mortality: A collaborative meta-analysis of individual data from 980,793 adults from 68 prospective studies. Lancet Diabetes Endocrinol. 2018, 6, 538–546. [Google Scholar] [CrossRef]
- Kaseta, J.R.; Skafar, D.F.; Ram, J.L.; Jacober, S.J.; Sowers, J.R. Cardiovascular Disease in the Diabetic Woman. J. Clin. Endocrinol. Metab. 1999, 84, 1835–1838. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Gaiz, A.; Mosawy, S.; Colson, N.; Singh, I. Thrombotic and cardiovascular risks in type two diabetes; Role of platelet hyperactivity. Biomed. Pharmacother. 2017, 94, 679–686. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane Biosynthesis and Platelet Function in Type II Diabetes Mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef]
- Davì, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In Vivo Formation of 8-Iso-Prostaglandin F 2α and Platelet Activation in Diabetes Mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Knebel, S.M.; Sprague, R.S.; Stephenson, A.H. Prostacyclin receptor expression on platelets of humans with type 2 diabetes is inversely correlated with hemoglobin A1c levels. Prostaglandins Other Lipid Mediat. 2015, 116, 131–135. [Google Scholar] [CrossRef]
- Hu, L.; Chang, L.; Zhang, Y.; Zhai, L.; Zhang, S.; Qi, Z.; Yan, H.; Yan, Y.; Luo, X.; Zhang, S.; et al. Platelets Express Activated P2Y12Receptor in Patients with Diabetes Mellitus. Circulation 2017, 136, 817–833. [Google Scholar] [CrossRef]
- Gligorijevic, N.; Robajac, D.; Nedic, O. Enhanced Platelet Sensitivity to IGF-1 in Patients with Type 2 Diabetes Mellitus. Biochemistry 2019, 84, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Hers, I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kα pathway. Blood 2007, 110, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Przygodzki, T.; Luzak, B.; Kassassir, H.; Mnich, E.; Boncler, M.; Siewiera, K.; Kosmalski, M.; Szymanski, J.; Watala, C. Diabetes and Hyperglycemia Affect Platelet GPIIIa Expression: Effects on Adhesion Potential of Blood Platelets from Diabetic Patients under In Vitro Flow Conditions. Int. J. Mol. Sci. 2020, 21, 3222. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Cifuentes-Pagano, E.; Pagano, P.J.; Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica 2019, 104, 1879–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renné, T.; Schröder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arter. Thromb. Vasc. Biol. 2021, 41, 683–697. [Google Scholar] [CrossRef]
- Pignatelli, P.; Violi, F. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb. Haemost. 2014, 111, 817–823. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Yngen, M.; Östenson, C.-G.; Hu, H.; Li, N.; Hjemdahl, P.; Wallén, N.H. Enhanced P-selectin expression and increased soluble CD40 Ligand in patients with Type 1 diabetes mellitus and microangiopathy: Evidence for platelet hyperactivity and chronic inflammation. Diabetologia 2004, 47, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Varo, N.; Libby, P.; Nuzzo, R.; Italiano, J.; Doria, A.; Schönbeck, U. Elevated release of sCD40L from platelets of diabetic patients by thrombin, glucose and advanced glycation end products. Diabetes Vasc. Dis. Res. 2005, 2, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Campanella, M.; Pula, G. The novel NOX inhibitor 2-acetylphenothiazine impairs collagen-dependent thrombus formation in a GPVI-dependent manner. Br. J. Pharmacol. 2012, 168, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Kilhovd, B.K.; Berg, T.J.; Birkeland, K.I.; Thorsby, P.; Hanssen, K.F. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care 1999, 22, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.; Rojas, A.; Palomo, I. Role of multiligand/RAGE axis in platelet activation. Thromb. Res. 2014, 133, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [Green Version]
- Razmara, M.; Hjemdahl, P.; Östenson, C.; Li, N. Platelet hyperprocoagulant activity in Type 2 diabetes mellitus: Attenuation by glycoprotein IIb/IIIa inhibition. J. Thromb. Haemost. 2008, 6, 2186–2192. [Google Scholar] [CrossRef]
- Ferreira, I.A.; Mocking, A.I.; Feijge, M.A.; Gorter, G.; Van Haeften, T.W.; Heemskerk, J.W.; Akkerman, J.-W.N. Platelet Inhibition by Insulin Is Absent in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, R.; Krishnan, L.K. Increased Platelet Cholesterol and Decreased Percentage Volume of Platelets as a Secondary Risk Factor for Coronary Artery Disease. Pathophysiol. Haemost. Thromb. 2007, 36, 45–51. [Google Scholar] [CrossRef]
- Frostegård, J. Immune Mechanisms in Atherosclerosis, Especially in Diabetes Type 2. Front. Endocrinol. 2013, 4, 162. [Google Scholar] [CrossRef] [Green Version]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2–mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.-I.; Edelstein, D.; Du, X.-L.; Brownlee, M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.Y.; Min, J.H.; Bin Wang, S.; Chae, Y.H.; Baek, J.Y.; Kim, M.; Ryu, J.-S.; Chang, T.-S. Resveratrol inhibits collagen-induced platelet stimulation through suppressing NADPH oxidase and oxidative inactivation of SH2 domain-containing protein tyrosine phosphatase-2. Free. Radic. Biol. Med. 2015, 89, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Bin Wang, S.; Park, S.J.; Oh, G.T.; Lee, S.-H.; Ho, Y.-S.; Chang, T.-S. Reactive Oxygen Species Play a Critical Role in Collagen-Induced Platelet ActivationviaSHP-2 Oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Jang, J.Y.; Chae, Y.H.; Min, J.H.; Baek, J.Y.; Kim, M.; Park, Y.; Hwang, G.S.; Ryu, J.S.; Chang, T.S. Kaempferol suppresses collagen-induced platelet activation by inhibiting NADPH oxidase and protecting SHP-2 from oxidative inactivation. Free Radic. Biol. Med. 2015, 83, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2019, 9, 3076. [Google Scholar] [CrossRef]
- Carr, M.E. Diabetes mellitus: A hypercoagulable state. J. Diabetes Complicat. 2001, 15, 44–54. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, J.E.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S. High coagulation factor levels and low protein C levels contribute to enhanced thrombin generation in patients with diabetes who do not have macrovascular complications. J. Diabetes Complicat. 2014, 28, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Sommeijer, D.W.; Hansen, H.R.; Van Oerle, R.; Hamulyák, K.; Van Zanten, A.P.; Meesters, E.; Spronk, H.M.H.; Cate, H.T. Soluble tissue factor is a candidate marker for progression of microvascular disease in patients with Type 2 diabetes. J. Thromb. Haemost. 2006, 4, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.W.; Kohler, H.P.; Ariens, R.A.; McCormack, L.J.; Grant, P.J. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care 2000, 23, 703–705. [Google Scholar] [CrossRef] [Green Version]
- Song, D.Y.; Gu, J.-Y.; Yoo, H.J.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Activation of Factor XII and Kallikrein-kinin System Combined with Neutrophil Extracellular Trap Formation in Diabetic Retinopathy. Exp. Clin. Endocrinol. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Boden, G.; Rao, A.K. Tissue factor and Toll-like receptor (TLR)4 in hyperglycaemia-hyperinsulinaemia. Effects in healthy subjects, and type 1 and type 2 diabetes mellitus. Thromb. Haemost. 2015, 113, 750–758. [Google Scholar] [CrossRef] [Green Version]
- Yasuma, T.; Yano, Y.; Alessandro-Gabazza, C.N.D.; Toda, M.; Gil-Bernabe, P.; Kobayashi, T.; Nishihama, K.; Hinneh, J.A.; Mifuji-Moroka, R.; Roeen, Z.; et al. Amelioration of Diabetes Mellitus by Protein S. Diabetes 2016, 65, 1940–1951. [Google Scholar] [CrossRef] [Green Version]
- Wieczór, R.; Wieczór, A.M.; Kulwas, A.; Rość, D. Type 2 Diabetes and Cardiovascular Factors Contrasted with Fibrinolysis Disorders in the Blood of Patients with Peripheral Arterial Disease. Medicina 2019, 55, 395. [Google Scholar] [CrossRef] [Green Version]
- Bryk, A.H.; Konieczynska, M.; Rostoff, P.; Broniatowska, E.; Hohendorff, J.; Malecki, M.T.; Undas, A. Plasma Protein Oxidation as a Determinant of Impaired Fibrinolysis in Type 2 Diabetes. Thromb. Haemost. 2019, 119, 213–222. [Google Scholar] [CrossRef]
- Maatman, B.T.; Schmeisser, G.; Kreutz, R.P. Fibrin Clot Strength in Patients with Diabetes Mellitus Measured by Thrombelastography. J. Diabetes Res. 2018, 2018, 4543065. [Google Scholar] [CrossRef] [Green Version]
- Bryk, A.H.; Konieczyńska, M.; Polak, M.; Plicner, D.; Bochenek, M.; Undas, A. Plasma fibrin clot properties and cardiovascular mortality in patients with type 2 diabetes: A long-term follow-up study. Cardiovasc. Diabetol. 2021, 20, 47. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Gamlen, T.; Standeven, K.F.; Mughal, S.; Hess, K.; Smith, K.A.; Dunn, E.J.; Anwar, M.M.; Rabbani, N.; Thornalley, P.J.; et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 2013, 122, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczynska, M.; Fil, K.; Bazanek, M.; Undas, A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb. Haemost. 2014, 111, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, Y.; Okumura, K.-I.; Yoshida, N.; Tayama, K.; Takemura, Y.; Inukai, T. Enhancement of Fibrinolysis in Poorly Controlled, Hospitalized Type 2 Diabetic Patients by Short-Term Metabolic Control: Association with a Decrease in Plasminogen Activator Inhibitor 1. Exp. Clin. Endocrinol. Diabetes 2004, 112, 175–180. [Google Scholar] [CrossRef]
- Verkleij, C.J.N.; De Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.; Meijers, J.C.M.; Marx, P.F. The Hemostatic System in Patients with Type 2 Diabetes with and Without Cardiovascular Disease. Clin. Appl. Thromb. 2010, 17, E57–E63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherif, E.M.; Elbarbary, N.S.; Al Aziz, M.M.A.; Mohamed, S.F. Plasma thrombin-activatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: Possible relation to diabetic microvascular complications. Blood Coagul. Fibrinolysis 2014, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum alpha2-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [PubMed]
- Fattah, M.A.; Shaheen, M.H.; Mahfouz, M.H. Disturbances of Haemostasis in Diabetes Mellitus. Dis. Markers 2004, 19, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erem, C.; Hacıhasanoğlu, A.; Çelik, Ş.; Ovalı, E.; Ersöz, H.Ö.; Ukinç, K.; Deger, O.; Telatar, M. Coagulation and Fibrinolysis Parameters in Type 2 Diabetic Patients with and without Diabetic Vascular Complications. Med. Princ. Pract. 2005, 14, 22–30. [Google Scholar] [CrossRef]
- Wu, M.D.; Atkinson, T.M.; Lindner, J.R. Platelets and von Willebrand factor in atherogenesis. Blood 2017, 129, 1415–1419. [Google Scholar] [CrossRef]
- Urano, T.; Castellino, F.J.; Suzuki, Y. Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 2018. [Google Scholar] [CrossRef]
- Peng, X.; Wang, X.; Fan, M.; Zhao, J.; Lin, L.; Liu, J. Plasma levels of von Willebrand factor in type 2 diabetes patients with and without cardiovascular diseases: A meta-analysis. Diabetes Metab. Res. Rev. 2020, 36, e3193. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.; Chow, W.-S.; Ai, V.H.; Metz, C.; Bucala, R.; Lam, K.S. Advanced glycation end products and endothelial dysfunction in type 2 diabetes. Diabetes Care 2002, 25, 1055–1059. [Google Scholar] [CrossRef] [Green Version]
- Kemeny, S.F.; Figueroa, D.S.; Andrews, A.M.; Barbee, K.A.; Clyne, A.M. Glycated collagen alters endothelial cell actin alignment and nitric oxide release in response to fluid shear stress. J. Biomech. 2011, 44, 1927–1935. [Google Scholar] [CrossRef]
- Khan, G.; Aftab, M.F.; Bano, B.; Khan, K.M.; Murtaza, M.; Siddiqui, S.; Rehman, M.H.; Waraich, R.S. A new indanedione derivative alleviates symptoms of diabetes by modulating RAGE-NF-kappaB pathway in db/db mice. Biochem. Biophys. Res. Commun. 2018, 501, 863–870. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1–RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetologia 2014, 57, 1047–1056. [Google Scholar] [CrossRef]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, N.; Ranalli, P.; Cuccurullo, C.; Davì, G. Diabetes mellitus and thrombosis. Thromb. Res. 2012, 129, 371–377. [Google Scholar] [CrossRef]
- Folli, F.; Corradi, D.; Fanti, P.; Davalli, A.; Paez, A.; Giaccari, A.; Perego, C.; Muscogiuri, G. The Role of Oxidative Stress in the Pathogenesis of Type 2 Diabetes Mellitus Micro- and Macrovascular Complications: Avenues for a Mechanistic-Based Therapeutic Approach. Curr. Diabetes Rev. 2011, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Kosugi, S.; Sano, F. Improvement of High Serum Levels of Biomarkers of Endothelial Injury (Vascular Cell Adhesion Molecule-1) and Inflammation (Tumor Necrosis Factor Receptor Type I) after Allogeneic Hematopoietic Stem Cell Transplantation with Sinusoidal Obstruction Syndrome Using Defibrotide. Am. J. Ther. 2020. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.-L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef]
- Elmariah, S.; Mauri, L.; Doros, G.; Galper, B.Z.; O’Neill, K.E.; Steg, P.G.; Kereiakes, D.J.; Yeh, R.W. Extended duration dual antiplatelet therapy and mortality: A systematic review and meta-analysis. Lancet 2015, 385, 792–798. [Google Scholar] [CrossRef] [Green Version]
- The ASCEND Study Collaborative Group; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Seidu, S.; Kunutsor, S.K.; Sesso, H.D.; Gaziano, J.M.; Buring, J.E.; Roncaglioni, M.C.; Khunti, K. Aspirin has potential benefits for primary prevention of cardiovascular outcomes in diabetes: Updated literature-based and individual participant data meta-analyses of randomized controlled trials. Cardiovasc. Diabetol. 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas Rios, J.R.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Diabetes and antiplatelet therapy: From bench to bedside. Cardiovasc. Diagn. Ther. 2018, 8, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.N.; Deyell, J.S.; Sharma, S.N.; Barseghian, A. Role of and Recent Evidence for Antiplatelet Therapy in Prevention of Cardiovascular Disease in Diabetes. Curr. Cardiol. Rep. 2019, 21, 78. [Google Scholar] [CrossRef]
- Meredith, I.T.; Tanguay, J.-F.; Kereiakes, D.J.; Cutlip, D.E.; Yeh, R.W.; Garratt, K.N.; Lee, D.P.; Steg, P.G.; Weaver, W.D.; Holmes, D.R., Jr.; et al. Diabetes Mellitus and Prevention of Late Myocardial Infarction After Coronary Stenting in the Randomized Dual Antiplatelet Therapy Study. Circulation 2016, 133, 1772–1782. [Google Scholar] [CrossRef]
- Calderone, D.; Capodanno, D.; Angiolillo, D.J. An updated drug profile of ticagrelor with considerations on the treatment of patients with coronary artery disease and diabetes mellitus. Expert Rev. Cardiovasc. Ther. 2020, 18, 449–464. [Google Scholar] [CrossRef]
- Neri Serneri, G.G.; Coccheri, S.; Marubini, E.; Violi, F.; Drug Evaluation in Atherosclerotic Vascular Disease in Diabetics (DAVID) Study Group. Picotamide, a combined inhibitor of thromboxane A2 synthase and receptor, reduces 2-year mortality in diabetics with peripheral arterial disease: The DAVID study. Eur. Heart J. 2004, 25, 1845–1852. [Google Scholar] [CrossRef]
- Stone, G.W.; Bertrand, M.; Colombo, A.; Dangas, G.; Farkouh, M.E.; Feit, F.; Lansky, A.J.; Lincoff, A.; Mehran, R.; Moses, J.W.; et al. Acute Catheterization and Urgent Intervention Triage strategY (ACUITY) trial: Study design and rationale. Am. Heart J. 2004, 148, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, D.; Xanthopoulou, I.; Mavronasiou, E.; Stavrou, K.; Siapika, A.; Tsoni, E.; Davlouros, P. Randomized Assessment of Ticagrelor Versus Prasugrel Antiplatelet Effects in Patients with Diabetes. Diabetes Care 2013, 36, 2211–2216. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Nawaz, M.I.; Siddiquei, M.M.; Abu El-Asrar, A.M. Apocynin ameliorates NADPH oxidase 4 (NOX4) induced oxidative damage in the hypoxic human retinal Müller cells and diabetic rat retina. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Kwon, G.; Uddin, M.J.; Lee, G.; Jiang, S.; Cho, A.; Lee, J.H.; Lee, S.R.; Bae, Y.S.; Moon, S.H.; Lee, S.J.; et al. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: Possible role of peroxisomal and mitochondrial biogenesis. Oncotarget 2017, 8, 74217–74232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.S.; Kim, H.M.; Lee, S.H.; Ha, K.B.; Bae, Y.S.; Lee, S.J.; Moon, S.H.; Lee, E.Y.; Lee, J.H.; Chung, C.H. APX-115, a pan-NADPH oxidase inhibitor, protects development of diabetic nephropathy in podocyte specific NOX5 transgenic mice. Free Radic. Biol. Med. 2020, 161, 92–101. [Google Scholar] [CrossRef]
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; De Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH Oxidase 1 Plays a Key Role in Diabetes Mellitus–Accelerated Atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; Van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [Green Version]
- De Livera, A.M.; Reutens, A.; Cooper, M.; Thomas, M.; Jandeleit-Dahm, K.; Shaw, J.E.; Salim, A. Evaluating the efficacy and safety of GKT137831 in adults with type 1 diabetes and persistently elevated urinary albumin excretion: A statistical analysis plan. Trials 2020, 21, 459. [Google Scholar] [CrossRef]
- Carnevale, R.; Loffredo, L.; Pignatelli, P.; Nocella, C.; Bartimoccia, S.; Di Santo, S.; Martino, F.; Catasca, E.; Perri, L.; Violi, F. Dark chocolate inhibits platelet isoprostanes via NOX2 down-regulation in smokers. J. Thromb. Haemost. 2012, 10, 125–132. [Google Scholar] [CrossRef]
- Wang, Y.; Chun, O.K.; Song, W.O. Plasma and Dietary Antioxidant Status as Cardiovascular Disease Risk Factors: A Review of Human Studies. Nutrients 2013, 5, 2969–3004. [Google Scholar] [CrossRef] [Green Version]
- Mega, J.L.; Braunwald, E.; Wiviott, S.D.; Bassand, J.-P.; Bhatt, D.L.; Bode, C.; Burton, P.; Cohen, M.; Cook-Bruns, N.; Fox, K.A.; et al. Rivaroxaban in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 366, 9–19. [Google Scholar] [CrossRef] [Green Version]
- January, C.T.; Wann, L.S.; Calkins, H.; Chen, L.Y.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; Furie, K.L.; et al. 2019 AHA/ACC/HRS Focused Update of the 2014 AHA/ACC/HRS Guideline for the Management of Patients with Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society in Collaboration with the Society of Thoracic Surgeons. Circulation 2019, 140, e125–e151. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-H.; Lee, H.-F.; Li, P.-R.; Liu, J.-R.; Chao, T.-F.; Wu, L.-S.; Chang, S.-H.; Yeh, Y.-H.; Kuo, C.-T.; See, L.-C.; et al. Effectiveness, safety, and major adverse limb events in atrial fibrillation patients with concomitant diabetes mellitus treated with non-vitamin K antagonist oral anticoagulants. Cardiovasc. Diabetol. 2020, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Ezekowitz, J.A.; Lewis, B.S.; Lopes, R.D.; Wojdyla, D.M.; McMurray, J.J.; Hanna, M.; Atar, D.; Bahit, M.C.; Keltai, M.; Lopez-Sendon, J.L.; et al. Clinical outcomes of patients with diabetes and atrial fibrillation treated with apixaban: Results from the ARISTOTLE trial. Eur. Heart J. Cardiovasc. Pharmacother. 2015, 1, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambatti, M.; Darius, H.; Oldgren, J.; Clemens, A.; Noack, H.H.; Brueckmann, M.; Yusuf, S.; Wallentin, L.; Ezekowitz, M.D.; Connolly, S.J.; et al. Comparison of dabigatran versus warfarin in diabetic patients with atrial fibrillation: Results from the RE-LY trial. Int. J. Cardiol. 2015, 196, 127–131. [Google Scholar] [CrossRef]
- De Caterina, R.; Patti, G.; Westerbergh, J.; Horowitz, J.; Ezekowitz, J.A.; Lewis, B.S.; Lopes, R.D.; McMurray, J.J.V.; Atar, D.; Bahit, M.C.; et al. Heterogeneity of diabetes as a risk factor for major adverse cardiovascular events in anticoagulated patients with atrial fibrillation: An analysis of the ARISTOTLE trial. Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]
Figure 1.
NOX1 upregulation in platelets from DM patients with poor glycemic control. Human platelets from patients with HbA1C > 7.0% were isolated and their levels of NOX1 expression were assessed by immunoblotting as previously described [23]. Actin was co-immunostained as a loading control. NOX1 expression was quantified by densitometry and expression as NOX1/actin ratio (Image J, 47v, Wayne Rasband, National Institute of Health, USA). (A) Representative example is shown in (A), with the statistical analysis shown in (B) (Student’s t-test, * p < 0.05, n = 10).
Figure 1.
NOX1 upregulation in platelets from DM patients with poor glycemic control. Human platelets from patients with HbA1C > 7.0% were isolated and their levels of NOX1 expression were assessed by immunoblotting as previously described [23]. Actin was co-immunostained as a loading control. NOX1 expression was quantified by densitometry and expression as NOX1/actin ratio (Image J, 47v, Wayne Rasband, National Institute of Health, USA). (A) Representative example is shown in (A), with the statistical analysis shown in (B) (Student’s t-test, * p < 0.05, n = 10).

Figure 2.
The genetic silencing of NOXs in platelets abolishes thrombotic carotid occlusion induced by ferric chloride. Local application of 5% w/v ferric chloride induced carotid occlusion (A). Doppler ultrasound scanning measured carotid blood flow and complete occlusion times were plotted (B). C57BL6/J (WT) were compared to Nox1-/-/Nox2-/-/Nox4-/- (3KO) and thrombocytopenic 3KO mice that received infusion of WT platelets. Platelet depletion was induced in 3KO mice by IV injection of the anti-GPIbα antibody R300 (0.2 μg/g body weight). Twelve hours after antibody injection, 6 × 108 platelets from WT mice were IV injected into thrombocytopenic mice (thrombocytopenia was confirmed by blood platelet counting). Data are mean ± SEM and statistical analysis was performed by one-way ANOVA with Bonferroni post-test (** p < 0.01, n = 4–7).
Figure 2.
The genetic silencing of NOXs in platelets abolishes thrombotic carotid occlusion induced by ferric chloride. Local application of 5% w/v ferric chloride induced carotid occlusion (A). Doppler ultrasound scanning measured carotid blood flow and complete occlusion times were plotted (B). C57BL6/J (WT) were compared to Nox1-/-/Nox2-/-/Nox4-/- (3KO) and thrombocytopenic 3KO mice that received infusion of WT platelets. Platelet depletion was induced in 3KO mice by IV injection of the anti-GPIbα antibody R300 (0.2 μg/g body weight). Twelve hours after antibody injection, 6 × 108 platelets from WT mice were IV injected into thrombocytopenic mice (thrombocytopenia was confirmed by blood platelet counting). Data are mean ± SEM and statistical analysis was performed by one-way ANOVA with Bonferroni post-test (** p < 0.01, n = 4–7).

Figure 3.
DM-dependent mechanisms underlying vascular damage. Platelet hyperactivity, blood hypercoagulability, and endothelial cell dysfunction are independent drivers of cardiovascular risk in DM patients. Abbreviation list: VWF: Von Willebrand factor; PAI-1: Plasminogen activator inhibitor; NO: Nitric oxide; eNOS: Endothelial nitric oxide synthase; NFκB: Nuclear factor kappa B; Rac-1: Ras-related C3 botulinum toxin substrate 1; TIAM-1: T cell lymphoma invasion and metastasis 1; AGE: Advanced glycation end product; RAGE: Receptor for advanced glycation end products; ROS: Reactive oxygen species; ICAM-1: Intercellular adhesion molecule 1; VCAM-1: Vascular cell adhesion molecule; Ox-LDL: Oxidized low-density lipoprotein; CD36: Cluster of Differentiation 36; NOX1: NADPH oxidase1; NOX2: NADPH oxidase 2; CD40L (Cluster Differentiation 40 Ligand: CD154 (Cluster of Differentiation 154); PTP: Protein tyrosine phosphatase; SHP-2: Src homology region 2 domain-containing phosphatase-2; cAMP: Cyclic adenosine monophosphate; cGMP: Cyclic guanosine monophosphate; PGI2R: Receptor of prostacyclin; IGF-1R: Insulin-like growth factor receptor; PK: Pre-kallikrein; TAFI: Thrombin-activatable fibrinolysis inhibitor.
Figure 3.
DM-dependent mechanisms underlying vascular damage. Platelet hyperactivity, blood hypercoagulability, and endothelial cell dysfunction are independent drivers of cardiovascular risk in DM patients. Abbreviation list: VWF: Von Willebrand factor; PAI-1: Plasminogen activator inhibitor; NO: Nitric oxide; eNOS: Endothelial nitric oxide synthase; NFκB: Nuclear factor kappa B; Rac-1: Ras-related C3 botulinum toxin substrate 1; TIAM-1: T cell lymphoma invasion and metastasis 1; AGE: Advanced glycation end product; RAGE: Receptor for advanced glycation end products; ROS: Reactive oxygen species; ICAM-1: Intercellular adhesion molecule 1; VCAM-1: Vascular cell adhesion molecule; Ox-LDL: Oxidized low-density lipoprotein; CD36: Cluster of Differentiation 36; NOX1: NADPH oxidase1; NOX2: NADPH oxidase 2; CD40L (Cluster Differentiation 40 Ligand: CD154 (Cluster of Differentiation 154); PTP: Protein tyrosine phosphatase; SHP-2: Src homology region 2 domain-containing phosphatase-2; cAMP: Cyclic adenosine monophosphate; cGMP: Cyclic guanosine monophosphate; PGI2R: Receptor of prostacyclin; IGF-1R: Insulin-like growth factor receptor; PK: Pre-kallikrein; TAFI: Thrombin-activatable fibrinolysis inhibitor.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vaidya, A.R.; Wolska, N.; Vara, D.; Mailer, R.K.; Schröder, K.; Pula, G. Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants 2021, 10, 706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050706
AMA Style
Vaidya AR, Wolska N, Vara D, Mailer RK, Schröder K, Pula G. Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants. 2021; 10(5):706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050706
Chicago/Turabian StyleVaidya, Aishwarya R., Nina Wolska, Dina Vara, Reiner K. Mailer, Katrin Schröder, and Giordano Pula. 2021. "Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress" Antioxidants 10, no. 5: 706. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10050706
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.