
Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibody
2.2. Cell Culture and Viability Assay
2.3. Analysis of Viability and Dead Cells Populations
2.4. Molecular Docking
2.5. Measurement of IL-1β by ELISA
2.6. Western Blotting
2.7. Reverse Transcription-Polymerase Chain Reactions (RT-PCR) Using Mouse Specific Primers
2.8. Immunostaining of p65 and p62
2.9. Analysis of mtROS
2.10. Mitochondrial Membrane Depolarization
2.11. Transfection of p62 Small Interfering RNA (siRNA)
2.12. Maintenance of Zebrafish Embryo and Larvae
2.13. Cardiac Toxicity Evaluation
2.14. Neutral Red Staining
2.15. Isolation of Total Zebrafish mRNA and RT-PCR
2.16. Statistical Analysis
3. Results
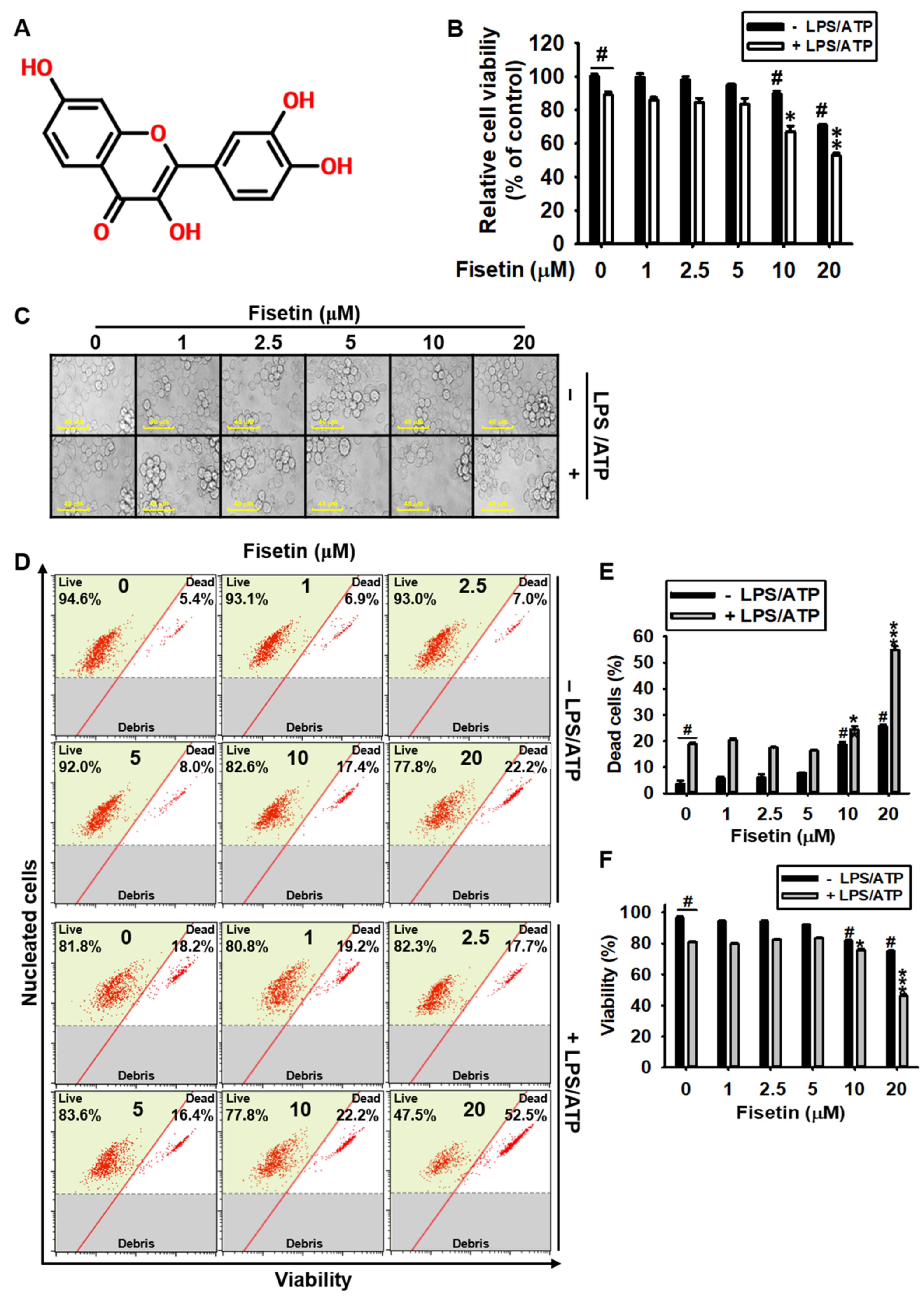
3.1. High Concentrations of Fisetin Is Cytotoxic to BV2 Microglial Cells
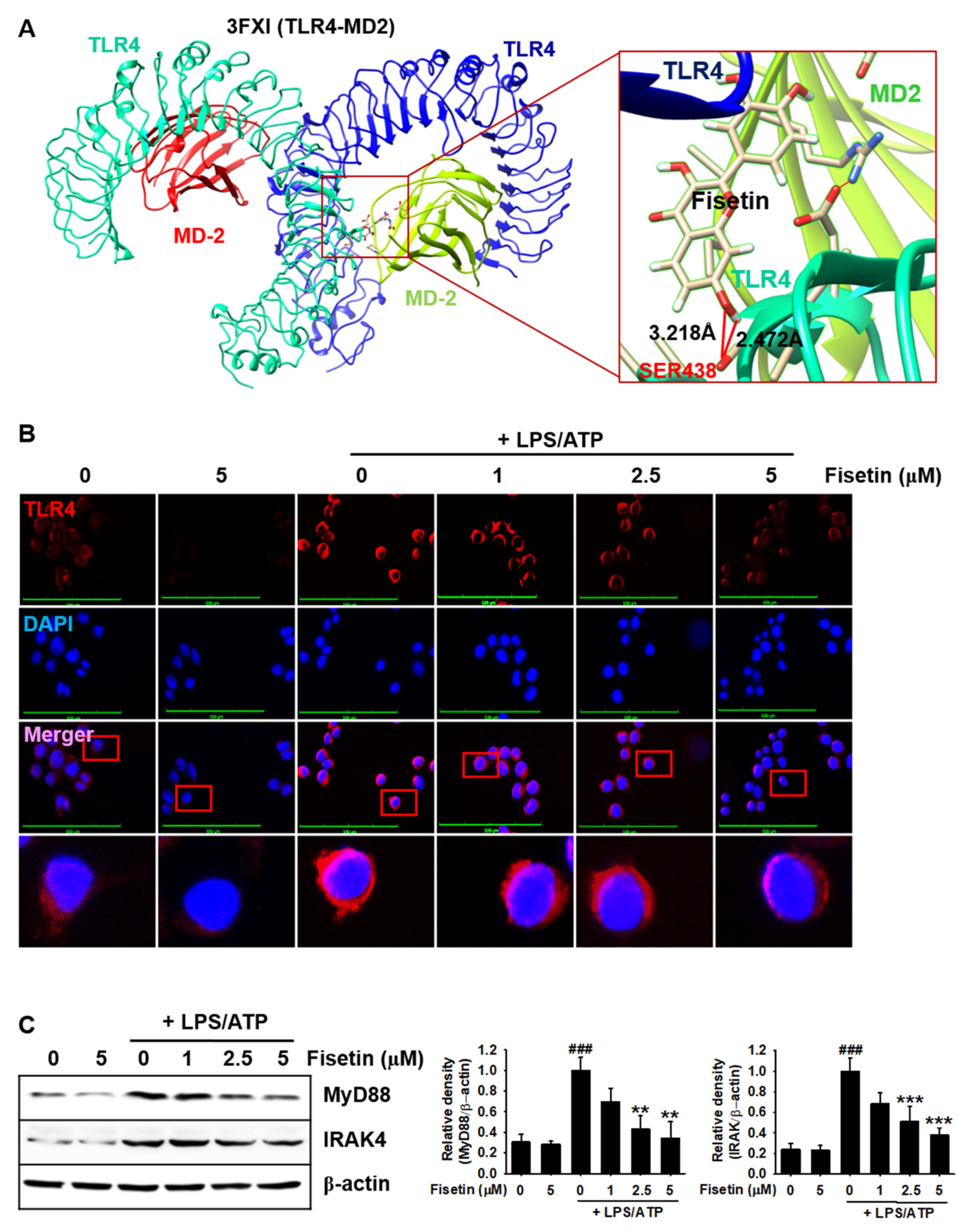
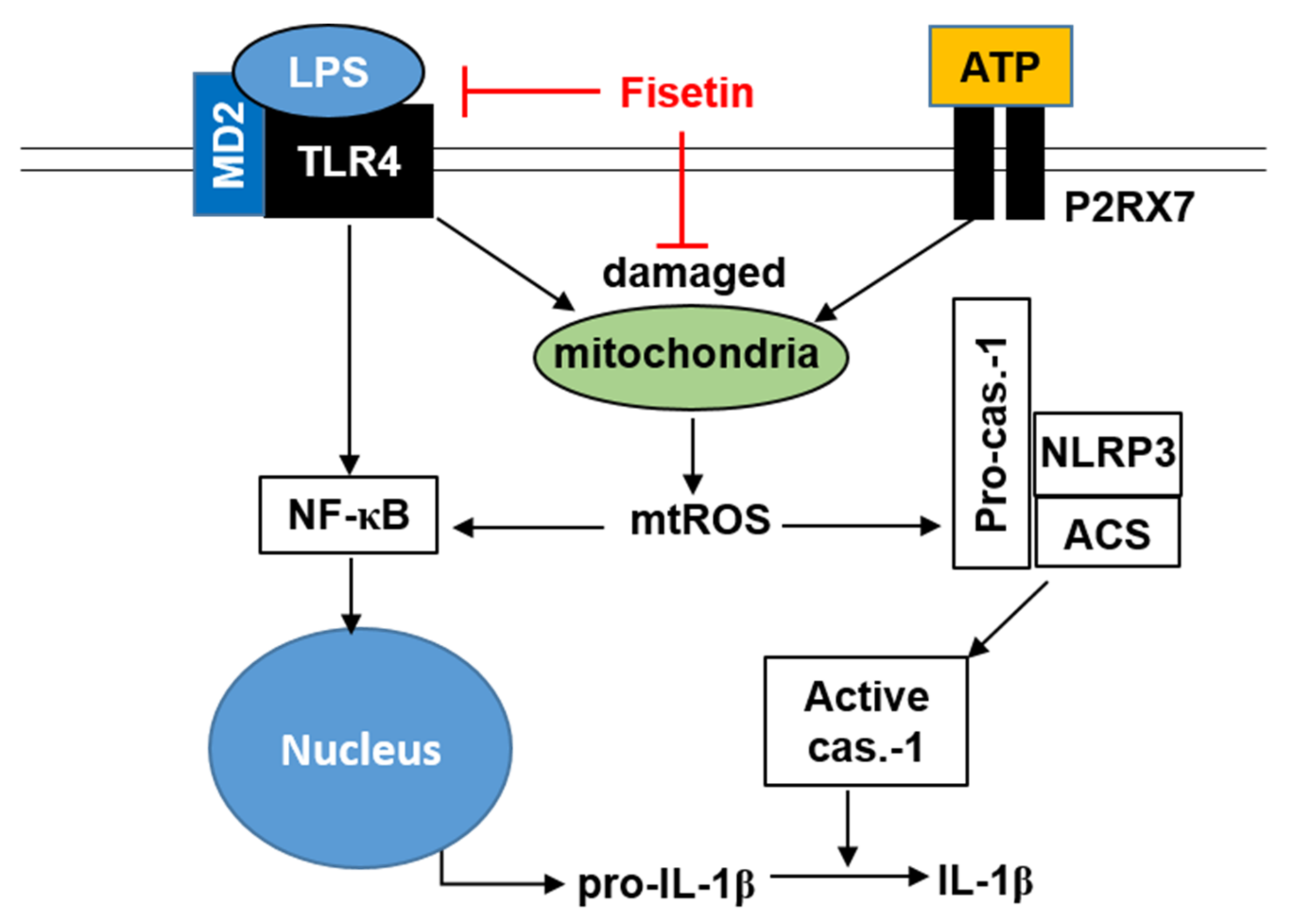
3.2. Fisetin Binds to the TLR4/MD2 Complex and Inhibits the Downstream Signaling Pathway
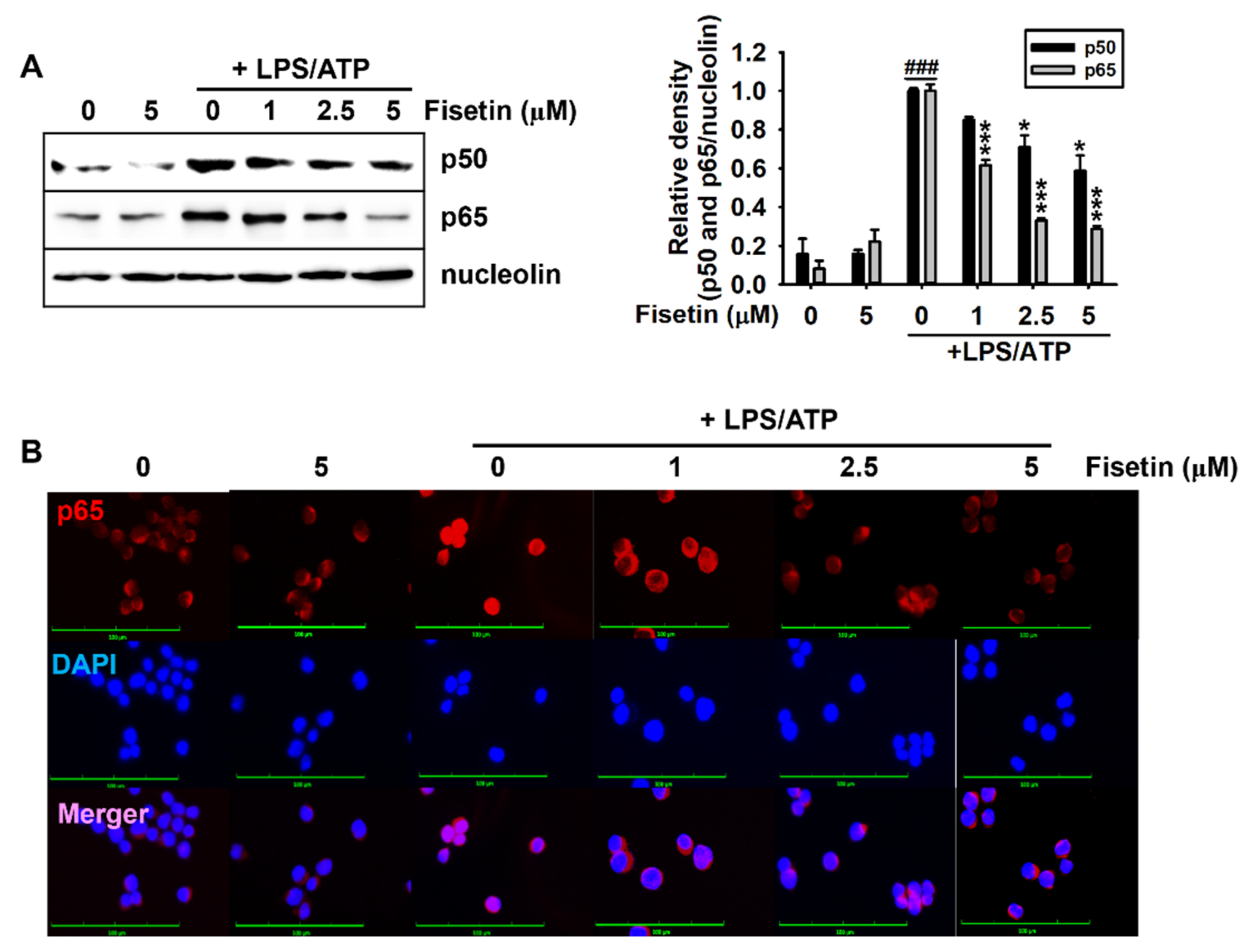
3.3. Fisetin Inhibits the NF-κB Signaling Pathway
3.4. Fisetin Inhibits the Expression of the Components of the NLRP3 Inflammasome
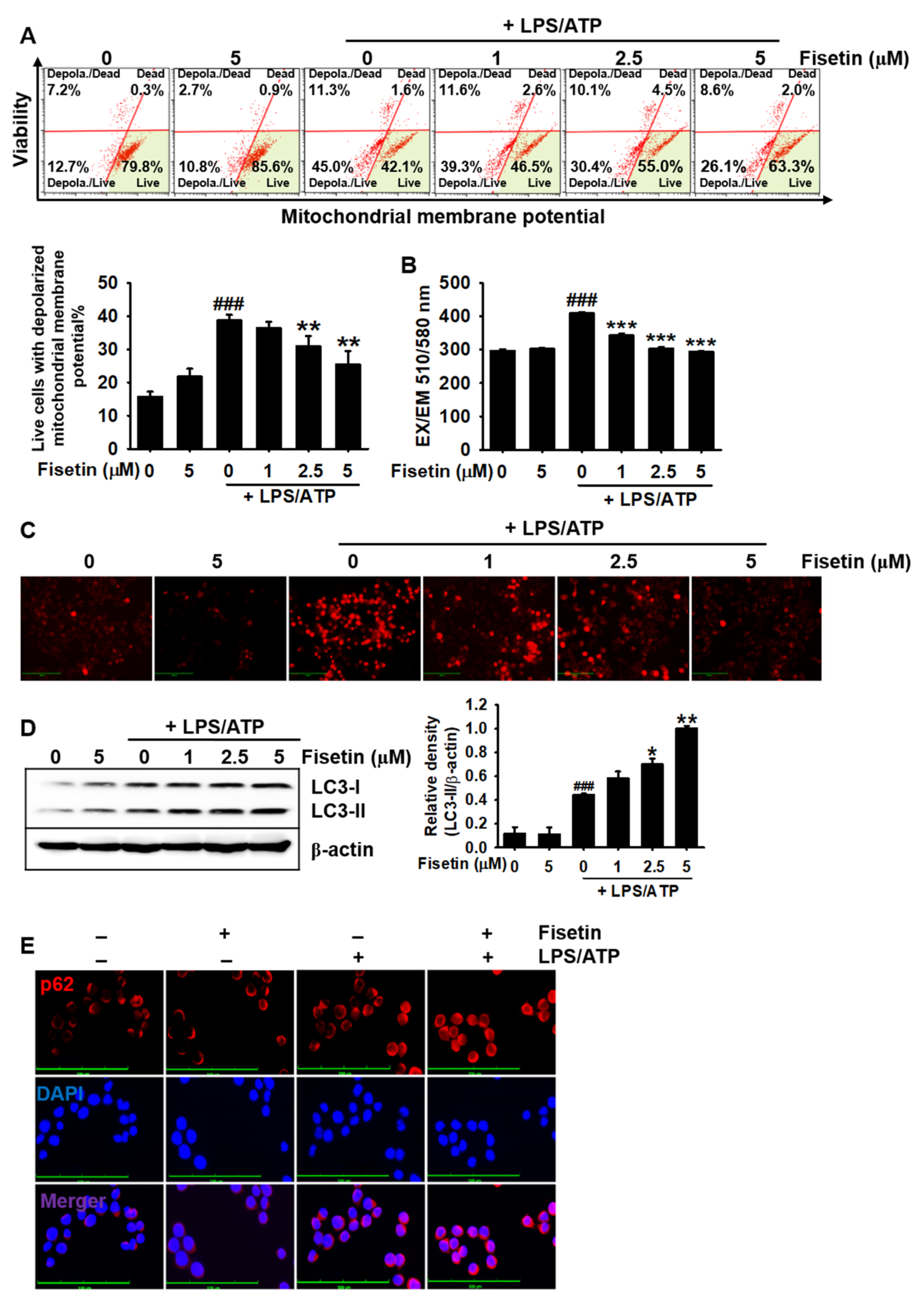
3.5. Fisetin Downregulates Mitochondrial Membrane Depolarization and mtROS Production
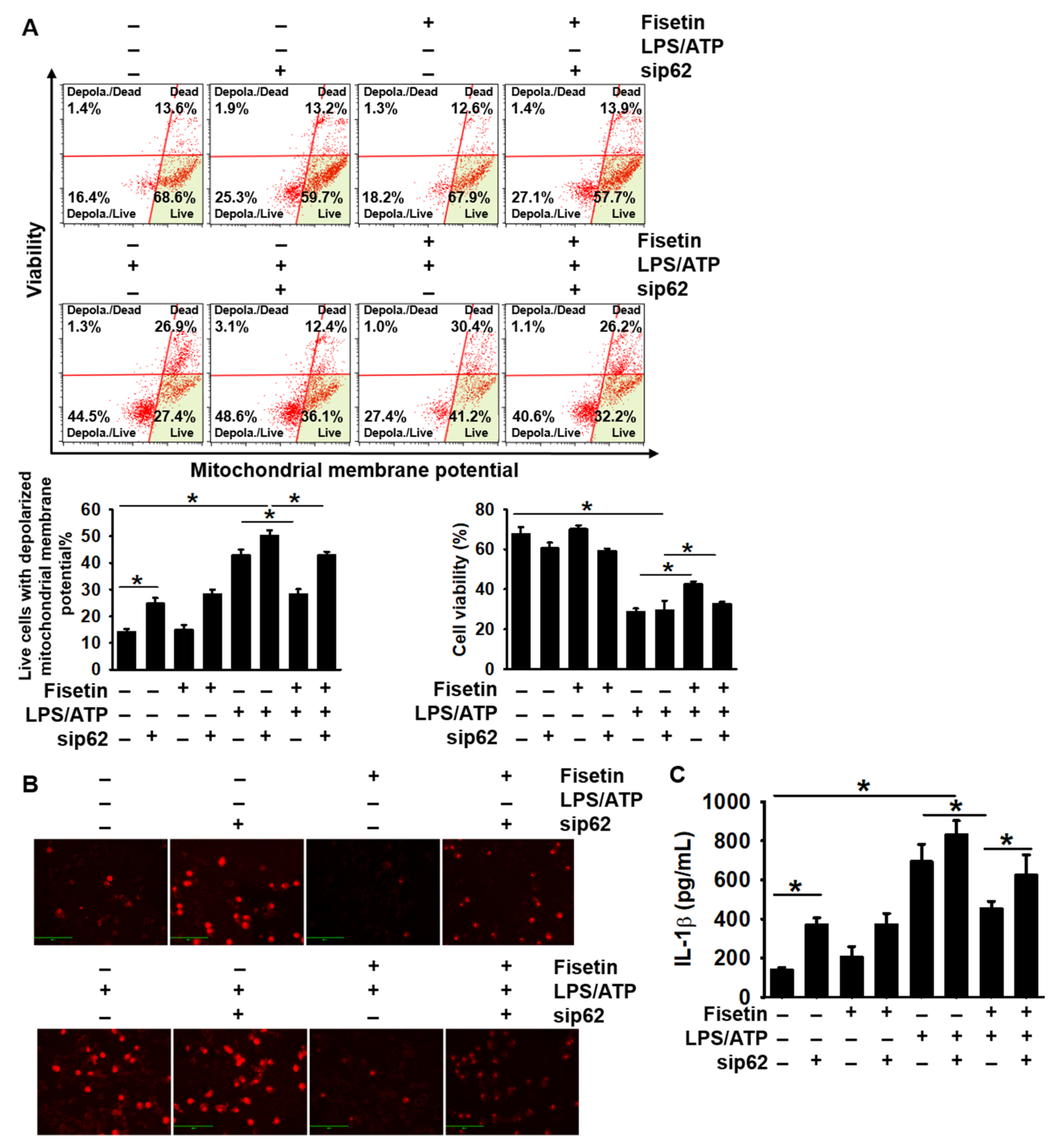
3.6. Transient Knockdown of p62 Reverses Fisetin-Induced Mitophagy and Formation of the NLRP3 Inflammasome
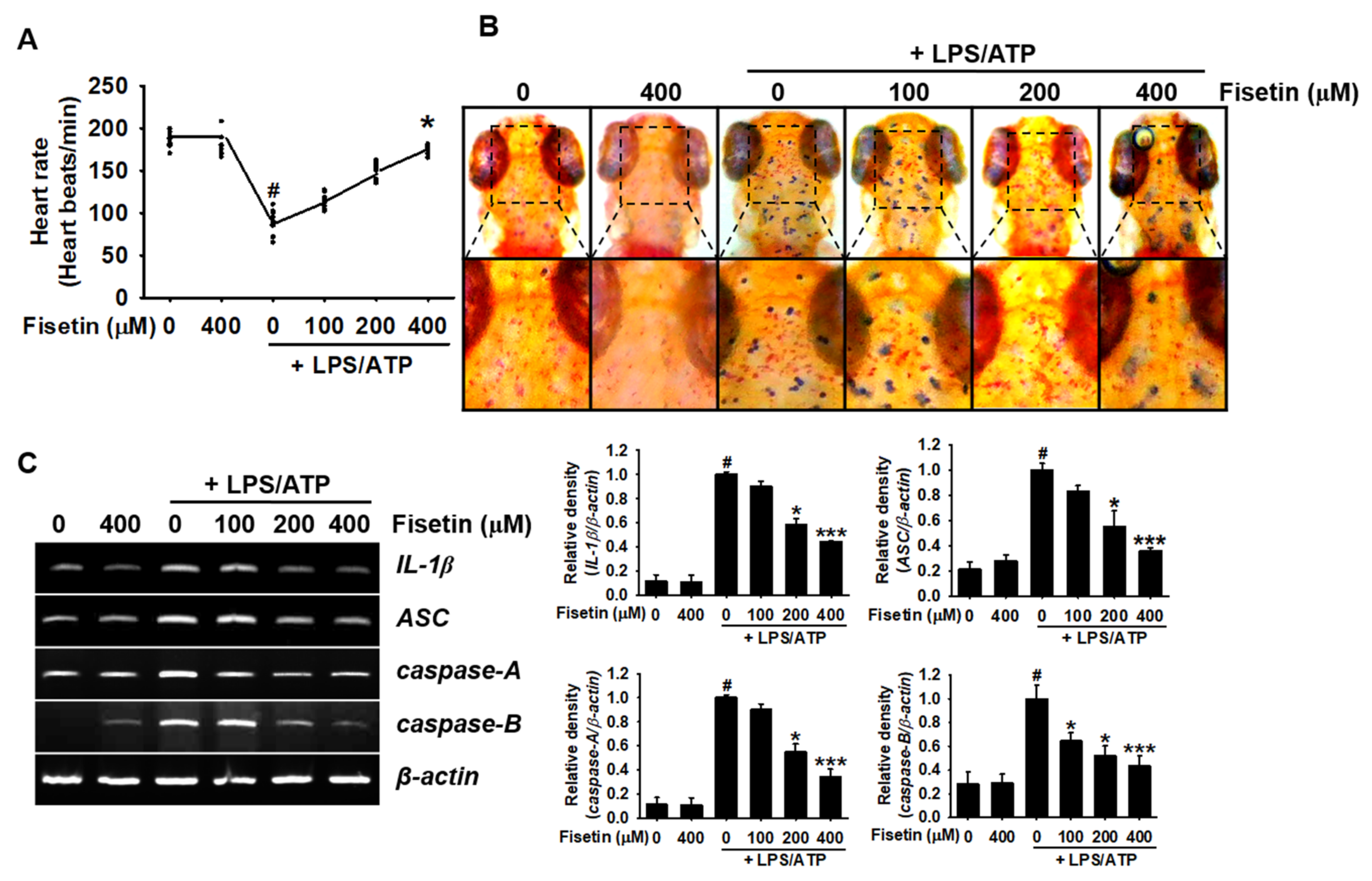
3.7. Fisetin Inhibits Activation of the NLRP3 Inflammasome in Zebrafish Larvae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, A.; Kim, S.H.; Arifuzzaman, S.; Yoon, T.; Chai, J.C.; Lee, Y.S.; Park, K.S.; Jung, K.H.; Chai, Y.G. Transcriptome sequencing reveals that LPS-triggered transcriptional responses in established microglia BV2 cell lines are poorly representative of primary microglia. J. Neuroinflamm. 2016, 13, 182. [Google Scholar] [CrossRef] [Green Version]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Cappellano, G.; Carecchio, M.; Fleetwood, T.; Magistrelli, L.; Cantello, R.; Dianzani, U.; Comi, C. Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 89–107. [Google Scholar]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Allan, S.M.; Tyrrell, P.J.; Rothwell, N.J. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005, 5, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Torres, V.; Kielian, T. Inflammasome activation and IL-1β/IL-18 processing are influenced by distinct pathways in microglia. J. Neurochem. 2011, 119, 736–748. [Google Scholar] [CrossRef] [PubMed]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. CMLS 2011, 68, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernucci, E.; Tomino, C.; Molinari, F.; Limongi, D.; Aventaggiato, M.; Sansone, L.; Tafani, M.; Russo, M.A. Mitophagy and oxidative stress in cancer and aging: Focus on sirtuins and nanomaterials. Oxid. Med. Cell. Longev. 2019, 2019, 6387357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Teaching the basics of autophagy and mitophagy to redox biologists—Mechanisms and experimental approaches. Redox Biol. 2015, 4, 242–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Misrani, A.; Tang, B.-l.; Chen, J.; Yang, L.; Long, C. Jujuboside A prevents sleep loss-induced disturbance of hippocampal neuronal excitability and memory impairment in young APP/PS1 mice. Sci. Rep. 2019, 9, 4512. [Google Scholar] [CrossRef]
- Kicinska, A.; Jarmuszkiewicz, W. Flavonoids and mitochondria: Activation of cytoprotective pathways? Molecules 2020, 25, 3060. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Jiang, Z.-Y.; Sun, B.; Fu, J.; Li, T.-Z. Fisetin alleviates lipopolysaccharide-induced acute lung injury via TLR4-mMediated NF-κB signaling pathway in rats. Inflammation 2016, 39, 148–157. [Google Scholar] [CrossRef]
- Suh, Y.; Afaq, F.; Khan, N.; Johnson, J.J.; Khusro, F.H.; Mukhtar, H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis 2010, 31, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanneken, A.; Lin, F.F.; Maher, P. Flavonoids protect human retinal pigment epithelial cells from oxidative stress–induced death. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1634. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Karunarathne, W.A.H.M.; Park, S.R.; Choi, Y.H.; Park, E.K.; Jin, C.-Y.; Yu, H.; Jo, W.S.; Lee, K.T.; Kim, G.-Y. GSK-3β-targeting fisetin promotes melanogenesis in B16F10 melanoma cells and zebrafish larvae through β-catenin activation. Int. J. Mol. Sci. 2020, 21, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-C.; Kang, S.-H.; Jeong, S.-J.; Kim, S.-H.; Ko, H.S.; Kim, S.-H. Inhibition of c-Jun N-terminal kinase and nuclear factor κB pathways mediates fisetin-exerted anti-inflammatory activity in lipopolysccharide-treated RAW264. 7 cells. Immunopharmacol. Immunotoxicol. 2012, 34, 645–650. [Google Scholar] [CrossRef]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Jayasingha, J.; Choi, Y.H.; Jayasooriya, R.; Kang, C.H.; Kim, G.Y. Fisetin inhibits lipopolysaccharide-induced inflammatory response by activating b-catenin, leading to a decrease in endotoxic shock. Sci. Rep. 2021, 11, 8377. [Google Scholar] [CrossRef]
- Molagoda, I.M.N.; Lee, S.; Jayasooriya, R.; Jin, C.Y.; Choi, Y.H.; Kim, G.Y. Deoxynivalenol enhances IL-1b expression in BV2 microglial cells through activation of the NF-kB pathway and the ASC/NLRP3 inflammasome. EXCLI J. 2019, 18, 356–369. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]
- Karunarathne, W.; Lee, K.T.; Choi, Y.H.; Jin, C.Y.; Kim, G.Y. Anthocyanins isolated from Hibiscus syriacus L. attenuate lipopolysaccharide-induced inflammation and endotoxic shock by inhibiting the TLR4/MD2-mediated NF-kB signaling pathway. Phytomedicine 2020, 76, 153237. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, X.; Wu, H.; Xiao, J.; Gao, Y.; Zhang, Y. Role of intestinal inflammation in predisposition of Edwardsiella tarda infection in zebrafish (Danio rerio). Fish Shellfish Immunol. 2014, 41, 271–278. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An Overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Demchuk, O.M. New perspectives for fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef]
- Sundarraj, K.; Raghunath, A.; Perumal, E. A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed. Pharm. 2018, 97, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rodriguez, A.B.; Hennessy, E.; Murray, C.L.; Nazmi, A.; Delaney, H.J.; Healy, D.; Fagan, S.G.; Rooney, M.; Stewart, E.; Lewis, A.; et al. Acute systemic inflammation exacerbates neuroinflammation in Alzheimer’s disease: IL-1b drives amplified responses in primed astrocytes and neuronal network dysfunction. Alzheimers Dement. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sang, X.; Wang, H.; Chen, Y.; Guo, Q.; Lu, A.; Zhu, X.; Meng, G. Vitamin C inhibits the activation of the NLRP3 inflammasome by scavenging mitochondrial ROS. Inflammasome 2016, 2, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Shiau, C.E.; Monk, K.R.; Joo, W.; Talbot, W.S. An Anti-inflammatory NOD-like Receptor Is Required for Microglia Development. Cell Rep. 2013, 5, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Hu, L.; Li, D.; Wang, Y.; He, Y.; Zhu, L.; Ren, W. MD-2 regulates LPS-induced NLRP3 inflammasome activation and IL-1b secretion by a MyD88/NF-kB-dependent pathway in alveolar macrophages cell line. Mol. Immunol. 2017, 90, 1–10. [Google Scholar] [CrossRef]
- Lima, C.X.; Souza, D.G.; Amaral, F.A.; Fagundes, C.T.; Rodrigues, I.P.S.; Alves-Filho, J.C.; Kosco-Vilbois, M.; Ferlin, W.; Shang, L.; Elson, G.; et al. Therapeutic effects of treatment with anti-TLR2 and anti-TLR4 monoclonal antibodies in polymicrobial sepsis. PLoS ONE 2015, 10, e0132336. [Google Scholar] [CrossRef] [Green Version]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Taxman, D.J.; Lei, Y.; Zhang, S.; Holley-Guthrie, E.; Offenbacher, S.; Ting, J.P. ASC-dependent RIP2 kinase regulates reduced PGE2 production in chronic periodontitis. J. Dent. Res. 2012, 91, 877–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, C.; Anand, P.K. Right place, right time: Localisation and assembly of the NLRP3 inflammasome. F1000Res 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to caspase-1–dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Sakai, E.; Shimada-Sugawara, M.; Yamaguchi, Y.; Sakamoto, H.; Fumimoto, R.; Fukuma, Y.; Nishishita, K.; Okamoto, K.; Tsukuba, T. Fisetin inhibits osteoclastogenesis through prevention of RANKL-induced ROS production by Nrf2-mediated up-regulation of phase II antioxidant enzymes. J. Pharmacol. Sci. 2013, 121, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Rodius, S.; de Klein, N.; Jeanty, C.; Sánchez-Iranzo, H.; Crespo, I.; Ibberson, M.; Xenarios, I.; Dittmar, G.; Mercader, N.; Niclou, S.P.; et al. Fisetin protects against cardiac cell death through reduction of ROS production and caspases activity. Sci. Rep. 2020, 10, 2896. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Yoon, S.-O.; Lee, H.J.; Oh, J.Y. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget 2017, 8, 40817–40831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.-S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.-I.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Gene * | Primer Sequence(5′-3′) | Accesion Number | Size | Tm | Cycle No. |
|---|---|---|---|---|---|---|
| mouse | NLRP3 | F: 5′-ATTACCCGCCCGAGAAAGG-3′ | NM_001359638 | 141 bp | 58 °C | 27 |
| R: 5′-TCGCAGCAAAGATCCACACAG-3′ | ||||||
| ASC | F: 5′-GCAACTGCGAGAAGGCTAT-3′ | NM_023258 | 256 bp | 58 °C | 27 | |
| R: 5′-CTGGTCCACAAAGTGTCCTG-3′ | ||||||
| IL-1β | F: 5′-GCCCATCCTCTGTGACTCAT-3′ | NM_008361 | 230 bp | 65 °C | 27 | |
| R: 5′-AGGCCACAGGTATTTTGTCG-3′ | ||||||
| caspase-1 | F: 5′-CTGACTGGGACCCTCAAG-3′ | NM_009807 | 529 bp | 63 °C | 27 | |
| R: 5′-CCTCTTCAGAGTCTCTTACTG-3′ | ||||||
| GAPDH | F: 5′-ACCACAGTCCATGCCATCAC-3′ | NM_001289726 | 450 bp | 63 °C | 23 | |
| R: 5′-CACCACCCTGTTGCTGTAGC-3′ | ||||||
| zebrafish | IL-1β | F 5′-TGGACTTCGCAGCACAAAATG-3′ | NM_212844 | 149 bp | 59 °C | 27 |
| R 5′-GTTCACTTCACGCTCTTGGATG-3′ | ||||||
| ASC | F 5′-GGCGGAATCTTTCAAGGAGC-3′ | NM_131495 | 171 bp | 58 °C | 27 | |
| R 5′-ACGCCGACCATTAAATCAGC-3′ | ||||||
| caspase-A | F 5′-GAGAATTGTCCAGCTCTGCG -3′ | NM_131505 | 198 bp | 58 °C | 27 | |
| R 5′-GCCGGTAAGATTTGGTGTCC-3′ | ||||||
| caspase-B | F 5′-CCTCGAGGATCTTGTGGAGT-3′ | NM_152884 | 184 bp | 58 °C | 27 | |
| R 5′-GCTTGATTTTGCGCAGTGTC-3′ | ||||||
| β-actin | F 5′-CGAGCGTGGCTACAGCTTCA-3′ | NM_131031 | 155 bp | 61 °C | 23 | |
| R 5′-GACCGTCAGGCAGCTCATAG-3′ |
| Binding Pose | Binding Score | Binding to TLR4 | |
|---|---|---|---|
| Binding AA 1 | H-Bond 2 Distance (Å) | ||
| 1 | −6.6 | SER438 (OG) 3 | 3.218 |
| SER438 (OG) | 2.472 | ||
| 2 | −6.3 | LYS435 (NZ) | 3.440 |
| SER438 (N) | 2.862 | ||
| SER438 (OG) | 3.250 | ||
| 3 | −5.5 | HIS431 (ND1) | 2.250 |
| HIS456 (O) | 2.608 | ||
| 4 | −5.4 | SER455 (OG) | 2.932 |
| SER528 (OG) | 2.699 | ||
| SER528 (OG) | 1.863 | ||
| SER528 (OG) | 3.087 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molagoda, I.M.N.; Athapaththu, A.M.G.K.; Choi, Y.H.; Park, C.; Jin, C.-Y.; Kang, C.-H.; Lee, M.-H.; Kim, G.-Y. Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production. Antioxidants 2021, 10, 1215. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081215
Molagoda IMN, Athapaththu AMGK, Choi YH, Park C, Jin C-Y, Kang C-H, Lee M-H, Kim G-Y. Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production. Antioxidants. 2021; 10(8):1215. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081215
Chicago/Turabian StyleMolagoda, Ilandarage Menu Neelaka, Athapaththu Mudiyanselage Gihan Kavinda Athapaththu, Yung Hyun Choi, Cheol Park, Cheng-Yung Jin, Chang-Hee Kang, Mi-Hwa Lee, and Gi-Young Kim. 2021. "Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production" Antioxidants 10, no. 8: 1215. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081215