
Oxidative Stress Biomarkers in the Relationship between Type 2 Diabetes and Air Pollution

 , , ,
, , ,
Abstract
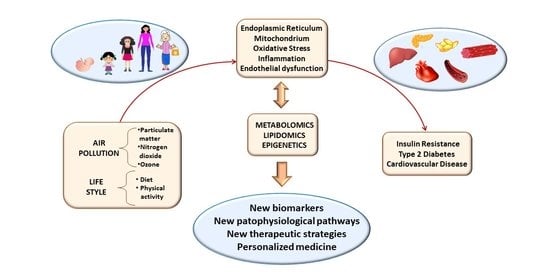
:
1. Introduction
2. Diabetes and Air Pollution
2.1. Mechanisms Underlying the Association between Diabetes and Air Pollution
2.2. Omics and Epigenetics: A New Frontier in the Association between Diabetes and Air Pollution
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ac | acetylation |
| BAT | brown adipose tissue |
| CVD | cardiovascular disease |
| IL | interleukin |
| IR | insulin resistance |
| MCP-1 | monocyte chemoattractant protein-1 |
| Me | methylation |
| miRNA | micro RNA |
| ncRNA | noncoding RNA |
| NO | nitric oxide |
| PM | particulate matter |
| T2D | type 2 diabetes |
| TNF-α | tumor necrosis factor alpha |
| UPR | unfolded protein response |
| WAT | white adipose tissue |
References
- WHO (World Health Organization). WHO Reveals Leading Causes of Death and Disability Worldwide: 2000–2019. 2020. Available online: https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019 (accessed on 2 June 2021).
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine 2014, 42, 698–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beulens, J.; Rutters, F.; Rydén, L.; Schnell, O.; Mellbin, L.; Hart, H.E.; Vos, R.C. Risk and management of pre-diabetes. Eur. J. Prev. Cardiol. 2019, 26, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. American Heart Association Council on Epidemiology and Prevention; Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Wang, Y.; Hou, B.; Lam, S.M.; Zhang, W.; Chen, R.; Shui, G.; Sun, Q.; Qiang, G.; Liu, C. Lipidomics insight into chronic exposure to ambient air pollution in mice. Environ. Pollut. 2020, 262, 114668. [Google Scholar] [CrossRef]
- Hill, B.G.; Rood, B.; Ribble, A.; Haberzettl, P. Fine particulate matter (PM2.5) inhalation-induced alterations in the plasma lipidome as promoters of vascular inflammation and insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1836–H1850. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Burnett, R.; Chen, H.; Szyszkowicz, M.; Fann, N.; Hubbell, B.; Pope, C.A., 3rd; Apte, J.S.; Brauer, M.; Cohen, A.; Weichenthal, S.; et al. Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter. Proc. Natl. Acad. Sci. USA 2018, 115, 9592–9597. [Google Scholar] [CrossRef] [Green Version]
- WHO (World Health Organization). Ambient (Outdoor) Air Pollution. Key Facts. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health (accessed on 6 June 2021).
- Hamanaka, R.B.; Mutlu, G.M. Particulate Matter Air Pollution: Effects on the Cardiovascular System. Front. Endocrinol. 2018, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Cesaroni, G.; Forastiere, F.; Stafoggia, M.; Andersen, Z.J.; Badaloni, C.; Beelen, R.; Caracciolo, B.; de Faire, U.; Erbel, R.; Eriksen, K.T.; et al. Long term exposure to ambient air pollution and incidence of acute coronary events: Prospective cohort study and meta-analysis in 11 European cohorts from the ESCAPE Project. BMJ 2014, 348, f7412. [Google Scholar] [CrossRef] [Green Version]
- Pope, C.A., 3rd; Turner, M.C.; Burnett, R.T.; Jerrett, M.; Gapstur, S.M.; Diver, W.R.; Krewski, D.; Brook, R.D. Relationships between fine particulate air pollution, cardiometabolic disorders, and cardiovascular mortality. Circ. Res. 2015, 116, 108–115. [Google Scholar] [CrossRef]
- Fu, P.; Guo, X.; Cheung, F.M.H.; Yung, K.K.L. The association between PM2.5 exposure and neurological disorders: A systematic review and meta-analysis. Sci. Total Environ. 2019, 655, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Berend, N. Contribution of air pollution to COPD and small airway dysfunction. Respirology 2016, 21, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Bloemsma, L.D.; Hoek, G.; Smit, L.A.M. Panel studies of air pollution in patients with COPD: Systematic review and meta-analysis. Environ. Res. 2016, 151, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope, C.A., 3rd; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 70, 460–479. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Brook, R.D. Air pollution and type 2 diabetes: Mechanistic insights. Diabetes 2012, 61, 3037–3045. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Ying, Z.; Harkema, J.; Sun, Q.; Rajagopalan, S. Epidemiological and experimental links between air pollution and type 2 diabetes. Toxicol. Pathol. 2013, 41, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.Y.; Fan, S.; Thiering, E.; Seissler, J.; Nowak, D.; Dong, G.H.; Heinrich, J. Ambient air pollution and diabetes: A systematic review and meta-analysis. Environ. Res. 2020, 180, 108817. [Google Scholar] [CrossRef]
- Balti, E.V.; Echouffo-Tcheugui, J.B.; Yako, Y.Y.; Kengne, A.P. Air pollution and risk of type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2014, 106, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Wang, W. Ambient Air Pollution and Type 2 Diabetes: A Systematic Review of Epidemiologic Research. Curr. Environ. Health Rep. 2014, 1, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Xu, D.; Jing, Z.; Liu, D.; Yan, S.; Wang, Y. Effect of long-term exposure to air pollution on type 2 diabetes mellitus risk: A systemic review and meta-analysis of cohort studies. Eur. J. Endocrinol. 2014, 171, R173–R182. [Google Scholar] [CrossRef] [Green Version]
- Dendup, T.; Feng, X.; Clingan, S.; Astell-Burt, T. Environmental Risk Factors for Developing Type 2 Diabetes Mellitus: A Systematic Review. Int. J. Environ. Res. Public Health 2018, 15, 78. [Google Scholar] [CrossRef] [Green Version]
- WHO (World Health Organization). The Global Health Observatory. 2020. Available online: https://www.who.int/data/gho/data/indicators/indicator-details/GHO/concentrations-of-fine-particulate-matter-(pm2-5) (accessed on 6 June 2021).
- Zuo, H.; Shi, Z.; Hussain, A. Prevalence, trends and risk factors for the diabetes epidemic in China: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2014, 104, 63–72. [Google Scholar] [CrossRef]
- Zhou, T.; Liu, X.; Liu, Y.; Li, X. Meta-analytic evaluation for the spatio-temporal patterns of the associations between common risk factors and type 2 diabetes in mainland China. Medicine 2019, 98, e15581. [Google Scholar] [CrossRef] [PubMed]
- Eze, I.C.; Hemkens, L.G.; Bucher, H.C.; Hoffmann, B.; Schindler, C.; Künzli, N.; Schikowski, T.; Probst-Hensch, N.M. Association between ambient air pollution and diabetes mellitus in Europe and North America: Systematic review and meta-analysis. Environ. Health Perspect. 2015, 123, 381–389. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Wu, S.; Zhao, H.; Qiu, H.; Fu, Y.; Li, X.; He, Y. Association between particulate matter 2.5 and diabetes mellitus: A meta-analysis of cohort studies. J. Diabetes Investig 2017, 8, 687–696. [Google Scholar] [CrossRef]
- Liu, F.; Chen, G.; Huo, W.; Wang, C.; Liu, S.; Li, N.; Mao, S.; Hou, Y.; Lu, Y.; Xiang, H. Associations between long-term exposure to ambient air pollution and risk of type 2 diabetes mellitus: A systematic review and meta-analysis. Environ. Pollut. 2019, 252 Pt B, 1235–1245. [Google Scholar] [CrossRef]
- Li, C.; Fang, D.; Xu, D.; Wang, B.; Zhao, S.; Yan, S.; Wang, Y. Main air pollutants and diabetes-associated mortality: A systematic review and meta-analysis. Eur. J. Endocrinol. 2014, 171, R183–R190. [Google Scholar] [CrossRef] [Green Version]
- Janghorbani, M.; Momeni, F.; Mansourian, M. Systematic review and metaanalysis of air pollution exposure and risk of diabetes. Eur. J. Epidemiol. 2014, 29, 231–242. [Google Scholar] [CrossRef]
- Tamagawa, E.; Bai, N.; Morimoto, K.; Gray, C.; Mui, T.; Yatera, K.; Zhang, X.; Xing, L.; Li, Y.; Laher, I.; et al. Particulate matter exposure induces persistent lung inflammation and endothelial dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L79–L85. [Google Scholar] [CrossRef] [Green Version]
- Riva, D.R.; Magalhães, C.B.; Lopes, A.A.; Lanças, T.; Mauad, T.; Malm, O.; Valença, S.S.; Saldiva, P.H.; Faffe, D.S.; Zin, W.A. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal. Toxicol. 2011, 23, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Roper, C.; Chubb, L.G.; Cambal, L.; Tunno, B.; Clougherty, J.E.; Fattman, C.; Mischler, S.E. Association of IL-6 with PM2.5 Components: Importance of Characterizing Filter-Based PM2.5 Following Extraction. Water Air Soil Pollut 2017, 228, 43. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, X.; Li, W.; Zu, Y.; Zhou, F.; Shou, Q.; Ding, Z. PM2.5 Exposure Induces Inflammatory Response in Macrophages via the TLR4/COX-2/NF-κB Pathway. Inflammation 2020, 43, 1948–1958. [Google Scholar] [CrossRef] [PubMed]
- Törnqvist, H.; Mills, N.L.; Gonzalez, M.; Miller, M.R.; Robinson, S.D.; Megson, I.L.; Macnee, W.; Donaldson, K.; Söderberg, S.; Newby, D.E.; et al. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am. J. Respir. Crit. Care Med. 2007, 176, 395–400. [Google Scholar] [CrossRef]
- Thompson, A.M.; Zanobetti, A.; Silverman, F.; Schwartz, J.; Coull, B.; Urch, B.; Speck, M.; Brook, J.R.; Manno, M.; Gold, D.R. Baseline repeated measures from controlled human exposure studies: Associations between ambient air pollution exposure and the systemic inflammatory biomarkers IL-6 and fibrinogen. Environ. Health Perspect. 2010, 118, 120–124. [Google Scholar] [CrossRef]
- Ostro, B.; Malig, B.; Broadwin, R.; Basu, R.; Gold, E.B.; Bromberger, J.T.; Derby, C.; Feinstein, S.; Greendale, G.A.; Jackson, E.A.; et al. Chronic PM2.5 exposure and inflammation: Determining sensitive subgroups in mid-life women. Environ. Res. 2014, 132, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Dabass, A.; Talbott, E.O.; Venkat, A.; Rager, J.; Marsh, G.M.; Sharma, R.K.; Holguin, F. Association of exposure to particulate matter (PM2.5) air pollution and biomarkers of cardiovascular disease risk in adult NHANES participants (2001–2008). Int. J. Hyg. Environ. Health 2016, 219, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Mills, N.L.; Törnqvist, H.; Gonzalez, M.C.; Vink, E.; Robinson, S.D.; Söderberg, S.; Boon, N.A.; Donaldson, K.; Sandström, T.; Blomberg, A.; et al. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N. Engl. J. Med. 2007, 357, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.H.; Hubbard, R.; Liu, S.L.; Shepherd, K.; Trenga, C.A.; Koenig, J.Q.; Chandler, W.L.; Kaufman, J.D. A community study of the effect of particulate matter on blood measures of inflammation and thrombosis in an elderly population. Environ. Health 2007, 6, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teichert, T.; Vossoughi, M.; Vierkötter, A.; Sugiri, D.; Schikowski, T.; Schulte, T.; Roden, M.; Luckhaus, C.; Herder, C.; Krämer, U. Association between traffic-related air pollution, subclinical inflammation and impaired glucose metabolism: Results from the SALIA study. PLoS ONE 2013, 8, e83042. [Google Scholar] [CrossRef] [Green Version]
- Park, K.W.; Halperin, D.S.; Tontonoz, P. Before they were fat: Adipocyte progenitors. Cell Metab. 2008, 8, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Spiegelman, B.M.; Flier, J.S. Obesity and the regulation of energy balance. Cell 2001, 104, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Waki, H.; Tontonoz, P. Endocrine functions of adipose tissue. Annu. Rev. Pathol. 2007, 2, 31–56. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Sun, Q.; Yue, P.; Deiuliis, J.A.; Lumeng, C.N.; Kampfrath, T.; Mikolaj, M.B.; Cai, Y.; Ostrowski, M.C.; Lu, B.; Parthasarathy, S.; et al. Ambient air pollution exaggerates adipose inflammation and insulin resistance in a mouse model of diet-induced obesity. Circulation 2009, 119, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, C.; Xu, Z.; Tzan, K.; Zhong, M.; Wang, A.; Lippmann, M.; Chen, L.C.; Rajagopalan, S.; Sun, Q. Long-term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol. Sci. 2011, 124, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.H.; Chou, C.C.; Lee, C.T.; Liu, J.Y.; Cheng, T.J. Enhanced insulin resistance in diet-induced obese rats exposed to fine particles by instillation. Inhal. Toxicol. 2011, 23, 507–519. [Google Scholar] [CrossRef]
- Esposito, K.; Petrizzo, M.; Maiorino, M.I.; Bellastella, G.; Giugliano, D. Particulate matter pollutants and risk of type 2 diabetes: A time for concern? Endocrine 2016, 51, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Münzel, T. Targeting vascular (endothelial) dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Gori, T.; Al-Kindi, S.; Deanfield, J.; Lelieveld, J.; Daiber, A.; Rajagopalan, S. Effects of gaseous and solid constituents of air pollution on endothelial function. Eur. Heart J. 2018, 39, 3543–3550. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Garcidueñas, L.; Villarreal-Calderon, R.; Valencia-Salazar, G.; Henríquez-Roldán, C.; Gutiérrez-Castrellón, P.; Torres-Jardón, R.; Osnaya-Brizuela, N.; Romero, L.; Torres-Jardón, R.; Solt, A.; et al. Systemic inflammation, endothelial dysfunction, and activation in clinically healthy children exposed to air pollutants. Inhal. Toxicol. 2008, 20, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.M.; Adar, S.D.; Szpiro, A.A.; Jorgensen, N.W.; Van Hee, V.C.; Barr, R.G.; O’Neill, M.S.; Herrington, D.M.; Polak, J.F.; Kaufman, J.D. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution). J. Am. Coll. Cardiol. 2012, 60, 2158–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, C.A., 3rd; Bhatnagar, A.; McCracken, J.P.; Abplanalp, W.; Conklin, D.J.; O’Toole, T. Exposure to Fine Particulate Air Pollution is Associated with Endothelial Injury and Systemic Inflammation. Circ. Res. 2016, 119, 1204–1214. [Google Scholar] [CrossRef] [Green Version]
- Riggs, D.W.; Zafar, N.; Krishnasamy, S.; Yeager, R.; Rai, S.N.; Bhatnagar, A.; O’Toole, T.E. Exposure to airborne fine particulate matter is associated with impaired endothelial function and biomarkers of oxidative stress and inflammation. Environ. Res. 2020, 180, 108890. [Google Scholar] [CrossRef]
- Hu, H.; Wu, J.; Li, Q.; Asweto, C.; Feng, L.; Yang, X.; Duan, F.; Duan, J.; Sun, Z. Fine particulate matter induces vascular endothelial activation via IL-6 dependent JAK1/STAT3 signaling pathway. Toxicol. Res. 2016, 5, 946–953. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Zhang, J.; Zhang, C.; Xu, Y.; Zhu, X.; Yao, C.; Liu, Y.; Li, T.; Cao, J. The injury of fine particulate matter from cooking oil fumes on umbilical cord blood vessels in vitro. Environ. Toxicol Pharmacol. 2017, 49, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Laing, S.; Wang, G.; Briazova, T.; Zhang, C.; Wang, A.; Zheng, Z.; Gow, A.; Chen, A.F.; Rajagopalan, S.; Chen, L.C.; et al. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am. J. Physiol. Cell Physiol. 2010, 299, C736–C749. [Google Scholar] [CrossRef] [Green Version]
- Haberzettl, P.; O’Toole, T.E.; Bhatnagar, A.; Conklin, D.J. Exposure to Fine Particulate Air Pollution Causes Vascular Insulin Resistance by Inducing Pulmonary Oxidative Stress. Environ. Health Perspect. 2016, 124, 1830–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Neas, L.; Herbst, M.C.; Case, M.; Williams, R.W.; Cascio, W.; Hinderliter, A.; Holguin, F.; Buse, J.B.; Dungan, K.; et al. Endothelial dysfunction: Associations with exposure to ambient fine particles in diabetic individuals. Environ. Health Perspect. 2008, 116, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Luyendyk, J.P.; Schoenecker, J.G.; Flick, M.J. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019, 133, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S. Inflammation and haemostasis. Biochem. Med. 2012, 22, 49–62. [Google Scholar] [CrossRef]
- Tang, H.; Cheng, Z.; Li, N.; Mao, S.; Ma, R.; He, H.; Niu, Z.; Chen, X.; Xiang, H. The short- and long-term associations of particulate matter with inflammation and blood coagulation markers: A meta-analysis. Environ. Pollut. 2020, 267, 115630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Wada, J.; Nakatsuka, A. Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med. Okayama 2016, 70, 151–158. [Google Scholar] [CrossRef]
- Dugan, L.L.; You, Y.H.; Ali, S.S.; Diamond-Stanic, M.; Miyamoto, S.; DeCleves, A.E.; Andreyev, A.; Quach, T.; Ly, S.; Shekhtman, G.; et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013, 123, 4888–4899. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K. Mitochondrial hormesis and diabetic complications. Diabetes 2015, 64, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Menzel, A.; Samouda, H.; Dohet, F.; Loap, S.; Ellulu, M.S.; Bohn, T. Common and Novel Markers for Measuring Inflammation and Oxidative Stress Ex Vivo in Research and Clinical Practice-Which to Use Regarding Disease Outcomes? Antioxidants 2021, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Vassalle, C. Oxidative stress and cardiovascular risk prediction: The long way towards a “radical” perspective. Int. J. Cardiol. 2018, 273, 252–253. [Google Scholar] [CrossRef]
- Milburn, M.V.; Lawton, K.A. Application of metabolomics to diagnosis of insulin resistance. Annu. Rev. Med. 2013, 64, 291–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.H.; Lee, H.S.; Yu, H.Y.; Kim, Y.J.; Jeon, H.J.; Oh, T.; Kim, B.J.; Choi, H.J.; Kim, J.M. Metabolomics profiles associated with HbA1c levels in patients with type 2 diabetes. PLoS ONE 2019, 14, e0224274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Gao, H.Y.; Fan, Z.Y.; He, Y.; Yan, Y.X. Metabolomics Signatures in Type 2 Diabetes: A Systematic Review and Integrative Analysis. J. Clin. Endocrinol. Metab. 2020, 105, dgz240. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, C.G.; Rusu, A.; Ciobanu, D.; Bucsa, C.; Roman, G. Amino Acid Signature of Oxidative Stress in Patients with Type 2 Diabetes: Targeted Exploratory Metabolomic Research. Antioxidants 2021, 10, 610. [Google Scholar] [CrossRef]
- Zhu, Y.; Tsai, M.Y.; Sun, Q.; Hinkle, S.N.; Rawal, S.; Mendola, P.; Ferrara, A.; Albert, P.S.; Zhang, C. A prospective and longitudinal study of plasma phospholipid saturated fatty acid profile in relation to cardiometabolic biomarkers and the risk of gestational diabetes. Am. J. Clin. Nutr. 2018, 107, 1017–1026. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.Y.; Kim, J.Y.; Paik, J.K.; Kim, O.Y.; Paik, Y.H.; Lee, E.J.; Lee, J.H. The association of specific metabolites of lipid metabolism with markers of oxidative stress, inflammation and arterial stiffness in men with newly diagnosed type 2 diabetes. Clin. Endocrinol. 2012, 76, 674–682. [Google Scholar] [CrossRef]
- Razquin, C.; Toledo, E.; Clish, C.B.; Ruiz-Canela, M.; Dennis, C.; Corella, D.; Papandreou, C.; Ros, E.; Estruch, R.; Guasch-Ferré, M.; et al. Plasma Lipidomic Profiling and Risk of Type 2 Diabetes in the PREDIMED Trial. Diabetes Care 2018, 41, 2617–2624. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Willinger, C.M.; Keefe, J.; Liu, J.; Fernández-Ortiz, A.; Ibáñez, B.; Peñalvo, J.; Adourian, A.; Chen, G.; Corella, D.; et al. Lipidomic profiling identifies signatures of metabolic risk. EBioMedicine 2020, 51, 102520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Berger, S.L. Epigenetics of aging and aging-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. S1), S17–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef] [PubMed]
- Gurha, P.; Marian, A.J. Noncoding RNAs in cardiovascular biology and disease. Circ. Res. 2013, 113, e115–e120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedman, Å.K.; Zilmer, M.; Sundström, J.; Lind, L.; Ingelsson, E. DNA methylation patterns associated with oxidative stress in an ageing population. BMC Med. Genom. 2016, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.J., 3rd; Reinberg, D. Is there a code embedded in proteins that is based on post-translational modifications? Nat. Rev. Mol. Cell Biol. 2008, 9, 815–820. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Tang, G.; Ozcan, S. Role of microRNAs in diabetes. Biochim. Biophys. Acta 2008, 1779, 697–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, M.U.; Ismail, M. The Impact of Traditional Food and Lifestyle Behavior on Epigenetic Burden of Chronic Disease. Glob. Chall. 2017, 1, 1700043. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2021, 70, 29–49. [Google Scholar] [CrossRef]
- Fiorito, G.; Vlaanderen, J.; Polidoro, S.; Gulliver, J.; Galassi, C.; Ranzi, A.; Krogh, V.; Grioni, S.; Agnoli, C.; Sacerdote, C.; et al. Oxidative stress and inflammation mediate the effect of air pollution on cardio- and cerebrovascular disease: A prospective study in nonsmokers. Environ. Mol. Mutagen. 2018, 59, 234–246. [Google Scholar] [CrossRef]
- Cantone, L.; Iodice, S.; Tarantini, L.; Albetti, B.; Restelli, I.; Vigna, L.; Bonzini, M.; Pesatori, A.C.; Bollati, V. Particulate matter exposure is associated with inflammatory gene methylation in obese subjects. Environ. Res. 2017, 152, 478–484. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Li, H.; Cai, J.; Wang, C.; Lin, Z.; Liu, C.; Niu, Y.; Zhao, Z.; Li, W.; Kan, H. Fine Particulate Air Pollution and the Expression of microRNAs and Circulating Cytokines Relevant to Inflammation, Coagulation, and Vasoconstriction. Environ. Health Perspect. 2018, 126, 017007. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jia, Y.; Nan, A.; Zhang, N.; Zhou, H.; Chen, L.; Pan, X.; Qiu, M.; Zhu, J.; Zhang, H.; et al. CircRNA104250 and lncRNAuc001.dgp.1 promote the PM2.5-induced inflammatory response by co-targeting miR-3607-5p in BEAS-2B cells. Environ. Pollut. 2020, 258, 113749. [Google Scholar] [CrossRef]
- Peng, C.; Bind, M.C.; Colicino, E.; Kloog, I.; Byun, H.M.; Cantone, L.; Trevisi, L.; Zhong, J.; Brennan, K.; Dereix, A.E.; et al. Particulate Air Pollution and Fasting Blood Glucose in Nondiabetic Individuals: Associations and Epigenetic Mediation in the Normative Aging Study, 2000–2011. Environ. Health Perspect. 2016, 124, 1715–1721. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Park, B.; Palanivel, R.; Vinayachandran, V.; Deiuliis, J.A.; Gangwar, R.S.; Das, L.; Yin, J.; Choi, Y.; Al-Kindi, S.; et al. Metabolic effects of air pollution exposure and reversibility. J. Clin. Investig. 2020, 130, 6034–6040. [Google Scholar] [CrossRef]
- Dimakakou, E.; Johnston, H.J.; Streftaris, G.; Cherrie, J.W. Exposure to Environmental and Occupational Particulate Air Pollution as a Potential Contributor to Neurodegeneration and Diabetes: A Systematic Review of Epidemiological Research. Int. J. Environ. Res. Public Health 2018, 15, 1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, J.; Ketzel, M.; Kakosimos, K.; Sørensen, M.; Jensen, S.S. Road traffic air and noise pollution exposure assessment—A review of tools and techniques. Sci. Total Environ. 2018, 634, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.C.; Thurston, G.D. Air Pollution, Oxidative Stress, and Diabetes: A Life Course Epidemiologic Perspective. Curr. Diab. Rep. 2019, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Vasile, V.C.; Donato, L.J.; Hurme, R.; Laaksonen, R. Ceramides and Ceramide Scores: Clinical Applications for Cardiometabolic Risk Stratification. Front. Endocrinol. (Lausanne) 2020, 11, 570628. [Google Scholar] [CrossRef] [PubMed]


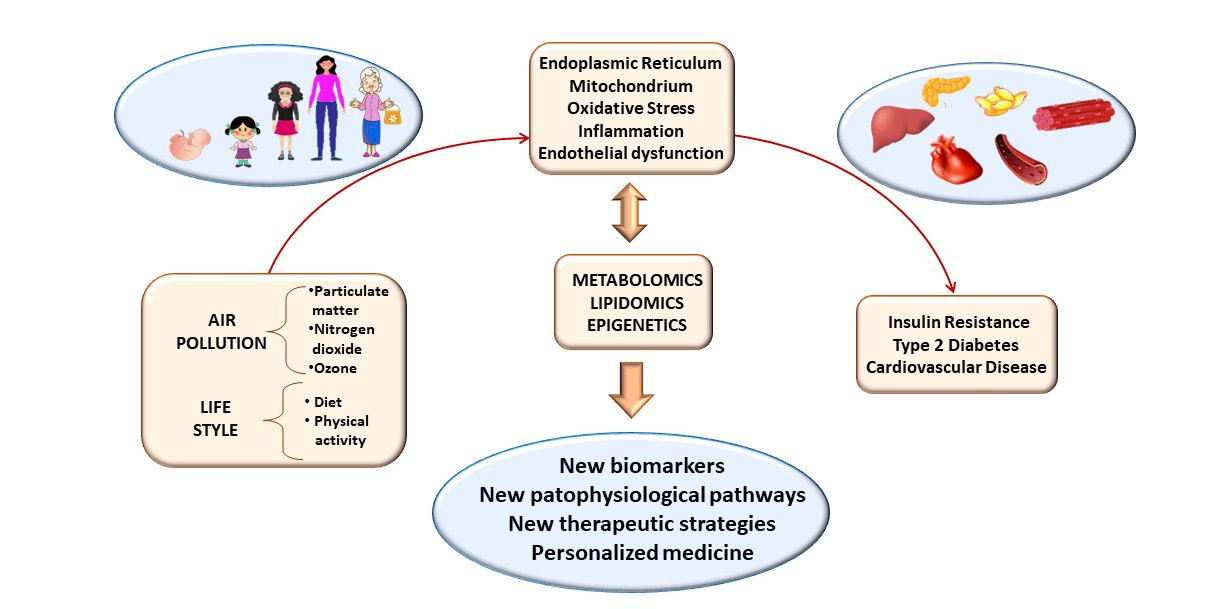{kind=link}
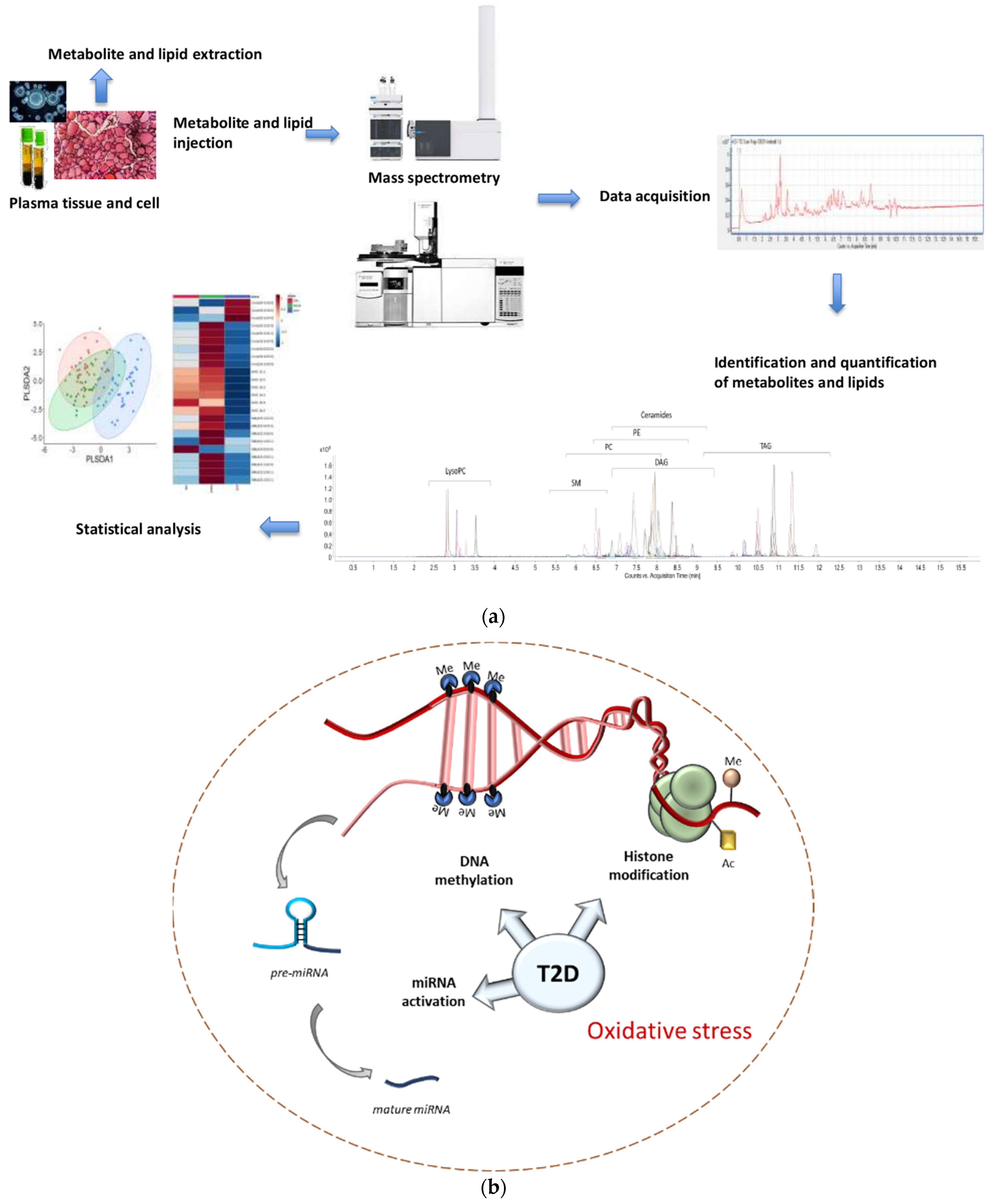{kind=link}
| ADA | WHO | |
|---|---|---|
| IFG (Impaired fasting glucose) | 5.6–7.0 mmol/L (100–125 mg/dL) | 6.1–7.0 mmol/L (110–125 mg/dL) |
| IGT (Impaired glucose tolerance in the two-hour OGTT) | 7.8–11.1 mmol/L (140–199 mg/dL) | 7.8–11.1 mmol/L (140–199 mg/dL) |
| High risk HbA1c | 39–46 mmol/mol (5.7–6.4%) | 42–46 mmol/mol (6.0–6.4%) |
| Reference | Abbreviation | Molecule | Effects |
|---|---|---|---|
| [36,37,38,39] | TNFα | Tumor necrosis factor alpha | Promotion of acute inflammation, apoptosis |
| [36,37,38,39] | IL-6 | Interleukin 6 | Induction of the acute phase response, immune and hematopoietic activities |
| [68] | IL-1 | Interleukin 1 | Regulation of immune and inflammatory responses |
| [36,37,38,39] | MCP-1 | Monocyte chemoattractant protein-1 | Regulation of migration/infiltration of monocytes/macrophages |
| [39] | NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells | Induction of the expression of various proinflammatory genes. Participation in inflammasome regulation. Regulation of the survival, activation and differentiation of innate immune cells and inflammatory T cells |
| [39] | COX-2 | Cyclooxygenase 2 | Production of the prostaglandins that contribute to pain, fever, and inflammation |
| [51] | Akt/PKB | Protein kinase N | Promotion of glucose metabolism, cell proliferation, transcription, migration, and apoptosis |
| [64,65] | NO | Nitric oxide | Control of vascular tone, dilation of blood vessels, reduction of blood pressure, inhibition of platelet aggregation (anti-thrombotic action) |
| [67] | - | Fibrin | Promotion of clot formation, fibrinolysis, cellular and matrix interactions, inflammation, and wound healing |
| [69] | - | Fibrinogen | During tissue and vascular injury, it is converted enzymatically by thrombin to fibrin |
| [68] | VWF | Von Willebrand factor | Promotion of platelet adhesion and, under high shear conditions, of platelet aggregation |
| [68] | TF | Tissue factor | Induction of blood coagulation |
| [68] | PAI-1 | Plasminogen activator inhibitor 1 | Inhibition of fibrinolysis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorini, F.; Sabatino, L.; Gaggini, M.; Chatzianagnostou, K.; Vassalle, C. Oxidative Stress Biomarkers in the Relationship between Type 2 Diabetes and Air Pollution. Antioxidants 2021, 10, 1234. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081234
Gorini F, Sabatino L, Gaggini M, Chatzianagnostou K, Vassalle C. Oxidative Stress Biomarkers in the Relationship between Type 2 Diabetes and Air Pollution. Antioxidants. 2021; 10(8):1234. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081234
Chicago/Turabian StyleGorini, Francesca, Laura Sabatino, Melania Gaggini, Kyriazoula Chatzianagnostou, and Cristina Vassalle. 2021. "Oxidative Stress Biomarkers in the Relationship between Type 2 Diabetes and Air Pollution" Antioxidants 10, no. 8: 1234. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081234