
Statins’ Regulation of the Virulence Factors of Helicobacter pylori and the Production of ROS May Inhibit the Development of Gastric Cancer


Abstract
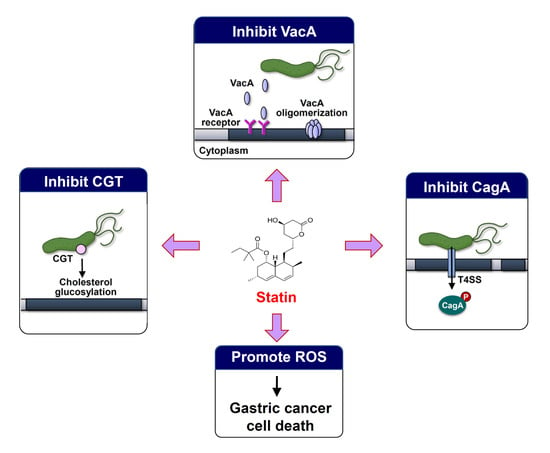
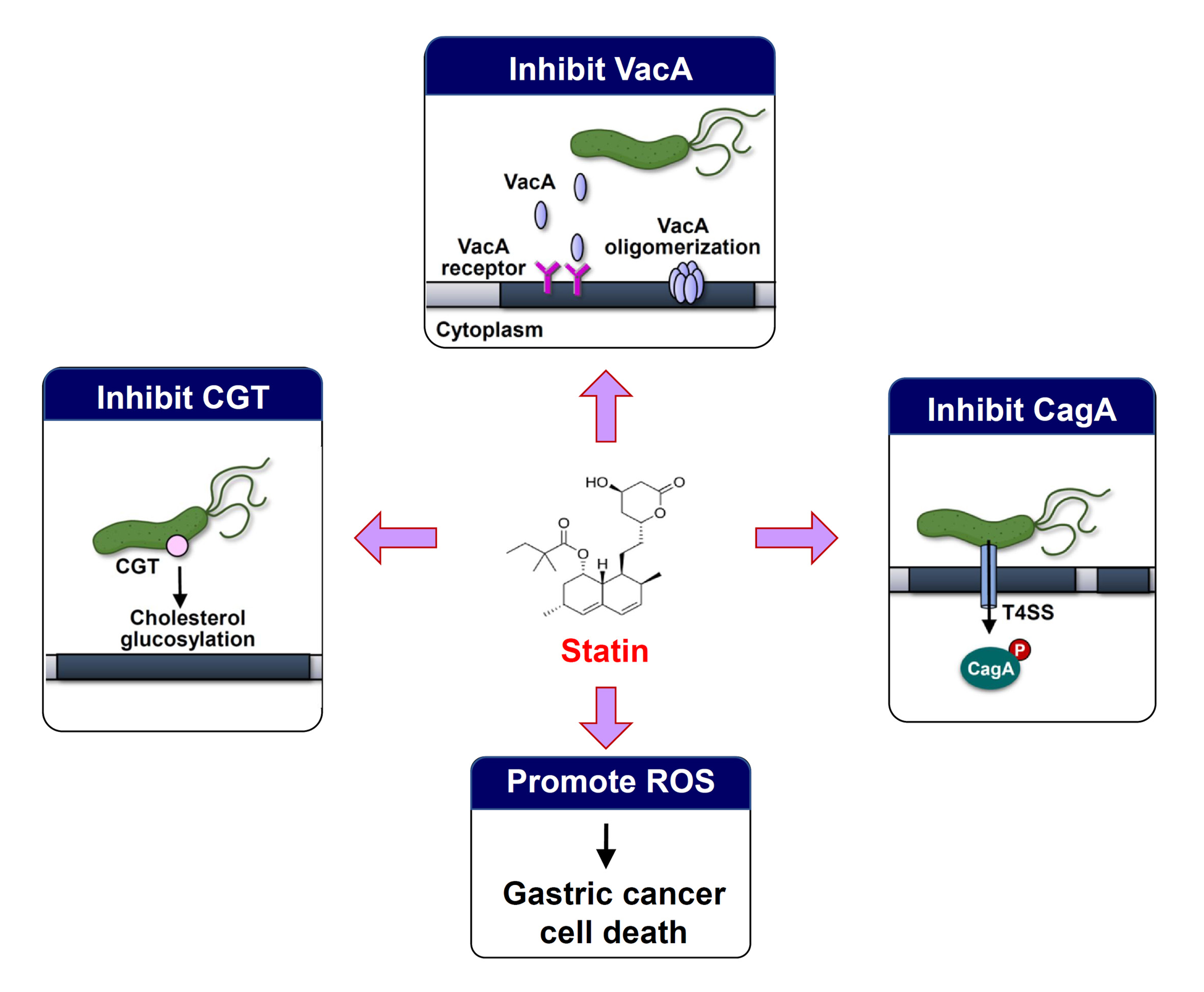
:
1. Introduction
2. H. pylori Virulence Factors Usurp Cholesterol and Lead to GC Development
3. Interplay between H. pylori and ROS Production to Induce Gastric Carcinogenesis
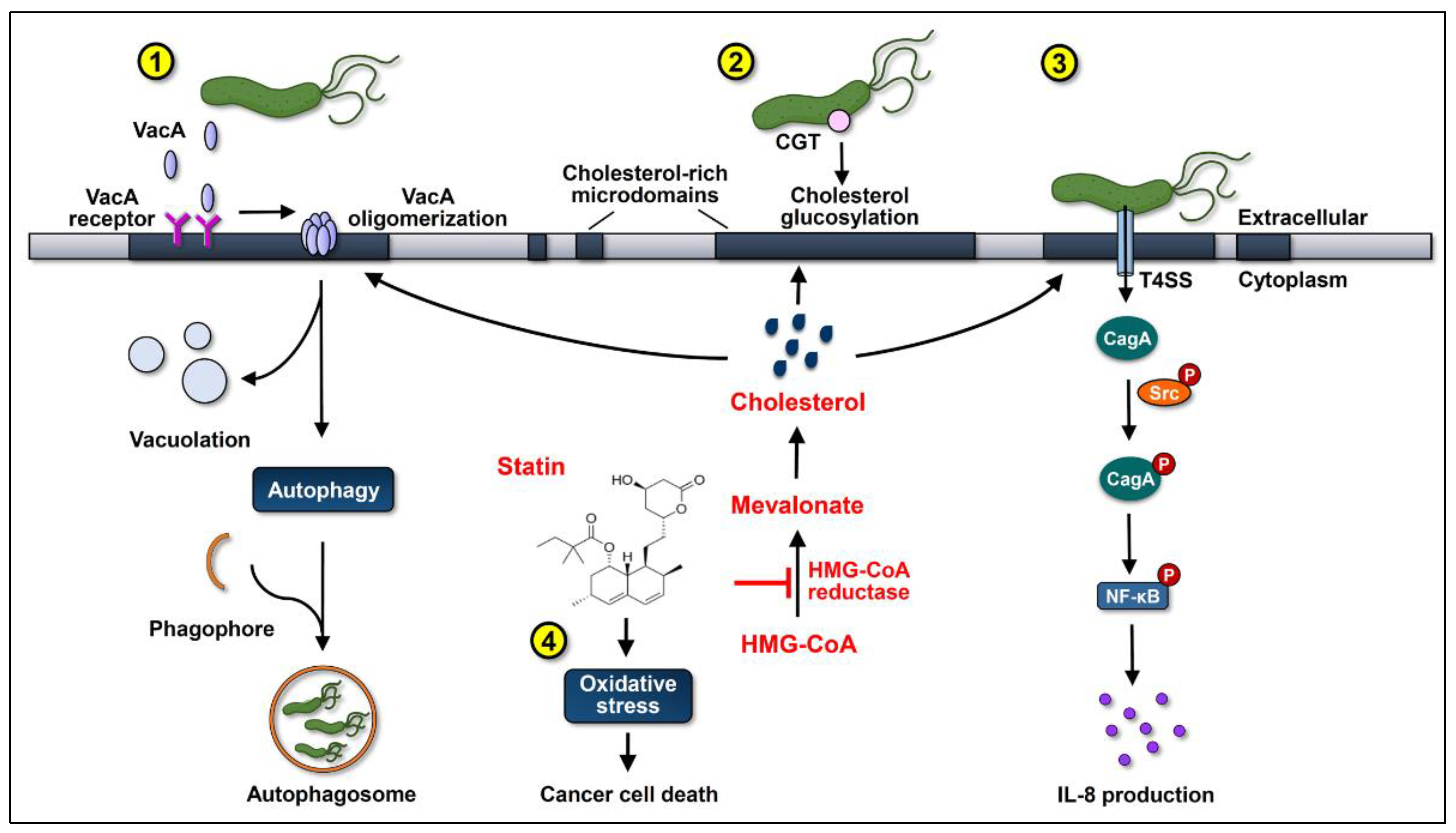
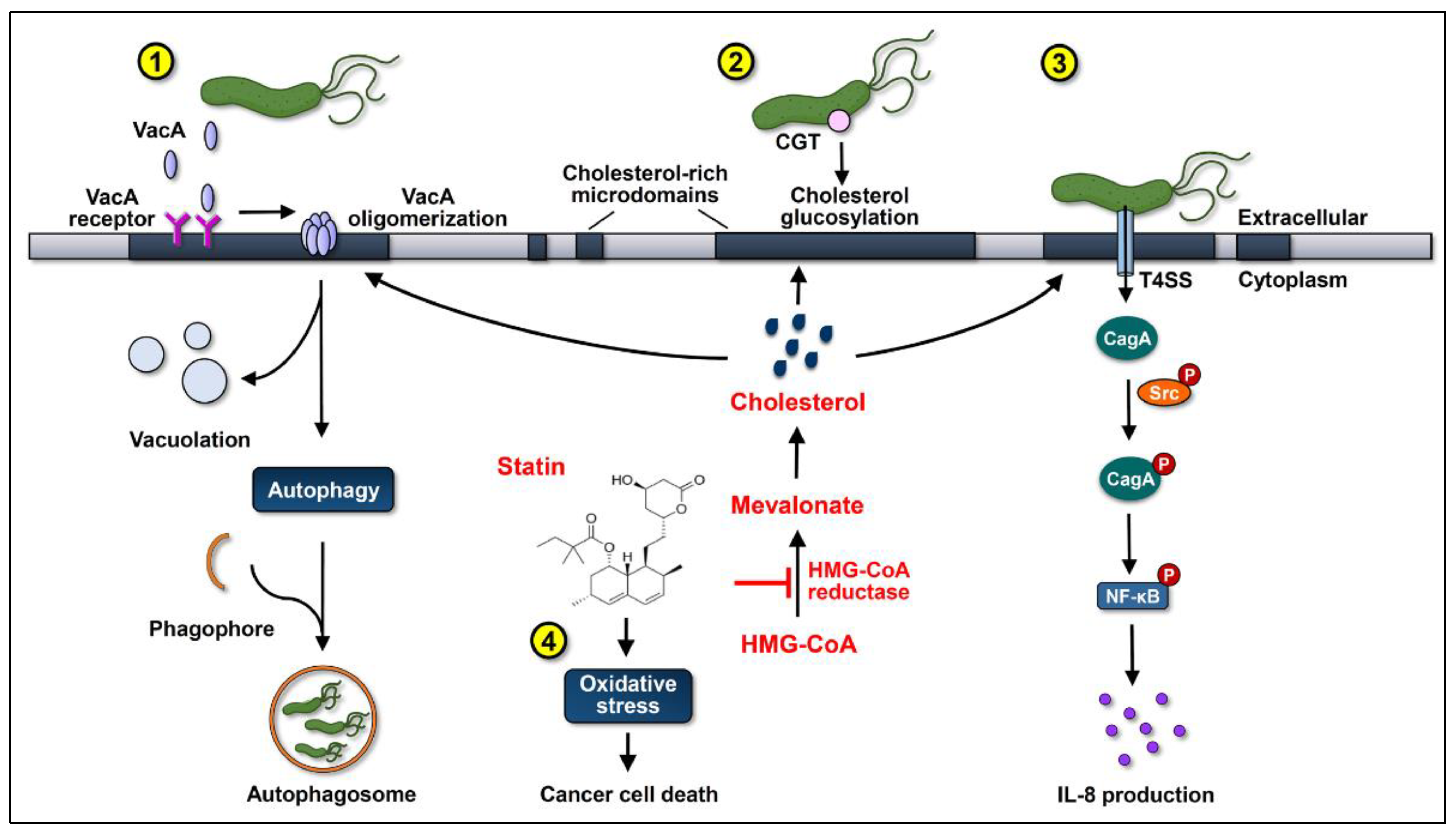
4. Statins Lower GC Risk by Reducing H. pylori Survival and Inhibition of Virulence Factor Actions
5. Statins Modulate MicroRNAs and Exosome Levels
6. Cholesterol-Independent Beneficial Effects of Statins in Cancer Therapy
7. Statin Use Reduces GC Risk
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Burkitt, M.D.; Duckworth, C.A.; Williams, J.M.; Pritchard, D.M. Helicobacter pylori-induced gastric pathology: Insights from in vivo and ex vivo models. Dis. Models Mech. 2017, 10, 89–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Weber, A.; Graham, D.Y. Age, period, and cohort effects on gastric cancer mortality. Dig. Dis. Sci. 2015, 60, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y. Helicobacter pylori update: Gastric cancer, reliable therapy, and possible benefits. Gastroenterology 2015, 148, 719–731.e713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrift, A.P.; El-Serag, H.B. Burden of gastric cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 534–542. [Google Scholar] [CrossRef]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Moehler, M.; Baltin, C.T.; Ebert, M.; Fischbach, W.; Gockel, I.; Grenacher, L.; Holscher, A.H.; Lordick, F.; Malfertheiner, P.; Messmann, H.; et al. International comparison of the german evidence-based s3-guidelines on the diagnosis and multimodal treatment of early and locally advanced gastric cancer, including adenocarcinoma of the lower esophagus. Gastric Cancer 2015, 18, 550–563. [Google Scholar] [CrossRef]
- Waddell, T.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; European Society for Medical Oncology (ESMO); European Society of Surgical Oncology (ESSO); European Society of Radiotherapy and Oncology (ESTRO). Gastric cancer: Esmo-esso-estro clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24 (Suppl. 6), 57–63. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Taylor, F.C.; Huffman, M.; Ebrahim, S. Statin therapy for primary prevention of cardiovascular disease. JAMA 2013, 310, 2451–2452. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Davis, A.M. Management of blood cholesterol. JAMA 2019, 321, 800–801. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Lin, C.J.; Liao, W.C.; Lin, H.J.; Hsu, Y.M.; Lin, C.L.; Chen, Y.A.; Feng, C.L.; Chen, C.J.; Kao, M.C.; Lai, C.H.; et al. Statins attenuate helicobacter pylori caga translocation and reduce incidence of gastric cancer: In vitro and population-based case-control studies. PLoS ONE 2016, 11, e0146432. [Google Scholar]
- Spence, A.D.; Busby, J.; Hughes, C.M.; Johnston, B.T.; Coleman, H.G.; Cardwell, C.R. Statin use and survival in patients with gastric cancer in two independent population-based cohorts. Pharmacoepidemiol. Drug Saf 2019, 28, 460–470. [Google Scholar] [CrossRef]
- Yang, P.R.; Tsai, Y.Y.; Chen, K.J.; Yang, Y.H.; Shih, W.T. Statin use improves overall survival of patients with gastric cancer after surgery and adjuvant chemotherapy in Taiwan: A nationwide matched cohort study. Cancers 2020, 12, 2055. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, W.; Jin, G.; Chu, P.; Li, H. Effect of statins on gastric cancer incidence: A meta-analysis of case control studies. J. Cancer Res. Ther. Oncol. 2014, 10, 859–865. [Google Scholar]
- Singh, P.P.; Singh, S. Statins are associated with reduced risk of gastric cancer: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.F.; Ho, S.C.; Chang, C.C.; Wu, T.N.; Yang, C.Y. Statins are associated with a reduced risk of gastric cancer: A population-based case-control study. Am. J. Gastroenterol. Suppl. 2011, 106, 2098–2103. [Google Scholar] [CrossRef]
- Wu, X.D.; Zeng, K.; Xue, F.Q.; Chen, J.H.; Chen, Y.Q. Statins are associated with reduced risk of gastric cancer: A meta-analysis. Eur. J. Clin. Pharmacol. 2013, 69, 1855–1860. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Brault, M.; Ray, J.; Gomez, Y.H.; Mantzoros, C.S.; Daskalopoulou, S.S. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism 2014, 63, 735–745. [Google Scholar] [CrossRef]
- Naci, H.; Brugts, J.; Ades, T. Comparative tolerability and harms of individual statins: A study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse effects of statin therapy: Perception vs. The evidence-focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.M.; Lee, Y.C.; El-Omar, E.M.; Wu, M.S. Efficacy and long-term safety of H. pylori eradication for gastric cancer prevention. Cancers 2019, 11, 593. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.T.; Ma, Y.; Sun, Y.; Bai, C.Q.; Ling, C.H.; Yuan, F.L. The protective effects of helicobacter pylori infection on allergic asthma. Int. Arch. Allergy Immunol. 2021, 182, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Huang, M.Z.; Wang, M.L.; Lin, C.J.; Lu, T.L.; Lo, H.R.; Pan, Y.J.; Sun, Y.C.; Kao, M.C.; Lim, H.J.; et al. Statin decreases helicobacter pylori burden in macrophages by promoting autophagy. Front. Cell Infect. Microbiol. 2016, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, S.H.; Hur, K.Y.; Woo, S.Y.; Kim, S.W.; Kang, W.K. Statins and the risk of gastric cancer in diabetes patients. BMC Cancer 2012, 12, 596. [Google Scholar] [CrossRef] [Green Version]
- You, H.S.; You, N.; Lee, J.W.; Lim, H.J.; Kim, J.; Kang, H.T. Inverse association between statin use and stomach cancer incidence in individuals with hypercholesterolemia, from the 2002-2015 nhis-heals data. Int. J. Environ. Res. Public Health 2020, 17, 1054. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Stoicov, C.; Li, H.; Cerny, J.; Houghton, J.M. How the study of helicobacter infection can contribute to the understanding of carcinoma development. Clin. Microbiol. Infect. 2009, 15, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Isomoto, H.; Moss, J.; Hirayama, T. Pleiotropic actions of helicobacter pylori vacuolating cytotoxin, vaca. Tohoku J. Exp. Med. 2010, 220, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, C.; de Bernard, M. Molecular and cellular mechanisms of action of the vacuolating cytotoxin (vaca) and neutrophil-activating protein (hp-nap) virulence factors of helicobacter pylori. Microbes Infect. 2003, 5, 715–721. [Google Scholar] [CrossRef]
- Capurro, M.I.; Greenfield, L.K.; Prashar, A.; Xia, S.; Abdullah, M.; Wong, H.; Zhong, X.Z.; Bertaux-Skeirik, N.; Chakrabarti, J.; Siddiqui, I.; et al. Vaca generates a protective intracellular reservoir for helicobacter pylori that is eliminated by activation of the lysosomal calcium channel trpml1. Nat. Microbiol. 2019, 4, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vacuolating cytotoxin and variants in atg16l1 that disrupt autophagy promote helicobacter pylori infection in humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, L.K.; Jones, N.L. Modulation of autophagy by helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 2013, 21, 602–612. [Google Scholar] [CrossRef]
- Capurro, M.I.; Prashar, A.; Jones, N.L. Mcoln1/trpml1 inhibition-a novel strategy used by. Autophagy 2020, 16, 169–170. [Google Scholar] [CrossRef]
- Montecucco, C.; de Bernard, M. Immunosuppressive and proinflammatory activities of the vaca toxin of helicobacter pylori. J. Exp. Med. 2003, 198, 1767–1771. [Google Scholar] [CrossRef]
- Nomura, A.M.; Lee, J.; Stemmermann, G.N.; Nomura, R.Y.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori caga seropositivity and gastric carcinoma risk in a japanese american population. J. Infect. Dis. 2002, 186, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Selbach, M.; Moese, S.; Hauck, C.R.; Meyer, T.F.; Backert, S. Src is the kinase of the helicobacter pylori caga protein in vitro and in vivo. J. Biol. Chem. 2002, 277, 6775–6778. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, M.; Higashi, H. Helicobacter pylori caga: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef]
- Roovers, K.; Assoian, R.K. Integrating the map kinase signal into the g1 phase cell cycle machinery. Bioessays 2000, 22, 818–826. [Google Scholar] [CrossRef]
- Meyer-ter-Vehn, T.; Covacci, A.; Kist, M.; Pahl, H.L. Helicobacter pylori activates mitogen-activated protein kinase cascades and induces expression of the proto-oncogenes c-fos and c-jun. J. Biol. Chem. 2000, 275, 16064–16072. [Google Scholar] [CrossRef] [Green Version]
- Lamb, A.; Chen, L.F. Role of the helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Brandt, S.; Kwok, T.; Hartig, R.; Konig, W.; Backert, S. Nf-kappab activation and potentiation of proinflammatory responses by the Helicobacter pylori caga protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun, A.H.; Wunder, C.; Hildebrand, J.; Churin, Y.; Zahringer, U.; Lindner, B.; Meyer, T.F.; Heinz, E.; Warnecke, D. Cloning of a cholesterol-alpha-glucosyltransferase from helicobacter pylori. J. Biol. Chem. 2006, 281, 27765–27772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunder, C.; Churin, Y.; Winau, F.; Warnecke, D.; Vieth, M.; Lindner, B.; Zahringer, U.; Mollenkopf, H.J.; Heinz, E.; Meyer, T.F. Cholesterol glucosylation promotes immune evasion by helicobacter pylori. Nat. Med. 2006, 12, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Huang, J.C.; Cheng, H.H.; Wu, M.C.; Huang, M.Z.; Hsu, H.Y.; Chen, Y.A.; Hsu, C.Y.; Pan, Y.J.; Chu, Y.T.; et al. Helicobacter pylori cholesterol glucosylation modulates autophagy for increasing intracellular survival in macrophages. Cell. Microbiol. 2018, 20, e12947. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Y.; Wang, H.J.; Cheng, H.H.; Chen, S.D.; Wang, L.H.; Wang, W.C. Cholesterol glucosylation by helicobacter pylori delays internalization and arrests phagosome maturation in macrophages. J. Microbiol. Immunol. Infect. 2016, 49, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Cheng, W.C.; Cheng, H.H.; Lai, C.H.; Wang, W.C. Helicobacter pylori cholesteryl glucosides interfere with host membrane phase and affect type iv secretion system function during infection in ags cells. Mol. Microbiol. 2012, 83, 67–84. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Caga protein of helicobacter pylori: A hijacker of gastric epithelial cell signaling. Biochem. Pharmacol. 2007, 73, 1697–1702. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Redox biology and gastric carcinogenesis: The role of helicobacter pylori. Redox Rep. 2011, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Wilson, K.T.; Ramanujam, K.S.; Mobley, H.L.; Musselman, R.F.; James, S.P.; Meltzer, S.J. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 1996, 111, 1524–1533. [Google Scholar] [CrossRef]
- Fu, S.; Ramanujam, K.S.; Wong, A.; Fantry, G.T.; Drachenberg, C.B.; James, S.P.; Meltzer, S.J.; Wilson, K.T. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in helicobacter pylori gastritis. Gastroenterology 1999, 116, 1319–1329. [Google Scholar] [CrossRef]
- Goto, T.; Haruma, K.; Kitadai, Y.; Ito, M.; Yoshihara, M.; Sumii, K.; Hayakawa, N.; Kajiyama, G. Enhanced expression of inducible nitric oxide synthase and nitrotyrosine in gastric mucosa of gastric cancer patients. Clin. Cancer Res. 1999, 5, 1411–1415. [Google Scholar]
- Nam, K.T.; Oh, S.Y.; Ahn, B.; Kim, Y.B.; Jang, D.D.; Yang, K.H.; Hahm, K.B.; Kim, D.Y. Decreased helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut 2004, 53, 1250–1255. [Google Scholar] [CrossRef]
- Huang, X.W.; Luo, R.H.; Zhao, Q.; Shen, Z.Z.; Huang, L.L.; An, X.Y.; Zhao, L.J.; Wang, J.; Huang, Y.Z. Helicobacter pylori induces mitochondrial DNA mutation and reactive oxygen species level in ags cells. Int. J. Med. Sci. 2011, 8, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, D.; Hussey, S.; Jones, N.L. Crohn disease atg16l1 polymorphism increases susceptibility to infection with helicobacter pylori in humans. Autophagy 2012, 8, 1387–1388. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of helicobacter pylori caga is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.Y.; Tang, C.H.; Chang, C.H.; Maa, M.C.; Fang, S.H.; Hsu, Y.M.; Lin, Y.H.; Lin, C.J.; Lee, W.C.; Lin, H.J.; et al. Helicobacter pylori attenuates lipopolysaccharide-induced nitric oxide production by murine macrophages. Innate Immun. 2012, 18, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; McGee, D.J.; Akhtar, M.; Mendz, G.L.; Newton, J.C.; Cheng, Y.; Mobley, H.L.; Wilson, K.T. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: A strategy for bacterial survival. Proc. Natl. Acad. Sci. USA 2001, 98, 13844–13849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, N.D.; Asim, M.; Barry, D.P.; Singh, K.; de Sablet, T.; Boucher, J.L.; Gobert, A.P.; Chaturvedi, R.; Wilson, K.T. Arginase ii restricts host defense to helicobacter pylori by attenuating inducible nitric oxide synthase translation in macrophages. J. Immunol. 2010, 184, 2572–2582. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, R.; Asim, M.; Lewis, N.D.; Algood, H.M.; Cover, T.L.; Kim, P.Y.; Wilson, K.T. l-arginine availability regulates inducible nitric oxide synthase-dependent host defense against helicobacter pylori. J. Immunol. 2007, 75, 4305–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Li, P.; Tao, J.; Shi, X.; Hu, B.; Chen, H.; Guo, X.H. Pylori escape host immunoreaction through inhibiting ilk expression by vaca. Cell Mol. Immunol. 2009, 6, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Istvan, E. Statin inhibition of hmg-coa reductase: A 3-dimensional view. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Lennernas, H.; Fager, G. Pharmacodynamics and pharmacokinetics of the hmg-coa reductase inhibitors. Similarities and differences. Clin. Pharmacokinet. 1997, 32, 403–425. [Google Scholar] [CrossRef]
- McKenney, J.M.; Jones, P.H.; Adamczyk, M.A.; Cain, V.A.; Bryzinski, B.S.; Blasetto, J.W.; Group, S.S. Comparison of the efficacy of rosuvastatin versus atorvastatin, simvastatin, and pravastatin in achieving lipid goals: Results from the stellar trial. Curr. Med. Res. Opin. 2003, 19, 689–698. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Patel, H.K.; Willhite, D.C.; Patel, R.M.; Ye, D.; Williams, C.L.; Torres, E.M.; Marty, K.B.; MacDonald, R.A.; Blanke, S.R. Plasma membrane cholesterol modulates cellular vacuolation induced by the helicobacter pylori vacuolating cytotoxin. Infect. Immun. 2002, 70, 4112–4123. [Google Scholar] [CrossRef] [Green Version]
- Mohajeri, M.; Banach, M.; Atkin, S.L.; Butler, A.E.; Ruscica, M.; Watts, G.F.; Sahebkar, A. Micrornas: Novel molecular targets and response modulators of statin therapy. Trends Pharmacol. Sci. 2018, 39, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, T.; Hirata, R.D.C.; Hirata, M.H.; Cerda, Á.; Salazar, L.A. Statins differentially modulate micrornas expression in peripheral cells of hyperlipidemic subjects: A pilot study. Eur. J. Pharm. Sci. 2018, 117, 55–61. [Google Scholar] [CrossRef]
- Moore, K.J.; Rayner, K.J.; Suárez, Y.; Fernández-Hernando, C. The role of micrornas in cholesterol efflux and hepatic lipid metabolism. Annu. Rev. Nutr. 2011, 31, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Kulshreshtha, A.; Singh, S.; Ahmad, M.; Khanna, K.; Ahmad, T.; Agrawal, A.; Ghosh, B. Simvastatin mediates inhibition of exosome synthesis, localization and secretion via multicomponent interventions. Sci. Rep. 2019, 9, 16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoces de la Guardia, A.; Staedel, C.; Kaafarany, I.; Clement, A.; Roubaud Baudron, C.; Megraud, F.; Lehours, P. Inflammatory cytokine and microrna responses of primary human dendritic cells cultured with helicobacter pylori strains. Front. Microbiol. 2013, 4, 236. [Google Scholar] [CrossRef] [Green Version]
- Sasaran, M.O.; Melit, L.E.; Dobru, E.D. Microrna modulation of host immune response and inflammation triggered by helicobacter pylori. Int. J. Mol. Sci. 2021, 22, 1406. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.G.; Wang, W.H.; Dai, Y.; Wang, S.J.; Chu, Y.X.; Li, J. Let-7b is involved in the inflammation and immune responses associated with helicobacter pylori infection by targeting toll-like receptor 4. PLoS ONE 2013, 8, e56709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres, E.; Bitarte, N.; Arias, F.; Agorreta, J.; Fortes, P.; Agirre, X.; Zarate, R.; Diaz-Gonzalez, J.A.; Ramirez, N.; Sola, J.J.; et al. Microrna-451 regulates macrophage migration inhibitory factor production and proliferation of gastrointestinal cancer cells. Clin. Cancer Res. 2009, 15, 2281–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Su, W.Y.; Xing, J.; Wang, Y.C.; Wang, P.; Chen, X.Y.; Shen, Z.Y.; Cao, H.; Lu, Y.Y.; Fang, J.Y. Mir-29a inhibits cell proliferation and induces cell cycle arrest through the downregulation of p42.3 in human gastric cancer. PLoS ONE 2011, 6, e25872. [Google Scholar] [CrossRef] [Green Version]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotamraju, S.; Williams, C.L.; Kalyanaraman, B. Statin-induced breast cancer cell death: Role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007, 67, 7386–7394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervin, S.; Singh, R.; Chaudhuri, G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (mda-mb-231): Potential role of cyclin d1. Proc. Natl. Acad. Sci. USA 2001, 98, 3583–3588. [Google Scholar] [CrossRef] [Green Version]
- Christie, C.F.; Fang, D.; Hunt, E.G.; Morris, M.E.; Rovini, A.; Heslop, K.A.; Beeson, G.C.; Beeson, C.C.; Maldonado, E.N. Statin-dependent modulation of mitochondrial metabolism in cancer cells is independent of cholesterol content. FASEB J. 2019, 33, 8186–8201. [Google Scholar] [CrossRef]
- Cherkas, A.; Zarkovic, N. 4-hydroxynonenal in redox homeostasis of gastrointestinal mucosa: Implications for the stomach in health and diseases. Antioxidants 2018, 7, 118. [Google Scholar]
- Ghaisas, M.M.; Dandawate, P.R.; Zawar, S.A.; Ahire, Y.S.; Gandhi, S.P. Antioxidant, antinociceptive and anti-inflammatory activities of atorvastatin and rosuvastatin in various experimental models. Inflammopharmacology 2010, 18, 169–177. [Google Scholar] [CrossRef]
- Stent, A.; Every, A.L.; Sutton, P. Helicobacter pylori defense against oxidative attack. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G579–G587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Chiba, T.; Marusawa, H. Helicobacter pylori-mediated genetic instability and gastric carcinogenesis. In Molecular Pathogenesis and Signal Transduction by Helicobacter Pylori; Tegtmeyer, N.M., Backert, S., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–323. [Google Scholar]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- McGregor, G.H.; Campbell, A.D.; Fey, S.K.; Tumanov, S.; Sumpton, D.; Blanco, G.R.; Mackay, G.; Nixon, C.; Vazquez, A.; Sansom, O.J.; et al. Targeting the metabolic response to statin-mediated oxidative stress produces a synergistic antitumor response. Cancer Res. 2020, 80, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Corcos, L.; Le Jossic-Corcos, C. Statins: Perspectives in cancer therapeutics. Dig. Liver Dis 2013, 45, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Gibot, L.; Follet, J.; Metges, J.P.; Auvray, P.; Simon, B.; Corcos, L.; Le Jossic-Corcos, C. Human caspase 7 is positively controlled by srebp-1 and srebp-2. Biochem. J. 2009, 420, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Follet, J.; Remy, L.; Hesry, V.; Simon, B.; Gillet, D.; Auvray, P.; Corcos, L.; Le Jossic-Corcos, C. Adaptation to statins restricts human tumour growth in nude mice. BMC Cancer 2011, 11, 491. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, T.; Tsukamoto, T.; Takasu, S.; Hirano, N.; Ban, H.; Shi, L.; Kumagai, T.; Tanaka, T.; Tatematsu, M. Pitavastatin fails to lower serum lipid levels or inhibit gastric carcinogenesis in helicobacter pylori-infected rodent models. Cancer Prev. Res. 2009, 2, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.W.; Liao, K.F.; Lin, C.Y.; Lin, C.L.; Sung, F.C. Statins on the risk of gastric cancer: A population-based observation in taiwan. Am. J. Gastroenterol. 2013, 108, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Seto, W.K.; Wong, I.C.K.; Leung, W.K. Statins were associated with a reduced gastric cancer risk in patients with eradicated helicobacter pylori infection: A territory-wide propensity score matched study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Lee, H.; Park, J.C.; Shin, S.K.; Lee, S.K.; Hyung, W.J.; Lee, Y.C.; Kang, M.W.; Noh, S.H. Long-term statin therapy improves oncological outcome after radical gastrectomy for stage ii and iii gastric cancer. Anticancer Res. 2014, 34, 355–361. [Google Scholar]
- Cho, M.H.; Yoo, T.G.; Jeong, S.M.; Shin, D.W. Association of aspirin, metformin, and statin use with gastric cancer incidence and mortality: A nationwide cohort study. Cancer Prev. Res. 2021, 14, 95–104. [Google Scholar] [CrossRef]
- Megraud, F.; Bessede, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 2015, 21, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Hartgrink, H.H.; Jansen, E.P.; van Grieken, N.C.; van de Velde, C.J. Gastric cancer. Lancet 2009, 374, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Schraw, W.; Li, Y.; McClain, M.S.; van der Goot, F.G.; Cover, T.L. Association of helicobacter pylori vacuolating toxin (vaca) with lipid rafts. J. Biol. Chem. 2002, 277, 34642–34650. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.H.; Chang, Y.C.; Du, S.Y.; Wang, H.J.; Kuo, C.H.; Fang, S.H.; Fu, H.W.; Lin, H.H.; Chiang, A.S.; Wang, W.C. Cholesterol depletion reduces helicobacter pylori caga translocation and caga-induced responses in ags cells. Infect. Immun. 2008, 76, 3293–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piscione, M.; Mazzone, M.; Di Marcantonio, M.C.; Muraro, R.; Mincione, G. Eradication of helicobacter pylori and gastric cancer: A controversial relationship. Front. Microbiol. 2021, 12, 630852. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Higuchi, T.; Hosomi, K.; Takada, M. Association between statin use and cancer: Data mining of a spontaneous reporting database and a claims database. Int. J. Med. Sci. 2015, 12, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Author and Study Year | Study Location | Type of Statin | Effect of Statins on Gastric Cancer | Experimental Studies | Analysis of H. pylori Status | Reference |
|---|---|---|---|---|---|---|
| Toyoda, 2009 | Japan | Pitavastatin | Pitavastatin was ineffective for chemoprevention of gastric carcinogenesis in gerbils | Rodent models | + | [94] |
| Chiu, 2011 | Taiwan | Lovastatin, pravastatin, rosuvastatin, fluvastatin, simvastatin, and atorvastatin | Any statins are associated with a reduction in gastric cancer risk | Clinical | + | [18] |
| Lee, 2012 | Korea | NA † | The longer prescription of statins, the more reduced risk of gastric cancer | Clinical | +/‒ ¶ | [27] |
| Lai, 2013 | Taiwan | Lovastatin, pravastatin, rosuvastatin, fluvastatin, simvastatin, and atorvastatin | Simvastatin significantly reduces gastric cancer risk with a dose-response relationship | Clinical | ‒ | [95] |
| Lin, 2016 | Taiwan | Simvastain and lovastatin | Statins reduce the risk of gastric cancer significantly | Clinical and in vitro | + | [13] |
| Cheung, 2019 | Hong Kong | NA | Statins lower the risk of H. pylori-eradicated gastric cancer | Clinical | ‒ | [96] |
| You, 2020 | Korea | Pravastatin, simvastatin, atorvastatin, cerivastatin, lovastatin, and fluvastatin | Statins decrease gastric cancer incidence in patients with hypercholesterolemia | Clinical | ‒ | [28] |
| Author and Study Year | Study Location | Type of Statin | Effect of Statins on Gastric Cancer | Experimental Studies | Analysis of H. pylori status | Reference |
|---|---|---|---|---|---|---|
| Nam, 2014 | Korea | Atorvastatin, rosuvastatin, simvastatin, pitavastatin, fluvastatin, and pravastatin | Statins prescribed more than six month was associated with increased survival | Clinical | ‒ | [97] |
| Spence, 2019 | UK | NA † | Statins decrease the mortality of gastric cancer | Clinical | ‒ | [14] |
| Yang, 2020 | Taiwan | NA | Statins increase overall survival of patients with gastric cancer after surgery and adjuvant chemotherapy | Clinical | ‒ | [15] |
| Cho, 2021 | Korea | NA | Statins lower the mortality of gastric cancer but fail to reduce the incidence | Clinical | ‒ | [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-Y.; Lan, W.-H.; Chiu, Y.-F.; Feng, C.-L.; Chiu, C.-H.; Kuo, C.-J.; Lai, C.-H. Statins’ Regulation of the Virulence Factors of Helicobacter pylori and the Production of ROS May Inhibit the Development of Gastric Cancer. Antioxidants 2021, 10, 1293. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081293
Lin T-Y, Lan W-H, Chiu Y-F, Feng C-L, Chiu C-H, Kuo C-J, Lai C-H. Statins’ Regulation of the Virulence Factors of Helicobacter pylori and the Production of ROS May Inhibit the Development of Gastric Cancer. Antioxidants. 2021; 10(8):1293. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081293
Chicago/Turabian StyleLin, Ting-Yu, Wen-Hsi Lan, Ya-Fang Chiu, Chun-Lung Feng, Cheng-Hsun Chiu, Chia-Jung Kuo, and Chih-Ho Lai. 2021. "Statins’ Regulation of the Virulence Factors of Helicobacter pylori and the Production of ROS May Inhibit the Development of Gastric Cancer" Antioxidants 10, no. 8: 1293. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox10081293