
Mitochondrial ROS, ER Stress, and Nrf2 Crosstalk in the Regulation of Mitochondrial Apoptosis Induced by Arsenite


{kind=link}
{kind=link}
Abstract
:1. Introduction
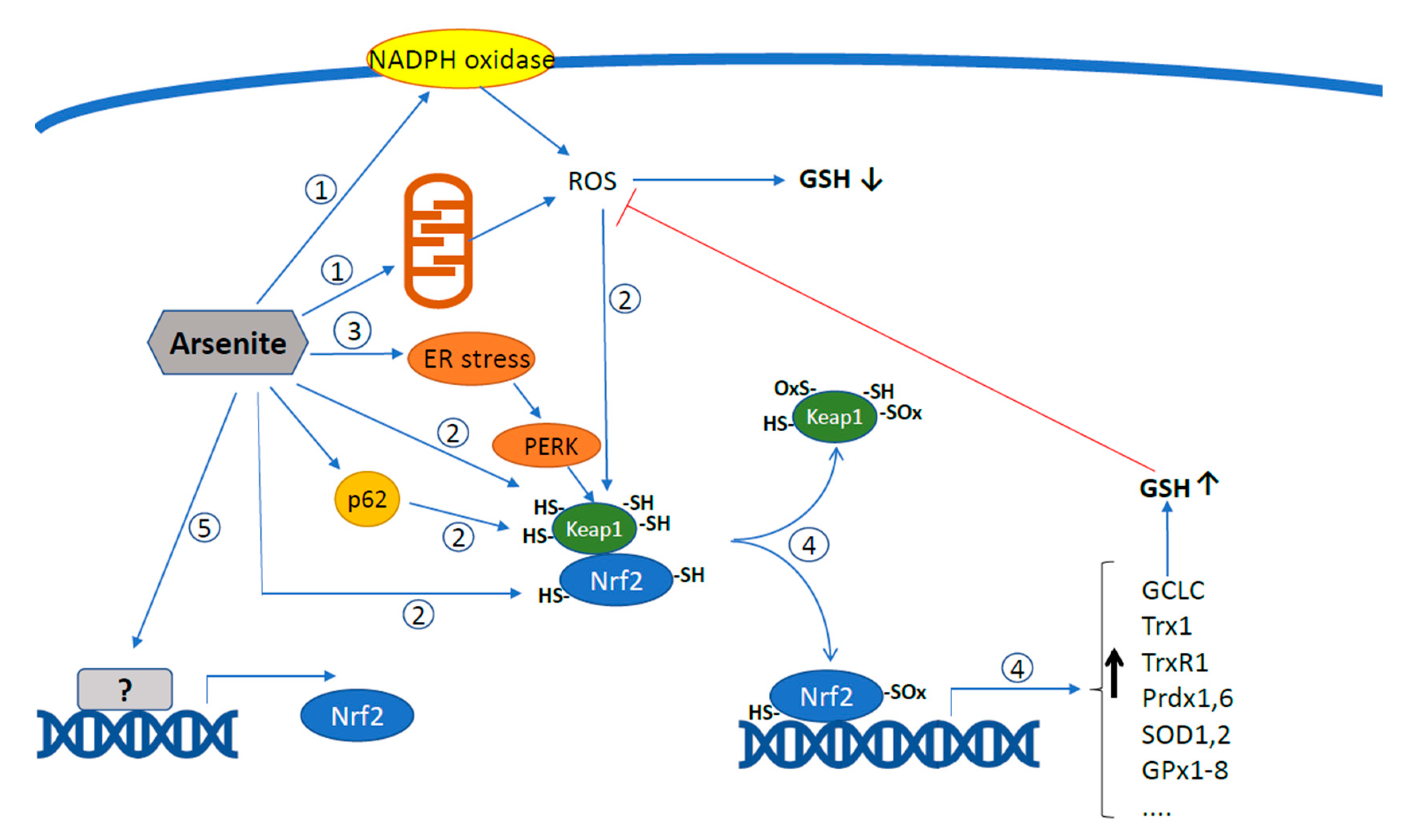
2. Arsenite Promotes the Mitochondrial Formation of Superoxide and Downstream Intra-Mitochondrial and Extra-Mitochondrial Effects
3. ER-Mitochondria Crosstalk Regulates Arsenite-Induced mitoO2•− Formation
4. Effect of Arsenic on Nrf2 and Its Target Genes
5. Arsenite-Dependent Regulation of Nrf2 by Mitochondrial ROS: Impact on Survival vs. Apoptotic Signalling
6. Crosstalk between Arsenite-Induced ER Stress, UPR, and Nrf2-Mediated Antioxidant Responses
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Masuda, H. Arsenic cycling in the Earth’s crust and hydrosphere: Interaction between naturally occurring arsenic and human activities. Prog. Earth Planet. Sci. 2018, 5, 68. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for Drinking-Water Quality [Electronic Resource]: Incorporating First Addendum; Recommendations; World Health Organization: Geneva, Switzerland, 2006; Volume 1. [Google Scholar]
- Shankar, S.; Shanker, U. Shikha Arsenic Contamination of Groundwater: A Review of Sources, Prevalence, Health Risks, and Strategies for Mitigation. Sci. World J. 2014, 2014, 304524. [Google Scholar] [CrossRef]
- Hong, Y.-S.; Song, K.-H.; Chung, J.-Y. Health Effects of Chronic Arsenic Exposure. J. Prev. Med. Public Health Yebang Uihakhoe Chi 2014, 47, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ro, S.-H.; Bae, J.; Jang, Y.; Myers, J.F.; Chung, S.; Yu, J.; Natarajan, S.K.; Franco, R.; Song, H.-S. Arsenic Toxicity on Metabolism and Autophagy in Adipose and Muscle Tissues. Antioxidants 2022, 11, 689. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Rai, C.B.; Kumari, N.; Sandhu, V.K.; Chandra, K.; Krishna, M.; Kota, S.H.; Anand, K.S.; Oudin, A. Air Pollution and Cognitive Impairment across the Life Course in Humans: A Systematic Review with Specific Focus on Income Level of Study Area. Int. J. Environ. Res. Public Health 2022, 19, 1405. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Sumi, D. Arsenic: Signal Transduction, Transcription Factor, and Biotransformation Involved in Cellular Response and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Hei, T.K.; Filipic, M. Role of oxidative damage in the genotoxicity of arsenic. Free Radic. Biol. Med. 2004, 37, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Reichard, J.F.; Puga, A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2010, 2, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.M.; Arnold, L.L.; Beck, B.D.; Lewis, A.S.; Eldan, M. Evaluation of the carcinogenicity of inorganic arsenic. Crit. Rev. Toxicol. 2013, 43, 711–752. [Google Scholar] [CrossRef]
- Hu, Y.; Li, J.; Lou, B.; Wu, R.; Wang, G.; Lu, C.; Wang, H.; Pi, J.; Xu, Y. The Role of Reactive Oxygen Species in Arsenic Toxicity. Biomolecules 2020, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Li, X.-F.; Cullen, W.R.; Weinfeld, M.; Le, X.C. Arsenic Binding to Proteins. Chem. Rev. 2013, 113, 7769–7792. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J. Arsenic-induced oxidative stress and its reversibility. Free Radic. Biol. Med. 2011, 51, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Prakash, C.; Soni, M.; Kumar, V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J. Appl. Toxicol. 2016, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic Exposure and Toxicology: A Historical Perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef] [Green Version]
- Nurchi, V.M.; Djordjevic, A.B.; Crisponi, G.; Alexander, J.; Bjørklund, G.; Aaseth, J. Arsenic Toxicity: Molecular Targets and Therapeutic Agents. Biomolecules 2020, 10, 235. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-X.; Davidson, M.M.; Tang, X.; Walker, W.F.; Athar, M.; Ivanov, V.; Hei, T.K. Mitochondrial Damage Mediates Genotoxicity of Arsenic in Mammalian Cells. Cancer Res. 2005, 65, 3236–3242. [Google Scholar] [CrossRef] [Green Version]
- Chavan, H.; Christudoss, P.; Mickey, K.; Tessman, R.; Ni, H.-M.; Swerdlow, R.; Krishnamurthy, P. Arsenite Effects on Mitochondrial Bioenergetics in Human and Mouse Primary Hepatocytes Follow a Nonlinear Dose Response. Oxidative Med. Cell. Longev. 2017, 2017, 9251303. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef]
- Chou, W.-C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Lemarie, A.; Bourdonnay, E.; Morzadec, C.; Fardel, O.; Vernhet, L. Inorganic arsenic activates reduced NADPH oxidase in human primary macrophages through a Rho kinase/p38 kinase pathway. J. Immunol. 2008, 180, 6010–6017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, A.C.; Clark, K.A.; Ross, M.A.; Chandra, A.G.; Li, S.; Gao, X.; Pagano, P.J.; Stolz, D.B.; Barchowsky, A. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase–generated superoxide. J. Clin. Investig. 2008, 118, 3980–3989. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Pachauri, V.; Mehta, A.; Mishra, D.; Flora, S.J. Arsenic induced neuronal apoptosis in guinea pigs is Ca2+ dependent and abrogated by chelation therapy: Role of voltage gated calcium channels. NeuroToxicology 2013, 35, 137–145. [Google Scholar] [CrossRef]
- Chen, A.; Cao, E.-H.; Zhang, T.-C.; Qin, J.-F. Arsenite-induced reactive oxygen species and the repression of alpha-tocopherol in the MGC-803 cells. Eur. J. Pharmacol. 2002, 448, 11–18. [Google Scholar] [CrossRef]
- Banerjee, C.; Goswami, R.; Datta, S.; Rajagopal, R.; Mazumder, S. Arsenic-induced alteration in intracellular calcium homeostasis induces head kidney macrophage apoptosis involving the activation of calpain-2 and ERK in Clarias batrachus. Toxicol. Appl. Pharmacol. 2011, 256, 44–51. [Google Scholar] [CrossRef]
- Bolt, A.M.; Zhao, F.; Pacheco, S.; Klimecki, W.T. Arsenite-induced autophagy is associated with proteotoxicity in human lymphoblastoid cells. Toxicol. Appl. Pharmacol. 2012, 264, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.-T.; Lin, H.-T.; Hwang, P.-A.; Wang, S.-T.; Hsieh, C.-H.; Hwang, D.-F. Taurine resumed neuronal differentiation in arsenite-treated N2a cells through reducing oxidative stress, endoplasmic reticulum stress, and mitochondrial dysfunction. Amino Acids 2015, 47, 735–744. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Harder, B.; Castro-Portuguez, R.; Rodrigues, S.D.; Wong, P.K.; Chapman, E.; Zhang, D.D. Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol. Appl. Pharmacol. 2018, 341, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, X.; Chen, Y.; Wang, H.; Zhang, R.; Zhang, Q.; Wei, Y.; Shi, S.; Li, X. Calreticulin regulated intrinsic apoptosis through mitochondria-dependent and independent pathways mediated by ER stress in arsenite exposed HT-22 cells. Chemosphere 2020, 251, 126466. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, A. ROS -mediated PERK-eIF2alpha-ATF4 pathway plays an important role in arsenite-induced L-02 cells apoptosis via regulating CHOP-DR5 signaling. Environ. Toxicol. 2020, 35, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gu, S.; Jiang, X.; Zhang, Z. Arsenite-induced endoplasmic reticulum-dependent apoptosis through disturbance of calcium homeostasis in HBE cell line. Environ. Toxicol. 2017, 32, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.-L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, P.; Da Conceicao, V.N.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Wang, H.; Xue, P.; Li, X.; Li, B.; Zheng, Q.; Sun, G. Arsenic induces mitochondria-dependent apoptosis by reactive oxygen species generation rather than glutathione depletion in Chang human hepatocytes. Arch. Toxicol. 2009, 83, 899–908. [Google Scholar] [CrossRef]
- Du, L.; Yu, Y.; Chen, J.; Liu, Y.; Xia, Y.; Chen, Q.; Liu, X. Arsenic induces caspase- and mitochondria-mediated apoptosis inSaccharomyces cerevisiae. FEMS Yeast Res. 2007, 7, 860–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranmandura, H.; Chen, X.; Tanaka, M.; Wang, W.W.; Rehman, K.; Xu, S.; Chen, Z.; Chen, S.Q.; Suzuki, N. Release of Apoptotic Cytochrome C From Mitochondria by Dimethylarsinous Acid Occurs Through Interaction With Voltage-Dependent Anion Channel In Vitro. Toxicol. Sci. 2012, 128, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peraza, M.A.; Cromey, D.W.; Carolus, B.; Carter, D.E.; Gandolfi, A.J. Morphological and functional alterations in human proximal tubular cell line induced by low level inorganic arsenic: Evidence for targeting of mitochondria and initiated apoptosis. J. Appl. Toxicol. 2006, 26, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yang, Y.; Shao, H.; Sun, W.; Gu, M.; Wang, H.; Jiang, L.; Qu, L.; Sun, D.; Gao, Y. Sodium Arsenite-Induced Learning and Memory Impairment Is Associated with Endoplasmic Reticulum Stress-Mediated Apoptosis in Rat Hippocampus. Front. Mol. Neurosci. 2017, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.F.; Chen, Y.H.; Chang, M.-M.; Chen, Y.C.; Huang, B.M. Arsenic compounds induce apoptosis through caspase pathway activation in MA-10 Leydig tumor cells. Oncol. Lett. 2019, 18, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-W.; Lin, P.-J.; Tsai, J.-S.; Lin, C.-Y.; Lin, L.-Y. Arsenite-induced apoptosis can be attenuated via depletion of mTOR activity to restore autophagy. Toxicol. Res. 2019, 8, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidarelli, A.; Fiorani, M.; Cerioni, L.; Cantoni, O. Calcium signals between the ryanodine receptor- and mitochondria critically regulate the effects of arsenite on mitochondrial superoxide formation and on the ensuing survival vs apoptotic signaling. Redox Biol. 2019, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Capellacci, V.; Cerioni, L.; Crinelli, R.; Cantoni, O. The dual role of mitochondrial superoxide in arsenite toxicity: Signaling at the boundary between apoptotic commitment and cytoprotection. Toxicol. Appl. Pharmacol. 2018, 345, 26–35. [Google Scholar] [CrossRef]
- Guidarelli, A.; Catalani, A.; Spina, A.; Varone, E.; Fumagalli, S.; Zito, E.; Fiorani, M.; Cantoni, O. Functional organization of the endoplasmic reticulum dictates the susceptibility of target cells to arsenite-induced mitochondrial superoxide formation, mitochondrial dysfunction and apoptosis. Food Chem. Toxicol. 2021, 156, 112523. [Google Scholar] [CrossRef]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Methods Enzymol. 2002, 349, 9–23. [Google Scholar] [CrossRef]
- Esposti, M.D. Inhibitors of NADH–ubiquinone reductase: An overview. Biochim. Biophys. Acta 1998, 1364, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, M.; Azzolini, C.; Cerioni, L.; Scotti, M.; Guidarelli, A.; Ciacci, C.; Cantoni, O. The mitochondrial transporter of ascorbic acid functions with high affinity in the presence of low millimolar concentrations of sodium and in the absence of calcium and magnesium. Biochim. Biophys. Acta 2015, 1848, 1393–1401. [Google Scholar] [CrossRef] [Green Version]
- Guidarelli, A.; Carloni, S.; Balduini, W.; Fiorani, M.; Cantoni, O. Mitochondrial ascorbic acid prevents mitochondrial O2.- formation, an event critical for U937 cell apoptosis induced by arsenite through both autophagic-dependent and independent mechanisms. Biofactors 2016, 42, 190–200. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Suriyo, T.; Watcharasit, P.; Thiantanawat, A.; Satayavivad, J. Arsenite promotes apoptosis and dysfunction in microvascular endothelial cells via an alteration of intracellular calcium homeostasis. Toxicol. Vitr. 2012, 26, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Guidarelli, A.; Fiorani, M.; Cantoni, O. Low Concentrations of Arsenite Target the Intraluminal Inositol 1, 4, 5-Trisphosphate Receptor/Ryanodine Receptor Crosstalk to Significantly Elevate Intracellular Ca(2). J. Pharmacol. Exp. Ther. 2018, 367, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Guidarelli, A.; Cerioni, L.; Fiorani, M.; Catalani, A.; Cantoni, O. Arsenite-Induced Mitochondrial Superoxide Formation: Time and Concentration Requirements for the Effects of the Metalloid on the Endoplasmic Reticulum and Mitochondria. J. Pharmacol. Exp. Ther. 2020, 373, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Spina, A.; Guidarelli, A.; Fiorani, M.; Varone, E.; Catalani, A.; Zito, E.; Cantoni, O. Crosstalk between ERO1alpha and ryanodine receptor in arsenite-dependent mitochondrial ROS formation. Biochem. Pharmacol. 2022, 198, 114973. [Google Scholar] [CrossRef]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Li, C.Y.-T.; Kong, A.-N.T. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharmacal Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobbagy, S.; Vitturi, D.A.; Salvatore, S.R.; Turell, L.; Pires, M.F.; Kansanen, E.; Batthyany, C.; Lancaster, J.R.; Freeman, B.A.; Schopfer, F.J. Electrophiles modulate glutathione reductase activity via alkylation and upregulation of glutathione biosynthesis. Redox Biol. 2019, 21, 101050. [Google Scholar] [CrossRef] [PubMed]
- Tejo, F.V.; Quintanilla, R. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Ryoo, I.-G.; Kwak, M.-K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Withers, C.M.; Bartz, R.R.; MacGarvey, N.C.; Fu, P.; Sweeney, T.E.; Welty-Wolf, K.E.; Suliman, H.B. Heme Oxygenase-1 Couples Activation of Mitochondrial Biogenesis to Anti-inflammatory Cytokine Expression. J. Biol. Chem. 2011, 286, 16374–16385. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Llères, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef] [Green Version]
- De La Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Reddy, S.P.; Debiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef]
- Uno, K.; Prow, T.W.; Bhutto, I.A.; Yerrapureddy, A.; McLeod, D.S.; Yamamoto, M.; Reddy, S.P.; Lutty, G.A. Role of Nrf2 in retinal vascular development and the vaso-obliterative phase of oxygen-induced retinopathy. Exp. Eye Res. 2010, 90, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, A.S.; Warren, A.J.; Barchowsky, A.; Temple, K.A.; Klei, L.; Soucy, N.V.; O’Hara, K.A.; Hamilton, J.W. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ. Health Perspect. 2003, 111, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Del Razo, L.M.; Quintanilla-Vega, B.; Brambila-Colombres, E.; Calderon-Aranda, E.S.; Manno, M.; Albores, A. Stress Proteins Induced by Arsenic. Toxicol. Appl. Pharmacol. 2001, 177, 132–148. [Google Scholar] [CrossRef]
- Ran, S.; Liu, J.; Li, S. A Systematic Review of the Various Effect of Arsenic on Glutathione Synthesis In Vitro and In Vivo. BioMed Res. Int. 2020, 2020, 9414196. [Google Scholar] [CrossRef]
- Wang, C.; Niu, Q.; Ma, R.; Song, G.; Hu, Y.; Xu, S.; Li, Y.; Wang, H.; Li, S.; Ding, Y. The Variable Regulatory Effect of Arsenic on Nrf2 Signaling Pathway in Mouse: A Systematic Review and Meta-analysis. Biol. Trace Element Res. 2019, 190, 362–383. [Google Scholar] [CrossRef]
- He, X.; Ma, Q. Critical Cysteine Residues of Kelch-Like ECH-Associated Protein 1 in Arsenic Sensing and Suppression of Nuclear Factor Erythroid 2-Related Factor 2. J. Pharmacol. Exp. Ther. 2010, 332, 66–75. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Q. NRF2 Cysteine Residues Are Critical for Oxidant/Electrophile-Sensing, Kelch-Like ECH-Associated Protein-1-Dependent Ubiquitination-Proteasomal Degradation, and Transcription Activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ling, M.; Chen, C.; Luo, F.; Yang, P.; Wang, D.; Chen, X.; Xu, H.; Xue, J.; Yang, Q.; et al. Impaired autophagic flux and p62-mediated EMT are involved in arsenite-induced transformation of L-02 cells. Toxicol. Appl. Pharmacol. 2017, 334, 75–87. [Google Scholar] [CrossRef]
- Shah, P.; Trinh, E.; Qiang, L.; Xie, L.; Hu, W.-Y.; Prins, G.S.; Pi, J.; He, Y.-Y. Arsenic Induces p62 Expression to Form a Positive Feedback Loop with Nrf2 in Human Epidermal Keratinocytes: Implications for Preventing Arsenic-Induced Skin Cancer. Molecules 2017, 22, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP Augments Promoter-Specific DNA Binding of Nrf2 during the Antioxidant Response. Mol. Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Wadgaonkar, P.; Chen, F. Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. 2021, 76, 258–266. [Google Scholar] [CrossRef]
- Sinha, D.; Biswas, J.; Bishayee, A. Nrf2-mediated redox signaling in arsenic carcinogenesis: A review. Arch. Toxicol. 2013, 87, 383–396. [Google Scholar] [CrossRef]
- Pi, J.; Qu, W.; Reece, J.M.; Kumagai, Y.; Waalkes, M.P. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: Involvement of hydrogen peroxide. Exp. Cell Res. 2003, 290, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Bjorkman, S.H.; Pereira, R.O. The Interplay Between Mitochondrial Reactive Oxygen Species, Endoplasmic Reticulum Stress, and Nrf2 Signaling in Cardiometabolic Health. Antioxid. Redox Signal. 2021, 35, 252–269. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, D.L.; Gladyshev, V.N. Hydrogen Peroxide Probes Directed to Different Cellular Compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef] [Green Version]
- Zito, E.; Melo, E.P.; Yang, Y.; Wahlander, Å.; Neubert, T.A.; Ron, D. Oxidative Protein Folding by an Endoplasmic Reticulum-Localized Peroxiredoxin. Mol. Cell 2010, 40, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Chernorudskiy, A.L.; Zito, E. Regulation of Calcium Homeostasis by ER Redox: A Close-Up of the ER/Mitochondria Connection. J. Mol. Biol. 2017, 429, 620–632. [Google Scholar] [CrossRef]
- Pozzer, D.; Varone, E.; Chernorudskiy, A.; Schiarea, S.; Missiroli, S.; Giorgi, C.; Pinton, P.; Canato, M.; Germinario, E.; Nogara, L.; et al. A maladaptive ER stress response triggers dysfunction in highly active muscles of mice with SELENON loss. Redox Biol. 2019, 20, 354–366. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha Regulates Ca(2+) Fluxes at the Endoplasmic Reticulum-Mitochondria Interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.S.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.M.Y.; Chao, P.L.; Fang, S.F.; Chi, C.W.; Yang, J.C.-H. Endoplasmic reticulum stress is involved in arsenite-induced oxidative injury in rat brain. Toxicol. Appl. Pharmacol. 2007, 224, 138–146. [Google Scholar] [CrossRef]
- Lin, A.M.Y.; Fang, S.F.; Chao, P.L.; Yang, J.C.-H. Melatonin attenuates arsenite-induced apoptosis in rat brain: Involvement of mitochondrial and endoplasmic reticulum pathways and aggregation of alpha-synuclein. J. Pineal Res. 2007, 43, 163–171. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.K. Role of Thiols in the Structure and Function of Inositol Trisphosphate Receptors. Curr. Top. Membr. 2010, 66, 299–322. [Google Scholar] [CrossRef]
- Anzai, K.; Ogawa, K.; Ozawa, T.; Yamamoto, H. Oxidative Modification of Ion Channel Activity of Ryanodine Receptor. Antioxid. Redox Signal. 2000, 2, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-A.; Wang, B.; Miyagi, M.; Hess, D.T.; Stamler, J.S. Oxygen-coupled Redox Regulation of the Skeletal Muscle Ryanodine Receptor/Ca2+ Release Channel (RyR1): Sites and Nature of Oxidative Modification. J. Biol. Chem. 2013, 288, 22961–22971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Hajnóczky, G. SR/ER–mitochondrial local communication: Calcium and ROS. Biochim. Biophys. Acta 2009, 1787, 1352–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbincius, J.F.; Elrod, J.W. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantoni, O.; Zito, E.; Guidarelli, A.; Fiorani, M.; Ghezzi, P. Mitochondrial ROS, ER Stress, and Nrf2 Crosstalk in the Regulation of Mitochondrial Apoptosis Induced by Arsenite. Antioxidants 2022, 11, 1034. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051034
Cantoni O, Zito E, Guidarelli A, Fiorani M, Ghezzi P. Mitochondrial ROS, ER Stress, and Nrf2 Crosstalk in the Regulation of Mitochondrial Apoptosis Induced by Arsenite. Antioxidants. 2022; 11(5):1034. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051034
Chicago/Turabian StyleCantoni, Orazio, Ester Zito, Andrea Guidarelli, Mara Fiorani, and Pietro Ghezzi. 2022. "Mitochondrial ROS, ER Stress, and Nrf2 Crosstalk in the Regulation of Mitochondrial Apoptosis Induced by Arsenite" Antioxidants 11, no. 5: 1034. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11051034