
Redox Mechanisms of Platelet Activation in Aging


1
Department of Laboratory Medicine, Yale School of Medicine, New Haven, CT 06511, USA
2
Department of Internal Medicine, University of Iowa Carver College of Medicine, Iowa City, IA 52242, USA
3
Iowa City VA Healthcare System, Iowa City, IA 52246, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2022, 11(5), 995; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050995
Submission received: 12 April 2022
/
Revised: 14 May 2022
/
Accepted: 17 May 2022
/
Published: 19 May 2022
(This article belongs to the Special Issue Oxidative Stress and Antioxidants in Aging)
{kind=link}
{kind=link}
Abstract
:Aging is intrinsically linked with physiologic decline and is a major risk factor for a broad range of diseases. The deleterious effects of advancing age on the vascular system are evidenced by the high incidence and prevalence of cardiovascular disease in the elderly. Reactive oxygen species are critical mediators of normal vascular physiology and have been shown to gradually increase in the vasculature with age. There is a growing appreciation for the complexity of oxidant and antioxidant systems at the cellular and molecular levels, and accumulating evidence indicates a causal association between oxidative stress and age-related vascular disease. Herein, we review the current understanding of mechanistic links between oxidative stress and thrombotic vascular disease and the changes that occur with aging. While several vascular cells are key contributors, we focus on oxidative changes that occur in platelets and their mediation in disease progression. Additionally, we discuss the impact of comorbid conditions (i.e., diabetes, atherosclerosis, obesity, cancer, etc.) that have been associated with platelet redox dysregulation and vascular disease pathogenesis. As we continue to unravel the fundamental redox mechanisms of the vascular system, we will be able to develop more targeted therapeutic strategies for the prevention and management of age-associated vascular disease.
1. Introduction
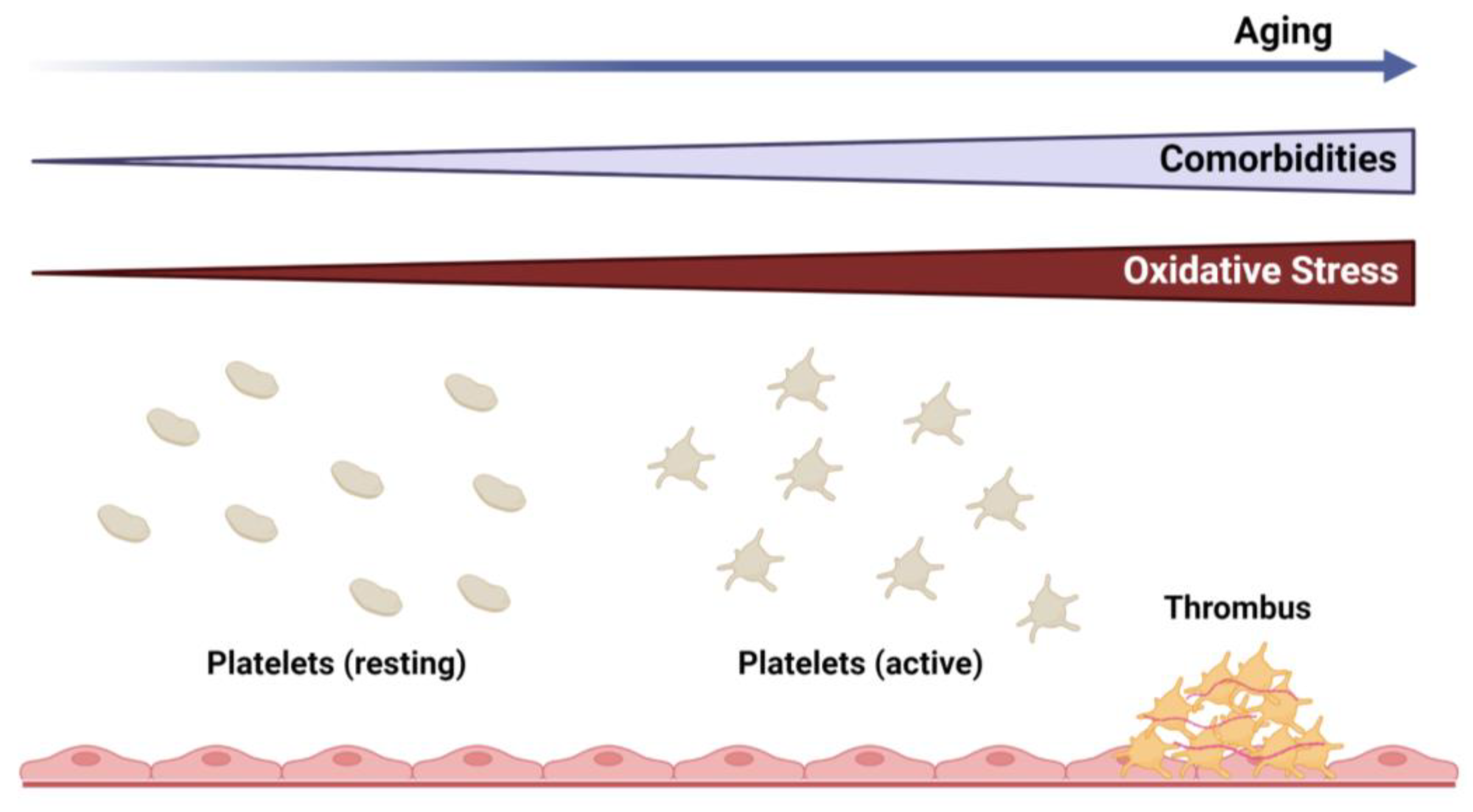
Aging is a biological phenomenon in living organisms that is characterized by a gradual decline in physical and mental capacity [1]. It represents an accumulation of adverse changes over time that increases the risk of disease and, ultimately, death. Cardiovascular disease is the leading cause of death in the United States [2] and worldwide [3], and it is well established that myocardial infarction and stroke increase significantly with age [4]. These observations are, in part, due to the high prevalence of associated comorbid conditions (e.g., diabetes, obesity, hypertension, hyperlipidemia, etc.) that are frequently observed in the elderly [5]. As the average human life expectancy and, consequently, the number of elderly in the population are expected to grow in the coming decades, the burden of chronic diseases, including cardiovascular and cerebrovascular disease, is projected to have a more significant impact on human health and healthcare costs [1,6,7]. While several cell types are known to contribute to vascular pathologies, this review will focus on platelets as the key mediators in the progression of thrombotic vascular disease (Figure 1).
2. Platelets and Thrombotic Vascular Disease
Platelets are anucleate cells derived from megakaryocytes that are key components of the vascular system, and they are traditionally recognized for their vital role in hemostasis through the enhancement of coagulation and thrombus formation at sites of vascular injury [8]. Platelet activation is central to thrombus formation and, thus, plays a critical role in the pathogenesis of thrombotic vascular diseases [9,10,11]. In addition to their fundamental roles in hemostasis and thrombosis, platelets can also act as mediators of inflammation and immunity through functional interactions with the vascular endothelium and circulating hematopoietic cells [10,12,13,14]. Altered platelet function and platelet hyperactivity have been associated with aging. Early epidemiological and functional studies suggested that platelet activity is enhanced in elderly individuals, although the precise mechanisms for these changes were not entirely clear [15,16,17,18]. Below, we discuss the recent literature on the redox mechanisms that regulate platelet activation while highlighting specific alterations that occur with age. In addition, we will discuss the role of platelets in cardiovascular comorbidities that are more prevalent in the elderly and can provide mechanistic insights into age-associated vascular disease. Understanding the fundamental cellular and molecular processes that occur in platelets with aging will provide opportunities to develop novel therapeutic strategies to prevent age-associated vascular pathologies and reduce the burden of disease in the elderly.
3. Oxidative and Antioxidative Mechanisms in Regulation of Platelets in Aging
It is well recognized that there is an age-associated increase in reactive oxygen species (ROS), and oxidative modifications to macromolecules (i.e., DNA, proteins, lipids) have been implicated as fundamental drivers of age-associated pathologies [19]. In the vascular system, accumulating evidence has demonstrated critical roles for ROS in the regulation of a variety of cellular and molecular processes, which, when dysregulated, can increase disease burden.
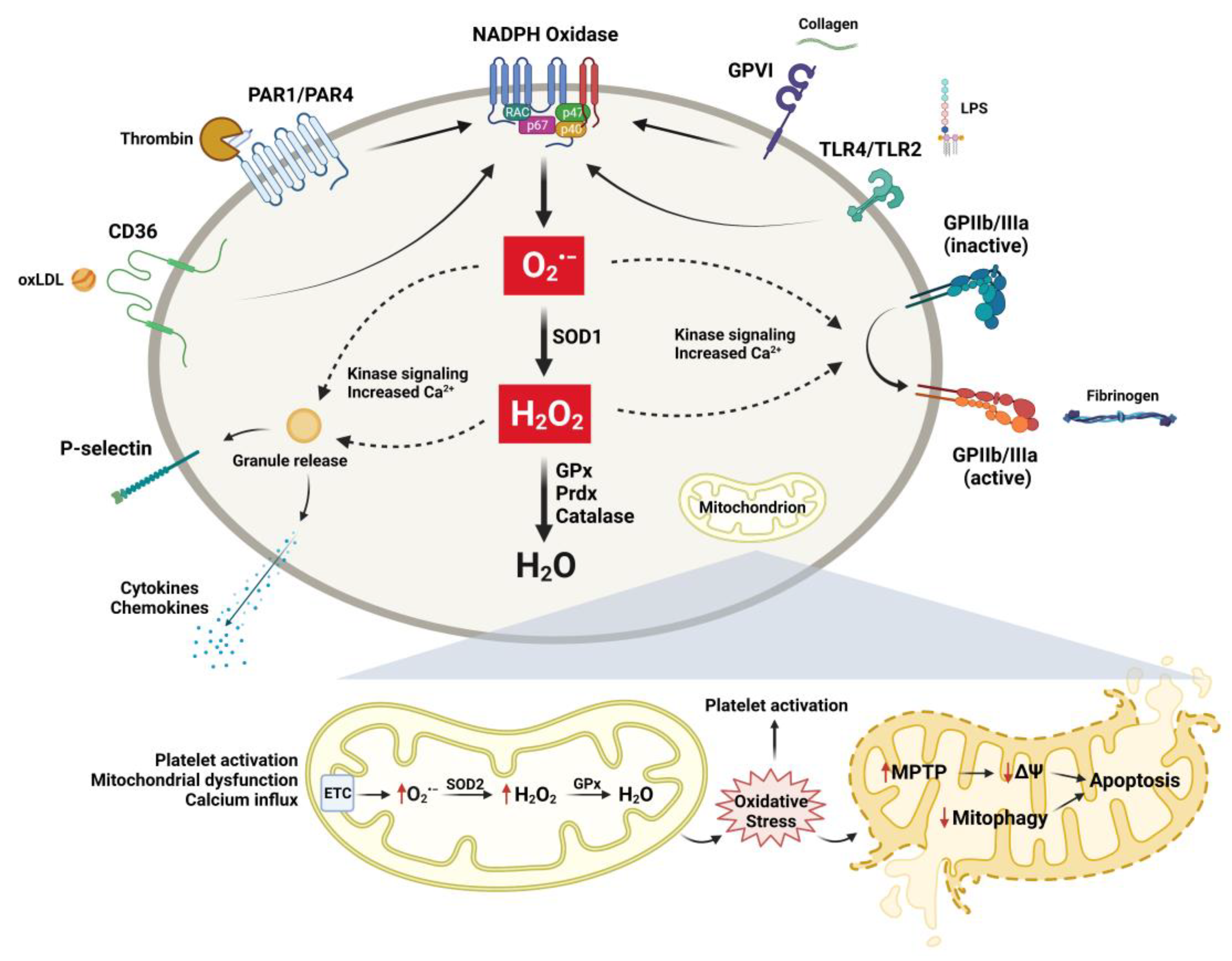
The relationship between oxidative stress and platelet activity has been a growing area of interest in studying the mechanisms of cardiovascular diseases and other age-related thrombotic vascular conditions. Initial studies in the 1970s first demonstrated the capacity of platelets to generate superoxide (O2●−) [20], a highly reactive species with a relatively short half-life. Several studies subsequently have demonstrated that exogenous and endogenous O2●− enhance platelet function, which can be reversed in the presence of SOD enzymes or O2●− scavengers [21,22]. Superoxide can be generated by several sources in the vasculature, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, uncoupled endothelial nitric oxide synthase (eNOS), xanthine oxidase, and mitochondrial sources [23]. Studies in our lab found that platelets isolated from aged mice displayed enhanced activation responses compared to those from young mice, which was prevented by the NADPH oxidase inhibitor apocynin [24]. Platelets from aged mice also had increased mRNA expression of the p47phox NADPH oxidase subunit. However, the mRNA levels for another subunit, Nox2, were not significantly different between platelets from aged and young mice, and the mRNA for Nox1 and Nox4 was not detectable in these studies [24]. The Nox family consists of seven isoforms, of which four (Nox1, 2, 4, and 5) are found in vascular cells [25]. Nox proteins are the catalytic component of the NADPH oxidase and, together with p22phox, comprise the large heterodimeric cytochrome subunit [26]. The Nox2 NADPH oxidase isoform consists of additional cytosolic regulatory subunits that include p47phox, p40phox, and p67phox. Further, studies in our lab found that Nox2 is not an essential source of platelet ROS and is not significantly involved in platelet activation and arterial thrombosis in young mice [27]. Some studies have reported a limited role of Nox2 in GPVI-dependent platelet responses [28], while other studies have found that Nox2 is important for collagen and thrombin-induced platelet responses in mice [29]. Using an EPR-based technique to measure intracellular platelet ROS, Vara et al. further elucidated the differential roles of Nox1 and Nox2 in platelet activation [30]. They found that Nox1 is important for collagen-induced platelet responses and that intracellular but not extracellular O2●− was critical for platelet activation by collagen, while Nox2 is important for thrombin-induced platelet activation [30]. Subsequent studies by the same group reported that a combined triple deficiency of Nox1, Nox2, and Nox4 in mice resulted in impaired platelet aggregation responses and decreased susceptibility to experimental thrombosis, while knockouts of single Nox isoforms showed no significant vascular effects in mice [31]. These studies indicate that Nox proteins are important for platelet activation and thrombosis in vivo, but there is redundancy in the O2●− generating system, given that single knockouts of individual Nox proteins produced minimal functional effects on platelets.
Studies have demonstrated that hydrogen peroxide (H2O2) can also be released from platelets [32]. H2O2 has a relatively long half-life and is an important intracellular signaling molecule, given its ability to diffuse across cellular membranes [33]. The conversion of O2●− to H2O2 is catalyzed by superoxide dismutase (SOD), which consists of three major isoforms in mammals, copper-zinc SOD (SOD1), manganese SOD (SOD2), and extracellular SOD (SOD3), that differ in their cellular localization and metal cofactors in the catalytic site [34]. The major antioxidant enzymes responsible for the catalytic inactivation of H2O2 include catalase, glutathione peroxidase (GPx), thioredoxin (Trx), and peroxiredoxin (Prdx). In humans, decreased activity of GPx-1 has been associated with both platelet hyperreactivity and an increased risk of cardiovascular events [35,36,37,38]. Studies in humans have also demonstrated that platelet activation is associated with the production of H2O2, and pre-treatment with catalase eliminated platelet H2O2 and inhibited collagen-induced platelet aggregation ex vivo [39]. Studies from our lab demonstrated that aged mice also exhibited increased susceptibility to thrombosis under experimental conditions and were protected from this phenotype by transgenic overexpression of intracellular GPx-1 [24]. Furthermore, platelets from aged mice displayed increased expression of not only the p47phox NADPH subunit but also SOD-1, providing a mechanistic explanation that age-dependent platelet hyperactivation is mediated by increased platelet O2●− generation and its conversion to H2O2 intracellularly [24]. These findings are in concordance with a more recent study using both human and mouse platelets that found that thrombin-induced platelet responses are dependent on the dismutation of O2●− to H2O2 [30]. In a separate study, Jin et al. demonstrated enhanced platelet aggregation responses and increased susceptibility to pulmonary thromboembolism and thrombotic stroke in mice with a deficiency of the circulating H2O2 metabolizing enzyme GPx-3 [40]. Together, these studies indicate a protective role for endogenous GPx in age-associated thrombotic consequences.
In a recent elegant study, Jain et al. stratified high cardiovascular risk patients into several age cohorts and measured the redox changes in platelets compared to younger healthy individuals [41]. A progressive age-associated increase in platelet reactivity and intracellular ROS was observed up until ~80 years of age, whereby a decline in platelet reactivity and ROS was seen. These findings were attributed to a decline in platelet antioxidants, including SOD-1, GPx-1, Prdx-6, and catalase, in patients aged 60–79 years and then an upregulation of these enzymes in people aged 80 or older that likely lowered the intracellular ROS burden. Using cross-sectional and longitudinal aging studies in mice, the authors recapitulated the findings of the human cohort [41]. These findings suggest that in the very elderly (i.e., 80 years or older), platelet antioxidant responses may be elicited as an adaptive mechanism to counterbalance the increase in oxidative stress and platelet hyperactivation associated with aging [42,43].
4. Platelet Mitochondria in Aging
Another important cellular source of ROS is mitochondria, which are key sites of the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. Oxygen consumption in the mitochondria is central to these processes. Its utilization to generate ATP through the electron transport chain is not completely efficient, and if electron transfers occur prematurely to oxygen, O2●− and its subsequent conversion to H2O2 can be generated as by-products [44]. Initially proposed by Denham Harman in the 1950s, the free radical theory of aging aimed to link aging with oxidative stress [45]. Subsequent revisions to this theory have focused on mitochondria as the primary source of ROS, and they are hypothesized to promote aging and age-related diseases through the accumulation of oxidative damage to protein, DNA, and lipid [46]. Although some studies have supported this theory [47,48], other observations have questioned a direct causal relationship between ROS and aging [49,50]. In fact, some studies have demonstrated increased lifespans in various model organisms with elevated mitochondrial ROS [51,52,53]. Thus, there is some controversy in the literature on the precise role of mitochondrial ROS in aging, which may reflect the different methods and types of model organisms used in the separate studies.
Despite not having a nucleus, platelets contain functional mitochondria and are metabolically active [54]. Platelets consume high levels of ATP, and mitochondrial aerobic respiration provides approximately 40% of the total basal energy requirements, while the remaining 60% is generated through glycolysis [54]. Studies have demonstrated that platelet activation is associated with alterations in mitochondrial processes such as the generation of ROS, induction of mitochondrial permeability transition pore (MPTP) formation, loss of mitochondrial membrane potential (ΔΨm), and induction of apoptosis [55,56,57,58]. Conversely, inhibition of mitochondrial respiratory function inhibits platelet aggregation and reduces clot formation [59]. A recent study provided evidence that platelet mitochondrial function is altered with age [60]. It was observed that older individuals (88 ± 2 years) display significant alterations in platelet bioenergetics, such as lower basal and ATP-linked respiration, compared to younger individuals (26 ± 5 years). Furthermore, an increased proton leak was observed in the platelet mitochondria of older individuals; this was attributed to the upregulation of an uncoupling protein (UCP). Previous studies have shown that UCPs are upregulated by superoxide and protected from ROS production through a decreased flow of electrons through the electron transport chain [61]. This process likely serves as an adaptive response to increased levels of mitochondrial ROS with advancing age in order to dissipate ROS generation [62,63]. Therefore, future studies could focus on the protective and harmful effects of key mitochondrial proteins regulating ROS within platelets during aging.
Mitochondrial superoxide overproduction has been shown to potentiate agonist-induced platelet activation under hyperglycemic conditions [64,65,66]. Moreover, hyperglycemia induces mitochondrial dysfunction and superoxide production in platelets that can induce both platelet hyperactivation and apoptosis, leading to increased susceptibility to experimental thrombosis in murine models of diabetes [66]. Despite observations that SOD2, the mitochondrial-specific antioxidant enzyme that dismutates O2●− to H2O2, is upregulated in patients with type 2 diabetes mellitus [67], studies failed to show significant alterations in platelet function or thrombotic susceptibility with platelet-specific knockouts of SOD2 in non-aged non-diabetic murine models [68]. Interestingly, these mice displayed increased mitochondrial superoxide production, while total intracellular ROS was unchanged compared to wild-type mice [68]. It is possible that the relatively low basal production of mitochondrial superoxide within platelets in young mice is insufficient to induce pathologic vascular changes, even with a concomitant deficiency of SOD2. Given the evidence that platelet activation and thrombotic risk are increased with age, it will be important for future studies using aging models to evaluate the effects of mitochondrial ROS and antioxidant enzymes in age-associated vascular disease and thrombosis.
Beyond its role in energy and redox metabolism, there is growing appreciation for mitochondria in other processes, including inflammation, stress response, and cell death, that can impact longevity and contribute to age-associated vascular diseases [69,70,71,72,73,74]. Autophagy is a fundamental cellular process that functions to degrade cellular contents and limit the accumulation of damaged biomacromolecules and organelles [75,76]. It helps in maintaining homeostasis during cellular stress and is presumed to prevent physiologic aging [77]. Autophagy has been demonstrated in platelets and has been shown to be important for platelet function and can impact hemostasis and thrombosis when dysregulated [72]. A pathway downstream to ROS in aging could be a mechanistic target of rapamycin complex 1 (mTORC1), which is involved in nutrient homeostasis and is closely integrated with the autophagy machinery [78]. A study showed that mTORC1 is upregulated in aged mice in a ROS-dependent pathway, and pharmacologic or genetic silencing of mTORC1 within platelets reduced the susceptibility to venous thrombosis in murine models [71].
Mitophagy is an autophagic process that is selective for removing damaged and dysfunctional mitochondria [79,80]. Alterations in mitophagy can impact platelet life span through direct interactions of apoptosis proteins with the mitophagic machinery [81]. Studies have demonstrated that platelets of diabetic patients are susceptible to oxidative stress, which can induce the phosphorylation of p53, resulting in mitochondrial dysfunction and apoptosis that can contribute to vascular thrombosis [66,82]. In parallel, increased ROS can induce selective mitophagy in human platelets, and this process serves as a protective mechanism against oxidative stress to remove damaged mitochondria and prevent apoptosis in the platelets of patients with diabetes [82]. In the same study, disruption of mitophagy using mice with a genetic deletion of Parkin or PINK1 produced platelets with increased susceptibility to H2O2-induced mitochondrial damage and apoptosis [82]. Moreover, genetic deletion of PINK1 in a diabetic mouse model produced platelets that were hyperactive with increased P-selectin surface expression, and the mice exhibited increased susceptibility to experimental carotid artery thrombosis [82].
Interestingly, platelet mitophagy can be regulated by methionine oxidation (MetO) [83], which is a reversible post-translational modification on proteins implicated in both aging and vascular disease [84,85,86,87,88]. In diabetic patients, increased oxidative stress was found to increase MetO-modified proteins. Specifically, Parkin Met192 can be oxidized and lead to protein aggregation and the disruption of mitophagy in human platelets [83]. In the same study, genetic ablation of the mitochondrial-specific methionine sulfoxide reductase MsrB2 also disrupted mitophagy and promoted the apoptosis of murine platelets [83]. Additional evidence indicates that MsrB2 is released from damaged mitochondria, reduces/reverses Parkin Met192 oxidation through direct interactions, and initiates mitophagy to prevent platelet apoptosis [83]. These findings represent a novel redox mechanism to regulate platelet mitophagy and apoptosis under conditions of increased oxidative stress, such as diabetes.
5. Platelets and Inflammation in Aging
The term “inflammaging” was a concept introduced in 2000 by Dr. Franceschi and is now commonly used to describe the pathologic consequences of chronic low-grade inflammation and physiologic stimulation of the innate immune system that occurs with advancing age [89]. This concept has been used to explain the higher prevalence of chronic disorders in the elderly, including obesity, type 2 diabetes mellitus, and cardiovascular diseases [89]. Platelets are also recognized as immune and inflammatory cells [13,90]. The immune functions of platelets are evidenced by their direct interactions with vascular and inflammatory cells and by the presence of various cytokines and chemokines contained in their granules and cytoplasm [13,90].
A recent article by Davizon-Castillo et al. provides compelling evidence of a link between inflammation and platelet activity [91], which is also discussed in several accompanying commentaries [92,93]. TNF-α is a key inflammatory cytokine that is highly correlated with age-associated cardiovascular disease and is intricately linked with ROS [94,95,96,97]. It was observed that both aged humans and mice exhibited elevated plasma TNF-α and platelet hyperactivation [91]. Utilizing several complementary murine models of TNF-α elevation or depletion, a direct functional effect of TNF-α in platelet hyperactivation during aging was demonstrated [91]. Single-cell transcriptome analysis of the bone marrow compartment showed significant reprogramming in platelet/megakaryocyte progenitor populations in aged mice that corresponded to alterations in mitochondrial function, oxidative phosphorylation, and inflammatory signaling pathways [91]. These findings are largely consistent with a prior study that also showed changes in platelet/megakaryocyte progenitors using murine models of aging [98]. In addition, Davizon-Castillo et al. observed increased mitochondrial mass and altered bioenergetics with increased oxygen consumption in platelets from aged mice that were mitigated with TNF-α blockage [91], which provides further evidence for an intriguing role of mitochondria in “inflammaging”. Phagosome maturation was one of the top pathways identified by Ingenuity Pathway Analysis software [91], and, as discussed above, autophagy/mitophagy is important for maintaining platelet function in the presence of Oxidative stress and mitochondrial damage [82]. Overall, Davizon-Castillo et al. provide substantial evidence that TNF-α is a crucial driver of platelet hyperreactivity during aging. Their findings provide a good rationale for future aging studies to examine the precise mechanisms of how inflammatory, mitochondrial, and oxidative pathways may converge to induce aberrant platelet hyperactivity and their pathological impact on age-associated thrombotic diseases.
In other studies, stimulation of TLRs on platelets has also been shown to modulate TNF-α production in vivo, likely through interactions with other immune or vascular cells [99]. Platelets express several toll-like receptor (TLR) family members [99,100] that have classical functions not only in mediating innate immune signaling but also in regulating platelet function [101,102,103,104]. Stimulation of TLR4 by lipopolysaccharide (LPS) enhances platelet activation and can promote direct interactions with inflammatory and immune cells such as neutrophils [101,105]. These effects are likely ROS-dependent, based on the observations that platelet ROS is increased with LPS stimulation, whereas treatment with antioxidant enzymes such as SOD or catalase prevents LPS-induced platelet activation [105]. Consistent with these findings, other studies have demonstrated that platelet TLR2 stimulation can also promote platelet activation and is associated with increased ROS production and platelet–neutrophil aggregates [106]. Given that LPS is a key outer membrane component of Gram-negative bacteria known to elicit robust inflammatory and immune responses, these findings may explain the observations of the increased risk of vascular events, such as MI and stroke, following acute infections that occur more commonly in the elderly population [107].
6. Oxidative Stress and Platelets in Chronic Diseases
Hyperlipidemia and atherosclerosis are significantly associated with advancing age and are major risk factors for the development of thrombotic vascular disease [5,108]. Early clinical studies observed enhanced platelet reactivity in patients with hyperlipidemia [109,110,111]. It was discovered that oxidized forms of lipids, such as oxidized low-density lipoprotein (oxLDL), can promote platelet activation and thrombosis in murine models of atherosclerosis via direct binding to CD36 [112], a multiligand scavenger receptor highly expressed on platelets as well as a broad range of other vascular cell types [113]. Redox-sensitive signaling pathways directly downstream of the CD36 receptor were identified to mediate oxLDL-induced platelet activation and thrombosis [114,115]. Specifically, the binding of oxLDL to the CD36 receptor on platelets induces sustained generation of intracellular ROS and promotes platelet aggregation, which was prevented by the pharmacologic inhibition of Nox2 in human platelets or the genetic ablation of NOX2 in murine platelets [115,116]. Other groups have shown that both NOX1 and NOX2 isoforms contribute to oxLDL-induced platelet activation [30]. It was demonstrated that a redox-sensitive protein, extracellular signal-regulated kinase 5 (ERK5) of the MAPK family, is directly activated by intracellular ROS generated through oxLDL-CD36 signaling and contributes to platelet activation, aggregation, and thrombus formation in vivo [117]. Moreover, this mechanistic pathway involving CD36 and ERK5 also promotes caspase activation and phosphatidylserine externalization on platelets that increase its procoagulant activity and support fibrin formation in vivo under conditions of dyslipidemia [118]. The CD36 signaling pathway can also activate innate immune signaling cascades in platelets through TLR activation, contributing to platelet hyperreactivity in the setting of hyperlipidemia [103]. The clinical relevance of the CD36 signaling pathway in thrombotic vascular disease is evidenced by human genetic studies identifying polymorphisms in the CD36 gene that are strongly associated with platelet surface CD36 expression and risk of acute myocardial infarction [119,120]. Platelet CD36 signaling has also been suggested to play a role in platelet hyperreactivity and thrombosis in other age-related conditions associated with increased cardiovascular risk, including diabetes mellitus [121].
Type 2 diabetes mellitus (T2DM) is a well-established cardiovascular risk factor and is significantly more prevalent in the elderly population [122]. Patients with T2DM exhibit platelet hyperreactivity, and several groups have provided evidence that hyperglycemia induces alterations in oxidative pathways in platelets. For instance, the expression of the P2Y12 receptor has been shown to be significantly increased in platelets from T2DM patients and is constitutively activated [123]. Stimulation of the P2Y12 receptor under high glucose conditions in animal models has been shown to induce pathways that increase platelet intracellular ROS, contributing to platelet hyperactivity and limiting certain antiplatelet therapies [123]. There is evidence that platelets from patients with T2DM with poor glycemic control express higher levels of Nox1 [124]. Hyperglycemia also can induce mitochondrial superoxide generation, potentiating collagen-induced platelet activation [64]. Urinary levels of platelet thromboxane (TX) metabolites are elevated in patients with T2DM [125], and it was demonstrated that aldose reductase is an important enzyme that mediates hyperglycemia and collagen-induced platelet activation and TX release through a pathway involving ROS generation [126]. Moreover, aldose reductase also contributes to mitochondrial dysfunction/damage and platelet apoptosis in the setting of hyperglycemia [66]. Other pathways of mitochondrial-ROS-driven platelet activation in diabetes have been discussed in the section on platelet mitochondria in aging (see above).
Cancer is another disease often associated with aging. The incidence of many cancers increases dramatically with age, and older individuals are at greater risk for the development of advanced disease [127]. Accumulating evidence has indicated that platelets and tumor cells can interact reciprocally through direct binding or through the secretion and uptake of cellular factors that can influence immune and vascular responses [128,129]. These interactions have been shown to alter the key pathological processes related to cancer tumorigenesis [130,131] and metastasis [132,133,134] and have also provided novel diagnostic tools for cancer detection [135,136]. Elevations in ROS have been detected in many subtypes of cancers, and redox dysregulation has been implicated in cellular signaling pathways associated with both cancer development and clearance [137,138]. Many of the growth factors and cytokines that can induce ROS generation and exert biological effects on cancer are abundant in platelets and can be secreted upon stimulation [90,139,140,141]. However, only a few studies have directly examined platelet-specific ROS in cancer and chemotherapeutics. A small study reported increased oxidative and nitrative modifications of platelet proteins from patients with breast cancer [142]. A separate study provided evidence that metabolites of tamoxifen can increase the production of ROS through the activation of NADPH oxidase and promote platelet aggregation [143]. Further studies elaborating on the specific platelet redox mechanisms in different cancer subtypes will better determine the role of platelets and ROS in cancer pathogenesis.
Smoking is significantly associated with increased risk and mortality from cardiovascular disease [144]. Several early clinical studies provided the initial evidence that smoking potentiates platelet aggregation responses [145,146]. Subsequent studies have shown that smoking can induce platelet thrombus formation ex vivo in patients with coronary artery disease who are taking aspirin [147]. Several mechanisms involving redox dysregulation and nitric oxide bioavailability have been implicated in smoking-induced platelet hyperactivation and thrombosis [148,149,150]. Interestingly, other studies have associated smoking with a paradoxical decrease in platelet activation and a reduced recurrence of cardiovascular events in patients who are taking oral P2Y12 inhibitors (i.e., clopidogrel) [151,152,153,154,155]. The precise mechanism of this protection is unclear but likely involves the effects of smoking on hepatic cytochrome P450 enzymes and the complex metabolism of clopidogrel [156,157]. Overall, the current evidence suggests that smoking increases platelet reactivity and thrombotic risk but enhances the efficacy of clopidogrel therapy after thrombotic events.
7. Platelets in COVID-19 Pathogenesis
Coronavirus disease 2019 (COVID-19) is caused by the infection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 primarily targets the lung epithelial cells to induce respiratory distress but can also cause a systemic inflammatory response and vascular thrombosis [158,159]. Age plays an important role in COVID-19 pathogenesis, with severe illness and mortality disproportionally affecting the elderly. This is compounded by the higher prevalence of other traditional cardiovascular risk factors with advancing age, including diabetes, obesity, and hypertension. Vascular thrombosis and thrombo-inflammatory complications are major causes of morbidity and mortality in COVID-19, and platelets have been implicated as a key contributor to disease pathogenesis [160]. SARS-CoV-2 mRNA can be detected in the platelets of subsets of COVID-19 patients, suggesting that platelets can uptake SARS-CoV-2 mRNA [161,162]. In a study evaluating 115 consecutive patients with COVID-19, platelets were shown to be hyperactive with increased adhesion, aggregation, degranulation, and extracellular vesicle release compared to healthy individuals, contributing to alterations in cytokine and growth factor release [161]. Other groups have also reported hyperactive platelets in COVID-19 and demonstrated that platelets can associate with monocytes through platelet surface P-selectin and αIIbβ3 binding, which induces tissue factor expression by monocytes [163]. In fact, platelets from COVID-19 patients exhibit increased interactions with multiple leukocyte subsets, including neutrophils, lymphocytes, and monocytes [162].
Interestingly, neutrophil extracellular traps (NETs), which are web-like chromatin structures containing DNA–histone complexes and antimicrobial proteins released upon neutrophil activation, are increased in the plasma of COVID-19 patients and correlated with disease severity in some studies [164,165,166]. In a small case series of COVID-19 patients who developed ST-elevated myocardial infarction (STEMI), increased incidence and density of NETs were observed in the coronary thrombi of COVID-19 patients undergoing primary coronary intervention [167]. NET formation can be induced by activated platelets [101,168,169,170], and there is evidence that platelets contribute to NET formation in COVID-19, with reports of NET-containing microthrombi associated with platelet deposition in COVID-19 autopsies [164]. These findings are supported by other studies demonstrating that NET formation can be triggered by platelet-rich plasma from COVID-19 patients [165]. There is evidence of redox dysregulation in COVID-19, with increased Nox2 activity detected by plasma metabolites [171]. Nox2 is able to regulate platelet activation and NET formation in the lung [172]. Another potential mechanism of redox dysregulation in COVID-19 is through altered iron homeostasis [173,174]. Studies early in the pandemic reported an association between increased serum ferritin and in-hospital mortality [175]. Iron overload is associated with ferroptosis, a form of regulated cell death characterized by the iron-dependent accumulation of lipid peroxides and redox dysregulation through the depletion of glutathione and the inhibition of GPx-4 [176,177]. Ferroptosis has been implicated in COVID-19 pathogenesis and is suggested to contribute to multiorgan damage [178]. The release of free iron from heme has been reported to increase lipid peroxidation and induce platelet activation and cell death through ferroptosis [179]. Nevertheless, the precise role of ROS and platelets in COVID-19 remains to be determined.
8. Conclusions
Platelets are fundamental vascular cells that regulate a myriad of physiologic processes through activation and interactions with immune and vascular cells. Although data are still emerging, accumulating evidence from clinical, translational, and basic science studies suggests that the process of aging results in alterations in redox biology and platelet function (summarized in Figure 2), which can have a significant impact on the development of vascular diseases. The regulation of these processes is complex and is impacted by a variety of changes that occur with aging, including inflammation, cellular stress, organelle dysfunction, and cell-to-cell interactions. Understanding the cellular and molecular redox mechanisms that drive these changes in platelets during aging will enhance our knowledge and will allow for the development of more targeted therapeutics.
Author Contributions
S.X.G. prepared the drafts and helped with the final preparation and editing of the manuscript. S.D. conceived the idea, designed the format, and co-wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Department of Veterans Affairs (I01CX001932) and the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (R01AI162778) to S.D.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; Elliott, P.; Kontis, V.; Ezzati, M. Worldwide Exposures to Cardiovascular Risk Factors and Associated Health Effects: Current Knowledge and Data Gaps. Circulation 2016, 133, 2314–2333. [Google Scholar] [CrossRef] [Green Version]
- Kontis, V.; Bennett, J.E.; Mathers, C.D.; Li, G.; Foreman, K.; Ezzati, M. Future life expectancy in 35 industrialised countries: Projections with a Bayesian model ensemble. Lancet 2017, 389, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, P.A.; Trogdon, J.G.; Khavjou, O.A.; Butler, J.; Dracup, K.; Ezekowitz, M.D.; Finkelstein, E.A.; Hong, Y.; Johnston, S.C.; Khera, A.; et al. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation 2011, 123, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Clemetson, K.J. Platelets and primary haemostasis. Thromb. Res. 2012, 129, 220–224. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Davi, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef]
- Jackson, S.P. Arterial thrombosis--insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.W.; Italiano, J.E., Jr.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Ramey, E.; Ramwell, P.W. Sex and age differences in human platelet aggregation. Nature 1975, 253, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Terres, W.; Weber, K.; Kupper, W.; Bleifeld, W. Age, cardiovascular risk factors and coronary heart disease as determinants of platelet function in men. A multivariate approach. Thromb. Res. 1991, 62, 649–661. [Google Scholar] [CrossRef]
- Jorgensen, K.A.; Dyerberg, J.; Olesen, A.S.; Stoffersen, E. Acetylsalicylic acid, bleeding time and age. Thromb. Res. 1980, 19, 799–805. [Google Scholar] [CrossRef]
- Meade, T.W.; Vickers, M.V.; Thompson, S.G.; Stirling, Y.; Haines, A.P.; Miller, G.J. Epidemiological characteristics of platelet aggregability. BMJ 1985, 290, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, A.; Idelchik, M.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.J.; Silk, S.T.; Safier, L.B.; Ullman, H.L. Superoxide production and reducing activity in human platelets. J. Clin. Investig. 1977, 59, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Handin, R.I.; Karabin, R.; Boxer, G.J. Enhancement of platelet function by superoxide anion. J. Clin. Investig. 1977, 59, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Begonja, A.J.; Gambaryan, S.; Geiger, J.; Aktas, B.; Pozgajova, M.; Nieswandt, B.; Walter, U. Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 2005, 106, 2757–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, C.F.; Laude, K.; McNally, J.S.; Harrison, D.G. ATVB in focus: Redox mechanisms in blood vessels. Arter. Thromb. Vasc. Biol. 2005, 25, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arter. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Sonkar, V.K.; Kumar, R.; Jensen, M.; Wagner, B.A.; Sharathkumar, A.A.; Miller, F.J., Jr.; Fasano, M.; Lentz, S.R.; Buettner, G.R.; Dayal, S. Nox2 NADPH oxidase is dispensable for platelet activation or arterial thrombosis in mice. Blood Adv. 2019, 3, 1272–1284. [Google Scholar] [CrossRef]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of Nox1 and Nox2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential Roles of the NADPH-Oxidase 1 and 2 in Platelet Activation and Thrombosis. Arter. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Vara, D.; Cifuentes-Pagano, E.; Pagano, P.J.; Pula, G. A novel combinatorial technique for simultaneous quantification of oxygen radicals and aggregation reveals unexpected redox patterns in the activation of platelets by different physiopathological stimuli. Haematologica 2019, 104, 1879–1891. [Google Scholar] [CrossRef] [Green Version]
- Vara, D.; Mailer, R.K.; Tarafdar, A.; Wolska, N.; Heestermans, M.; Konrath, S.; Spaeth, M.; Renne, T.; Schroder, K.; Pula, G. NADPH Oxidases Are Required for Full Platelet Activation In Vitro and Thrombosis In Vivo but Dispensable for Plasma Coagulation and Hemostasis. Arter. Thromb. Vasc. Biol. 2021, 41, 683–697. [Google Scholar] [CrossRef]
- Finazzi-Agro, A.; Menichelli, A.; Persiani, M.; Biancini, G.; Del Principe, D. Hydrogen peroxide release from human blood platelets. Biochim. Biophys. Acta 1982, 718, 21–25. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Blankenberg, S.; Rupprecht, H.J.; Bickel, C.; Torzewski, M.; Hafner, G.; Tiret, L.; Smieja, M.; Cambien, F.; Meyer, J.; Lackner, K.J.; et al. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N. Engl. J. Med. 2003, 349, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinola-Klein, C.; Rupprecht, H.J.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Munzel, T.; Blankenberg, S.; AtheroGene, I. Glutathione peroxidase-1 activity, atherosclerotic burden, and cardiovascular prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J.; Benoit, S.E.; Valeri, C.R.; Barnard, M.R.; Michelson, A.D. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J. Clin. Investig. 1996, 97, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voetsch, B.; Jin, R.C.; Bierl, C.; Benke, K.S.; Kenet, G.; Simioni, P.; Ottaviano, F.; Damasceno, B.P.; Annichino-Bizacchi, J.M.; Handy, D.E.; et al. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: A novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007, 38, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.C.; Mahoney, C.E.; Coleman Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.Y.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Jain, K.; Tyagi, T.; Patell, K.; Xie, Y.; Kadado, A.J.; Lee, S.H.; Yarovinsky, T.; Du, J.; Hwang, J.; Martin, K.A.; et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: Implications for CVD risk patients. EBioMedicine 2019, 44, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Iyer, K.S.; Dayal, S. Platelet antioxidants: A conundrum in aging. EBioMedicine 2019, 47, 29–30. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.; Tyagi, T.; Ionescu, C.N.; Hwa, J. Author’s response to “platelet antioxidants: A conundrum in aging”. EBioMedicine 2019, 47, 31–32. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Sanz, A.; Fernandez-Ayala, D.J.; Stefanatos, R.K.; Jacobs, H.T. Mitochondrial ROS production correlates with, but does not directly regulate lifespan in Drosophila. Aging 2010, 2, 200–223. [Google Scholar] [CrossRef]
- Yang, W.; Li, J.; Hekimi, S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 2007, 177, 2063–2074. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [Green Version]
- Yee, C.; Yang, W.; Hekimi, S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013, 41, 118–123. [Google Scholar] [CrossRef]
- Remenyi, G.; Szasz, R.; Friese, P.; Dale, G.L. Role of mitochondrial permeability transition pore in coated-platelet formation. Arter. Thromb. Vasc. Biol. 2005, 25, 467–471. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.J.; Salido, G.M.; Gomez-Arteta, E.; Rosado, J.A.; Pariente, J.A. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J. Thromb. Haemost. 2007, 5, 1283–1291. [Google Scholar] [CrossRef]
- Jobe, S.M.; Wilson, K.M.; Leo, L.; Raimondi, A.; Molkentin, J.D.; Lentz, S.R.; Di Paola, J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008, 111, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Choo, H.J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arter. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [Green Version]
- Barile, C.J.; Herrmann, P.C.; Tyvoll, D.A.; Collman, J.P.; Decreau, R.A.; Bull, B.S. Inhibiting platelet-stimulated blood coagulation by inhibition of mitochondrial respiration. Proc. Natl. Acad. Sci. USA 2012, 109, 2539–2543. [Google Scholar] [CrossRef] [Green Version]
- Braganza, A.; Corey, C.G.; Santanasto, A.J.; Distefano, G.; Coen, P.M.; Glynn, N.W.; Nouraie, S.M.; Goodpaster, B.H.; Newman, A.B.; Shiva, S. Platelet bioenergetics correlate with muscle energetics and are altered in older adults. JCI Insight 2019, 4, e128248. [Google Scholar] [CrossRef] [Green Version]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Divakaruni, A.S.; Jastroch, M.; Brand, M.D. Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 2010, 131, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Ulgherait, M.; Chen, A.; McAllister, S.F.; Kim, H.X.; Delventhal, R.; Wayne, C.R.; Garcia, C.J.; Recinos, Y.; Oliva, M.; Canman, J.C.; et al. Circadian regulation of mitochondrial uncoupling and lifespan. Nat. Commun. 2020, 11, 1927. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.I.; Edelstein, D.; Du, X.L.; Brownlee, M. Hyperglycemia potentiates collagen-induced platelet activation through mitochondrial superoxide overproduction. Diabetes 2001, 50, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, E.; Araya-Maturana, R.; Urra, F.A. Regulation of mitochondrial function as a promising target in platelet activation-related diseases. Free Radic. Biol. Med. 2019, 136, 172–182. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Avila, C.; Huang, R.J.; Stevens, M.V.; Aponte, A.M.; Tripodi, D.; Kim, K.Y.; Sack, M.N. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Exp. Clin. Endocrinol. Diabetes 2012, 120, 248–251. [Google Scholar] [CrossRef]
- Fidler, T.P.; Rowley, J.W.; Araujo, C.; Boudreau, L.H.; Marti, A.; Souvenir, R.; Dale, K.; Boilard, E.; Weyrich, A.S.; Abel, E.D. Superoxide Dismutase 2 is dispensable for platelet function. Thromb. Haemost. 2017, 117, 1859–1867. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, X.; Fan, X.; Xiao, M.; Yang, D.; Liang, B.; Dai, M.; Shan, L.; Lu, J.; Lin, Z.; et al. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood 2016, 128, 615–624. [Google Scholar] [CrossRef]
- Ouseph, M.M.; Huang, Y.; Banerjee, M.; Joshi, S.; MacDonald, L.; Zhong, Y.; Liu, H.; Li, X.; Xiang, B.; Zhang, G.; et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood 2015, 126, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Qi, B.; Hardwick, J.M. A Bcl-xL timer sets platelet life span. Cell 2007, 128, 1035–1036. [Google Scholar] [CrossRef] [Green Version]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ma, Q.; Siraj, S.; Ney, P.A.; Liu, J.; Liao, X.; Yuan, Y.; Li, W.; Liu, L.; Chen, Q. Nix-mediated mitophagy regulates platelet activation and life span. Blood Adv. 2019, 3, 2342–2354. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G.; et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, S.; Du, J.; Jain, K.; Ding, M.; Kadado, A.J.; Atteya, G.; Jaji, Z.; Tyagi, T.; Kim, W.H.; et al. Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol. Med. 2019, 11, e10409. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Van Remmen, H.; Richardson, A.; Wehr, N.B.; Levine, R.L. Methionine oxidation and aging. Biochim. Biophys. Acta 2005, 1703, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Stevens, J.W.; Lentz, S.R. Regulation of thrombosis and vascular function by protein methionine oxidation. Blood 2015, 125, 3851–3859. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.X.; Blokhin, I.O.; Wilson, K.M.; Dhanesha, N.; Doddapattar, P.; Grumbach, I.M.; Chauhan, A.K.; Lentz, S.R. Protein methionine oxidation augments reperfusion injury in acute ischemic stroke. JCI Insight 2016, 1, e86460. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of protein function by reversible methionine oxidation and the role of selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [Green Version]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef]
- Podrez, E.A. Platelet hyperreactivity: A new twist in old mice. Blood 2019, 134, 723–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.X.; Dayal, S. Inflammation mediated platelet hyperactivity in aging. Ann. Blood 2020, 134, 727. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends. Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Newman, A.B. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2011, 10, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Penninx, B.W.; Newman, A.B.; Kritchevsky, S.B.; Nicklas, B.J.; Sutton-Tyrrell, K.; Rubin, S.M.; Ding, J.; Simonsick, E.M.; Harris, T.B.; et al. Inflammatory markers and onset of cardiovascular events: Results from the Health ABC study. Circulation 2003, 108, 2317–2322. [Google Scholar] [CrossRef] [Green Version]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.; Patel, K.D.; Kubes, P. Platelets express functional Toll-like receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Vallance, T.M.; Ravishankar, D.; Albadawi, D.A.I.; Layfield, H.; Sheard, J.; Vaiyapuri, R.; Dash, P.; Patel, K.; Widera, D.; Vaiyapuri, S. Effect of ultrapure lipopolysaccharides derived from diverse bacterial species on the modulation of platelet activation. Sci. Rep. 2019, 9, 18258. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Zimman, A.; Gao, D.; Byzova, T.V.; Podrez, E.A. TLR2 Plays a Key Role in Platelet Hyperreactivity and Accelerated Thrombosis Associated With Hyperlipidemia. Circ. Res. 2017, 121, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, S.; Ma, Y.; Hong, L.; Gao, D.; West, X.Z.; Salomon, R.G.; Byzova, T.V.; Podrez, E.A. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ. Res. 2013, 112, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Smeeth, L.; Thomas, S.L.; Hall, A.J.; Hubbard, R.; Farrington, P.; Vallance, P. Risk of myocardial infarction and stroke after acute infection or vaccination. N. Engl. J. Med. 2004, 351, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.C.; Colman, R.W.; Lees, R.S. Platelet function in hyperlipoproteinemia. N. Engl. J. Med. 1974, 290, 434–438. [Google Scholar] [CrossRef]
- Davi, G.; Averna, M.; Catalano, I.; Barbagallo, C.; Ganci, A.; Notarbartolo, A.; Ciabattoni, G.; Patrono, C. Increased thromboxane biosynthesis in type IIa hypercholesterolemia. Circulation 1992, 85, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Cipollone, F.; Mezzetti, A.; Porreca, E.; Di Febbo, C.; Nutini, M.; Fazia, M.; Falco, A.; Cuccurullo, F.; Davi, G. Association between enhanced soluble CD40L and prothrombotic state in hypercholesterolemia: Effects of statin therapy. Circulation 2002, 106, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Febbraio, M.; Li, W.; Silverstein, R.L. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ. Res. 2008, 102, 1512–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.; Wraith, K.; Woodward, C.; Aburima, A.; Raslan, Z.; Hindle, M.S.; Moellmann, J.; Febbraio, M.; Naseem, K.M. Dyslipidemia-associated atherogenic oxidized lipids induce platelet hyperactivity through phospholipase Cgamma2-dependent reactive oxygen species generation. Platelets 2019, 30, 467–472. [Google Scholar] [CrossRef]
- Yang, M.; Cooley, B.C.; Li, W.; Chen, Y.; Vasquez-Vivar, J.; Scoggins, N.O.; Cameron, S.J.; Morrell, C.N.; Silverstein, R.L. Platelet CD36 promotes thrombosis by activating redox sensor ERK5 in hyperlipidemic conditions. Blood 2017, 129, 2917–2927. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kholmukhamedov, A.; Schulte, M.L.; Cooley, B.C.; Scoggins, N.O.; Wood, J.P.; Cameron, S.J.; Morrell, C.N.; Jobe, S.M.; Silverstein, R.L. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv. 2018, 2, 2848–2861. [Google Scholar] [CrossRef]
- Ghosh, A.; Murugesan, G.; Chen, K.; Zhang, L.; Wang, Q.; Febbraio, M.; Anselmo, R.M.; Marchant, K.; Barnard, J.; Silverstein, R.L. Platelet CD36 surface expression levels affect functional responses to oxidized LDL and are associated with inheritance of specific genetic polymorphisms. Blood 2011, 117, 6355–6366. [Google Scholar] [CrossRef] [Green Version]
- Knowles, J.W.; Wang, H.; Itakura, H.; Southwick, A.; Myers, R.M.; Iribarren, C.; Fortmann, S.P.; Go, A.S.; Quertermous, T.; Hlatky, M.A. Association of polymorphisms in platelet and hemostasis system genes with acute myocardial infarction. Am. Heart J. 2007, 154, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [Green Version]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chang, L.; Zhang, Y.; Zhai, L.; Zhang, S.; Qi, Z.; Yan, H.; Yan, Y.; Luo, X.; Zhang, S.; et al. Platelets Express Activated P2Y12 Receptor in Patients With Diabetes Mellitus. Circulation 2017, 136, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.R.; Wolska, N.; Vara, D.; Mailer, R.K.; Schroder, K.; Pula, G. Diabetes and Thrombosis: A Central Role for Vascular Oxidative Stress. Antioxidants 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges for aspirin and other antiplatelet agents. Blood 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015, 126, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci. Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.V.; Wurtzel, J.G.T.; Mao, G.F.; Rao, A.K.; Kolpakov, M.A.; Sabri, A.; Hoffman, N.E.; Rajan, S.; Tomar, D.; Madesh, M.; et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 2017, 130, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin alpha6beta1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 9024–9028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjors, G.J.G.; Wurdinger, T. Tumor-educated platelets. Blood 2019, 22, 2359–2364. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; GJG, S.; Vancura, A.; Muller, M.; Niemeijer, A.N.; Fejes, A.V.; Tjon Kon Fat, L.A.; Huis, A.E.; Leurs, C.; et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell 2017, 32, 238–252.e239. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Pauklin, S. ROS and TGFbeta: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 152. [Google Scholar] [CrossRef]
- Roy, D.; Sarkar, S.; Felty, Q. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front. Biosci. 2006, 11, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Kedzierska, M.; Olas, B.; Wachowicz, B.; Stochmal, A.; Oleszek, W.; Jeziorski, A.; Piekarski, J. The nitrative and oxidative stress in blood platelets isolated from breast cancer patients: The protectory action of aronia melanocarpa extract. Platelets 2010, 21, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Vitseva, O.; Flockhart, D.A.; Jin, Y.; Varghese, S.; Freedman, J.E. The effects of tamoxifen and its metabolites on platelet function and release of reactive oxygen intermediates. J. Pharmacol. Exp. Ther. 2005, 312, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Carter, B.D.; Feskanich, D.; Freedman, N.D.; Prentice, R.; Lopez, A.D.; Hartge, P.; Gapstur, S.M. 50-year trends in smoking-related mortality in the United States. N. Engl. J. Med. 2013, 368, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Levine, P.H. An acute effect of cigarette smoking on platelet function. A possible link between smoking and arterial thrombosis. Circulation 1973, 48, 619–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.I. Smoking, platelets and thrombosis. Nature 1972, 236, 450–452. [Google Scholar] [CrossRef]
- Hung, J.; Lam, J.Y.; Lacoste, L.; Letchacovski, G. Cigarette smoking acutely increases platelet thrombus formation in patients with coronary artery disease taking aspirin. Circulation 1995, 92, 2432–2436. [Google Scholar] [CrossRef]
- Ichiki, K.; Ikeda, H.; Haramaki, N.; Ueno, T.; Imaizumi, T. Long-term smoking impairs platelet-derived nitric oxide release. Circulation 1996, 94, 3109–3114. [Google Scholar] [CrossRef]
- Barua, R.S.; Ambrose, J.A.; Srivastava, S.; DeVoe, M.C.; Eales-Reynolds, L.J. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: An in vitro demonstration in human coronary artery endothelial cells. Circulation 2003, 107, 2342–2347. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Ikeda, H.; Haramaki, N.; Eguchi, H.; Imaizumi, T. Only two-week smoking cessation improves platelet aggregability and intraplatelet redox imbalance of long-term smokers. J. Am. Coll. Cardiol. 2005, 45, 589–594. [Google Scholar] [CrossRef] [Green Version]
- Bliden, K.P.; Dichiara, J.; Lawal, L.; Singla, A.; Antonino, M.J.; Baker, B.A.; Bailey, W.L.; Tantry, U.S.; Gurbel, P.A. The association of cigarette smoking with enhanced platelet inhibition by clopidogrel. J. Am. Coll. Cardiol. 2008, 52, 531–533. [Google Scholar] [CrossRef] [Green Version]
- Addad, F.; Chakroune, T.; Asma, A.; Abderazek, F.; Zohra, D.; Ghrissi, I.; Hassine, M.; Gamra, H.; Elalamy, I. Clopidogrel but not Aspirin prevents acute smoking-induced platelet aggregation in patients with stable coronary artery disease. Thromb. Res. 2009, 123, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.S.; Bhatt, D.L.; Steinhubl, S.R.; Shao, M.; Steg, P.G.; Montalescot, G.; Hacke, W.; Fox, K.A.; Lincoff, A.M.; Topol, E.J.; et al. Smoking, clopidogrel, and mortality in patients with established cardiovascular disease. Circulation 2009, 120, 2337–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, N.R.; Mega, J.L.; Jiang, S.; Cannon, C.P.; Sabatine, M.S. Interaction between cigarette smoking and clinical benefit of clopidogrel. J. Am. Coll. Cardiol. 2009, 53, 1273–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.H.; Cho, J.H.; Kang, M.K.; Koh, J.S.; Kim, I.S.; Park, Y.; Hwang, S.J.; Kwak, C.H.; Hwang, J.Y. Smoking at least 10 cigarettes per day increases platelet inhibition by clopidogrel in patients with ST-segment-elevation myocardial infarction. Thromb. Res. 2010, 126, e334–e338. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Baker, B.A.; Bailey, W.L.; Bliden, K.P.; Tantry, U.S. Unravelling the smokers’ paradox: Cigarette smoking, high-risk coronary artery disease and enhanced clinical efficacy of oral P2Y(1)(2) inhibitors. Thromb. Haemost. 2014, 111, 1187–1190. [Google Scholar] [CrossRef]
- Somaschini, A.; Cornara, S.; De Servi, S.; Crimi, G. Smoking, clopidogrel and platelet reactivity: Are we still missing something? Platelets 2020, 31, 968. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Zhang, L.; Feng, X.; Zhang, D.; Jiang, C.; Mei, H.; Wang, J.; Zhang, C.; Li, H.; Xia, X.; Kong, S.; et al. Deep Vein Thrombosis in Hospitalized Patients With COVID-19 in Wuhan, China: Prevalence, Risk Factors, and Outcome. Circulation 2020, 142, 114–128. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2020, 18, 194–209. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, A.; Coronado, M.J.; Hernandez-Terciado, F.; Martin, P.; Royuela, A.; Ramil, E.; Garcia, D.; Goicolea, J.; Del Trigo, M.; Ortega, J.; et al. Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol. 2020, 6, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.J.; Yeh, C.Y.; Hsu, R.B.; Lee, C.M.; Shun, C.T.; Chia, J.S. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation 2015, 131, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in Covid-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef]
- Hook, J.S.; Cao, M.; Potera, R.M.; Alsmadi, N.Z.; Schmidtke, D.W.; Moreland, J.G. Nox2 Regulates Platelet Activation and NET Formation in the Lung. Front. Immunol. 2019, 10, 1472. [Google Scholar] [CrossRef] [PubMed]
- Habib, H.M.; Ibrahim, S.; Zaim, A.; Ibrahim, W.H. The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed. Pharmacother. 2021, 136, 111228. [Google Scholar] [CrossRef] [PubMed]
- Muhoberac, B.B. What Can Cellular Redox, Iron, and Reactive Oxygen Species Suggest About the Mechanisms and Potential Therapy of COVID-19? Front. Cell Infect. Microbiol. 2020, 10, 569709. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Fratta Pasini, A.M.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants 2021, 10, 1677. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef]
Figure 1.
Overview of the relationship between aging and vascular disease, highlighting the pathogenic role of platelets. Cardiovascular comorbidities and oxidative stress are increased with advancing age, which can promote platelet activation and thrombus formation.
Figure 1.
Overview of the relationship between aging and vascular disease, highlighting the pathogenic role of platelets. Cardiovascular comorbidities and oxidative stress are increased with advancing age, which can promote platelet activation and thrombus formation.

Figure 2.
Selected mechanisms of platelet activation and ROS generation in aging and age-associated disease. A variety of extracellular ligands can associate with their respective platelet receptors and induce platelet activation and ROS generation. Multiple sources, including NADPH oxidases and mitochondria, can increase intracellular ROS in platelets. ROS generation in the cytoplasm can activate platelet surface receptors and induce granule release mediated by kinase signaling cascades and increased intracellular calcium. Dysfunctional mitochondria can produce increased ROS and result in mitochondrial permeability transition pore (MPTP) formation, loss of mitochondrial membrane potential (ΔΨm), and induction of platelet apoptosis. Antioxidant enzymes localized in both the cytoplasm and the mitochondria can reduce ROS and prevent platelet activation and apoptosis.
Figure 2.
Selected mechanisms of platelet activation and ROS generation in aging and age-associated disease. A variety of extracellular ligands can associate with their respective platelet receptors and induce platelet activation and ROS generation. Multiple sources, including NADPH oxidases and mitochondria, can increase intracellular ROS in platelets. ROS generation in the cytoplasm can activate platelet surface receptors and induce granule release mediated by kinase signaling cascades and increased intracellular calcium. Dysfunctional mitochondria can produce increased ROS and result in mitochondrial permeability transition pore (MPTP) formation, loss of mitochondrial membrane potential (ΔΨm), and induction of platelet apoptosis. Antioxidant enzymes localized in both the cytoplasm and the mitochondria can reduce ROS and prevent platelet activation and apoptosis.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gu, S.X.; Dayal, S. Redox Mechanisms of Platelet Activation in Aging. Antioxidants 2022, 11, 995. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050995
AMA Style
Gu SX, Dayal S. Redox Mechanisms of Platelet Activation in Aging. Antioxidants. 2022; 11(5):995. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050995
Chicago/Turabian StyleGu, Sean X., and Sanjana Dayal. 2022. "Redox Mechanisms of Platelet Activation in Aging" Antioxidants 11, no. 5: 995. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050995
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.