
Physiological Overview of the Potential Link between the UPS and Ca2+ Signaling


Department of Health Sciences and Technology, Lee Gil Ya Cancer and Diabetes Institute, Gachon Advanced Institute for Health Sciences & Technology, Gachon University, 155 Getbeolro, Yeonsu-gu, Incheon 21999, Korea
*
Author to whom correspondence should be addressed.
Antioxidants 2022, 11(5), 997; https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050997
Submission received: 21 March 2022
/
Revised: 10 May 2022
/
Accepted: 17 May 2022
/
Published: 19 May 2022
(This article belongs to the Special Issue Oxidative Stress Impact on Protein Synthesis and Protein Degradation Systems in Health and Disease)
Abstract
:The ubiquitin–proteasome system (UPS) is the main proteolytic pathway by which damaged target proteins are degraded after ubiquitination and the recruit of ubiquitinated proteins, thus regulating diverse physiological functions and the maintenance in various tissues and cells. Ca2+ signaling is raised by oxidative or ER stress. Although the basic function of the UPS has been extensively elucidated and has been continued to define its mechanism, the precise relationship between the UPS and Ca2+ signaling remains unclear. In the present review, we describe the relationship between the UPS and Ca2+ signaling, including Ca2+-associated proteins, to understand the end point of oxidative stress. The UPS modulates Ca2+ signaling via the degradation of Ca2+-related proteins, including Ca2+ channels and transporters. Conversely, the modulation of UPS is driven by increases in the intracellular Ca2+ concentration. The multifaceted relationship between the UPS and Ca2+ plays critical roles in different tissue systems. Thus, we highlight the potential crosstalk between the UPS and Ca2+ signaling by providing an overview of the UPS in different organ systems and illuminating the relationship between the UPS and autophagy.
1. Introduction
The homeostatic maintenance of protein levels or elimination of misfolded or oxidized proteins requires essential quality control processes such as the ubiquitin–proteasome system (UPS) and autophagy [1,2,3,4]. The UPS regulates the intracellular protein levels and mediates the cell cycle modulation, DNA repair, transcription, and apoptosis [5]. Gradationally, the UPS begins with sequential ubiquitination to produce a poly-ubiquitin chain on the target protein [6,7] and is mediated by ubiquitin ligases E1, E2, and E3, which bind ubiquitin to the lysine residues of the target protein [6]. Poly-ubiquitinated proteins are recruited to the 26S proteasome and degraded through proteasome complexes, including the 19S and 20S proteasome [8,9]. The 19S proteasome, which is called the cap of the proteasome, detects poly-ubiquitinated proteins leading to the 20S proteasome, where poly-ubiquitinated proteins are deubiquitinated to recycle ubiquitin proteins [8]. The 20S proteasome, which is composed of α and β subunits, degrades poly-ubiquitinated target proteins [9].
The crosstalk between UPS and oxidative stress has been addressed bilaterally. It is known that inhibitors of UPS induce oxidative or endoplasmic reticulum (ER) stress [10], and protein oxidation through proteasome malfunction has been suggested as a major cause of human diseases such as Alzheimer’s disease (AD) [11], osteoarthritis [12], asthma [13], atherosclerosis [14], and chronic obstructive pulmonary disease [15]. In addition, the activity of UPS is increased by oxidative stress for the degradation of oxidized proteins, and extensive oxidation impairs the components of the UPS [16]. For example, the H2O2-induced protein carbonyl group, which is the indicator of protein oxidation, is increased by the treatment of proteasome inhibitor MG-132 [17].
The increase of an intracellular Ca2+ concentration ([Ca2+]i) is a messenger signal of oxidative pathways. Oxidative stress triggers an increase of [Ca2+]i through the ER membrane Ca2+ channels [18], and on the contrary, Ca2+ influx induces the generation of reactive oxygen species (ROS) [19]. Ca2+ signaling has been extensively studied for the past several decades. Briefly, [Ca2+]i is initiated through two pathways via the release of Ca2+ from intracellular stores by various extracellular stimuli and the influx of Ca2+ through plasma membrane-associated Ca2+ channels, including voltage-gated Ca2+ channels (VGCCs), ligand-gated Ca2+ channels (LGCCs), and Ca2+ ATPases [20]. Increased [Ca2+]i induces versatile and universal Ca2+ signaling to regulate various cellular physiological functions [21], including muscle contraction [22], the release of neurotransmitters [23], T-cell development [24], and fluid secretion [25].
The disruption of proteasome triggers an imbalance of human health and diseases occur [26,27,28,29]. A change of the signaling messenger Ca2+ is an essential process in various diseases, including oxidative stress. Thus, we suggest that the studies of basic mechanisms for the UPS with Ca2+ establish the foundation for the therapy of proteasome-associated diseases. In the present review, we will discuss the current advances in the roles of Ca2+-related proteins and the pathways of Ca2+ signaling in the UPS. Although Ca2+ signaling and the UPS have critical roles in other organisms, including plants [30,31,32,33] and yeast [34,35,36,37], this review focuses on the mammalian UPS for relating to therapeutic potentials.
2. The Relationship between the UPS and Ca2+ Signaling
2.1. UPS-Mediated Degradation of Ca2+-Related Proteins
The UPS and Ca2+ signaling are interconnected, since each affects the other. The interconnected nature of these signals plays a critical role in regulating cellular functions. The UPS regulates Ca2+ signaling through the degradation of Ca2+-related proteins. Ca2+ channels and transporters are distributed on the membranes of intracellular organelles or the plasma membrane. In this section, we will discuss the relationship between these systems and how this affects protein degradation.
The endoplasmic reticulum (ER) is a major intracellular Ca2+ store. On the ER membrane, inositol 1,4,5-trisphosphate receptor (IP3R) releases Ca2+ to the cytosol via the binding of released IP3 from phosphatidylinositol 4,5-bisphosphate (PIP2) [38,39]. Generally, PIP2 is hydrolyzed to IP3 by phospholipase C (PLC), which is stimulated by the G-protein-coupled receptor, and IP3 subsequently activates IP3R to release ER Ca2+ [38,39]. Ubiquitin ligase ring finger protein 170 (RNF170), which has three membrane-spanning helices, is localized to the ER membrane and binds to IP3R [40]. Ubiquitin ligase RNF170-induced UPS downregulates IP3R in rat pancreas cells and CHO cells [41,42,43]. A deletion of endogenous RNF170 increased the expression of IP3R1 [40]. In other words, the knock-down of RNF170 inhibits IP3R ubiquitination and degradation [40]. In addition, reactive oxygen species are involved in the proteasome-associated degradation of IP3R. H2O2 treatment enhances the proteasome-induced degradation of IP3R in vascular smooth muscle cells [44]. The treatment of MG-132 recovers the H2O2-induced degradation of IP3Rs [44]. Other ER resident proteins: sarco-/endoplasmic reticulum Ca2+-ATPase (SERCAs) and ryanodine receptors (RyRs) are also degraded by the UPS [23,45]. SERCAs are family to the ER-localized P-type cation ATPase that transports cytosolic Ca2+ to the ER [46,47]. Inhibition of the UPS with MG-132 increases SERCA expression [23]. Type-2 RyR (RyR2), which contributes to cardiac excitation–contraction coupling, is degraded by the UPS [45]. In the simulated ischemia–reperfusion of mouse cardiomyocytes, RyR2 is degraded by the UPS following the activation of the Ca2+-dependent cysteine protease calpain, which is activated during ischemia/reperfusion [45]. Although the studies of the relationship between the UPS and intracellular organelle-releasing Ca2+ have been well-developed in the ER, it is meaningful to investigate the effect of the UPS on Ca2+ channels and transporters on other intracellular organelles, including the mitochondria and Golgi apparatus.
Regulatory channels of Ca2+ signaling, store-operated Ca2+ channels (SOCCs), are stimulated by changes in the Ca2+ store levels. When the concentration of ER Ca2+ is depleted, stromal interaction molecule (STIM) senses the depleted ER, elicits oligomerization, and forms a complex with the Orai channels to induce Ca2+ influx [48]. The overexpression of E3 ubiquitin ligase reduces the surface expression of STIM1, and the treatment with MG-132 increases the store-operated Ca2+ entry (SOCE), a Ca2+ homeostatic process to regulate cellular functions, by rescuing the STIM1 expression [49], whereas the inhibition of the proteasome degrades STIM1 and STIM2 through the complementary activation of autophagy [50]. To maintain the cellular activity by the degradation of proteins, autophagy and the UPS are known to communicate with each other [5]. If one is inhibited, the other is activated to degrade proteins [5]. Thus, the inhibition of the UPS complementally stimulates autophagic flux to maintain the [Ca2+]i level. The crosstalk between the UPS and autophagy with Ca2+ signaling will be discussed in Section 5.
The N-type Ca2+ channel voltage-gated calcium channel (CaV)2.2, which induces peripheral neuron neurotransmission [51], is degraded via the UPS to maintain the precise modulation of its expression [52,53]. For example, the overexpression of Parkin, which is an E3 ligase, decreases the current of CaV2.2 through proteasome-induced degradation [52], and proteasome inhibition through MG-132 increases the current of CaV2.2 [53]. The degradation of CaV2.2 is induced by the light chain of microtubule-associated protein B through ubiquitin-conjugating enzyme E2 L3 (UBE2L3)-mediated ubiquitination [54]. UBE2L3 is an E2-type ubiquitin ligase that is related to the occurrence of various diseases, including rheumatoid arthritis, celiac disease, and Crohn’s disease [55]. The β-subunit of CaV2.2 protects against the excessive degradation of CaV2.2 and even the formation of polyubiquitin chains but not from the binds of one to four ubiquitins [56,57]. CaV1.2 is expressed in the brain, cardiomyocytes, pancreas, adrenal medulla, and bladder smooth muscle [58] and specifically initiates cardiac excitation-contraction coupling [59] and triggers smooth muscle contractions [60]. Similar to CaV2.2, the β-subunit of CaV1.2 promotes the trafficking of CaV1.2 to the plasma membrane to avoid the UPS [61]. The aberrant splicing variant form of the CaV1.2 β-subunit increases the UPS-induced degradation of CaV1.2, which triggers cardiac hypertrophy [62]. Galectin-1 (Gal-1), which reduces the current density of CaV1.2 [63], induces the proteasome-induced degradation of CaV1.2 by disrupting the CaV1.2 β-subunit in HEK 293 cells [64]. Coupling between CaV1.2 and Gal-1 regulates the blood pressure, and Gal-1 deficiency triggers hypertension by activating CaV1.2 in spontaneously hypertensive rats [64]. In conclusion, adjustment of the UPS with the scope of the UPS to regulate Ca2+ signaling is proposed as a therapeutic strategy for Ca2+ channel-associated diseases, including cardiac hypertrophy and ischemia–reperfusion injury.
2.2. Ca2+ Signaling and Ca2+-Related Proteins Regulate UPS Activity
Ca2+ signaling regulates numerous cellular functions. In this section, we will elucidate Ca2+ signaling to regulate the UPS. For example, treatment with a Ca2+ ionophore (A23187) activates the proteasome within 10 min in ascidian and Xenopus eggs [65,66]. Increased proteasome activation is attenuated by the Ca2+-chelating agent 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetra acetic acid (BAPTA)-AM [65,66]. Furthermore, A23187-induced Ca2+ increasingly activates the UPS to degrade the signaling proteins, including cyclooxygenase-1 and islet-brain1/JNK interacting protein 1 [67,68]. In neuronal membrane proteasome-inhibited neurons, Ca2+ signaling is dominantly attenuated [69]. Similarly, [Ca2+]i increases by the constitutive activation of the epithelial sodium channel, which induces the aggregation and activation of caspase-8 to inhibit the proteasome, and activated caspase-8 induces cellular apoptosis [70].
The ER is a major source of increased [Ca2+]i that regulates the UPS. Acute ER stress increases the degradation of the amyloid precursor protein, a diagnostic marker of AD [71]. In contrast, human islet amyloid polypeptide aggregation induces ER stress and subsequently impairs the UPS [72]. Aggravated oxidative stress and ER stress produce misfolded proteins in pancreatic β cells and subsequently impair the β-cell function [73]. In summary, the mechanisms by which Ca2+-related proteins regulate the UPS can be used to elucidate the interplay between Ca2+ signaling and the UPS with the scope of Ca2+ signaling to regulate the UPS and may provide dynamic tools for potential therapeutic applications.
2.2.1. Membrane-Bound Proteins and the UPS
Membrane-bound Ca2+ channels are categorized into various subfamilies. In this section, we will discuss the membrane-associated Ca2+ channels, which regulate the UPS. First, the UPS is regulated by Ca2+ signaling from intracellular organelles, including the mitochondria and ER. For example, the treatment with curcumin, which may have anticancer properties [74,75], induces a mitochondrial Ca2+ increase, which inhibits the UPS and induces severe vacuolation, which is a marker of paraptosis, along with apoptotic signals, including cellular shrinkage and the generation of apoptotic bodies [76]. Plasma membrane-bound VGCCs are categorized into several subtypes, including L-, N-, P/Q-, R-, and T-type channels [77]. The T-type Ca2+ channel inhibitor NNC 55-0396 blocks angiogenesis in human umbilical vein endothelial cells through hypoxia-inducible factor-1 (HIF-1) degradation [78]. Under hypoxic conditions, NNC 55-0396 treatment induces the ubiquitination of HIF-1 and subsequent UPS degradation [78]. Thus, modulation of the T-type Ca2+ channels and the subsequent UPS may have therapeutic potential in treating cancer by inhibiting angiogenesis. Transient receptor potential (TRP) channels are nonselective Ca2+ channels with various functions and subtypes [79,80] that also regulate the UPS. In oxidative stress induced by ultraviolet irradiation, TRP vanilloid (TRPV)1 is activated and induces an increase in the Ca2+ levels in human dermal fibroblasts [81]. The activation of TRPV1 induces the ubiquitination of nuclear factor erythroid 2-related factor 2 (Nrf2), which is a key factor in oxidative stress [81]. In addition, the overexpression of TRPV1 increases the ubiquitination of the epidermal growth factor receptor (EGFR) to reduce EGFR expression [82]. Another plasma membrane channel, the Ca2+-sensing receptor (CaSR), which maintains Ca2+ homeostasis, also induces proteasome-induced degradation [83]. CaSR inhibits the TGF-beta-dependent phosphorylation of Smad2, which increases its proliferative effect in human embryonic kidney (HEK) 293 cells [83]. The mechanisms by which Ca2+ channels and transporters are activated are diverse, and their roles in regulating ubiquitination and the UPS should be studied in further detail. Although the importance of Ca2+ channels and transporters is being magnified, the study of the UPS for Ca2+ channels and transporters is still attractive.
2.2.2. Cytosolic Ca2+-Binding Proteins and the UPS
Cytosolic Ca2+-binding proteins involved with the UPS have emerged in various studies. In this section, we will discuss the accumulating evidence of the role of Ca2+-binding/related proteins in the modulation of the UPS. The secondary messenger Ca2+ delivers signals through Ca2+-binding proteins, such as calmodulin (CaM) [84]. CaM is stimulated by the binding of Ca2+ and activates Ca2+/calmodulin-dependent protein kinases (CaMK) to regulate a variety of physiological functions, including smooth muscle contraction [85], the activation of phosphorylase kinase [86], and activation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor [87]. In addition, CaM and CaMK regulate the UPS. E3 ligase, mahogunin ring finger 1 (MGRN1), and glycoprotein 78 (GP78) bind CaM under high [Ca2+]i, and the treatment with BAPTA attenuates the ubiquitination of MGRN1 and GP78 [88]. CaM bound to MGRN1 and GP78 activates the translocation of GP78 onto the ER membrane to induce ER-associated protein degradation [88]. In hippocampal neurons, the UPS induces an action potential that is inhibited by MG-132 [89]. The treatment with the Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor AIPII reduces the rate of protein degradation, while overexpression of the constitutively active form of CaMKII increases the protein degradation [89]. A recent study addressed a new T-type channel enhancer, ethyl-8-methyl-2,4-dioxo-2-(piperidin-1-yl)-2H-spiro[cyclopentane-1,3-imidazo [1,2-a]pyridin]-2-ene-3-carboxylate (SAK3), which has potential therapeutic effects against AD [90]. CaMKII is a scaffold protein that phosphorylates the proteasome subunit Rpt6 [91]. The administration of SAK3 increases CaMKII-Rpt6 signaling, which enhances the proteasome activity in dendritic cells [90]. In myotubules, the Ca2+ ionophore A23187 induces the UPS, while CaMKII inhibitors KN-62 and KN-93 dominantly attenuate the proteasome activity [92].
During muscle wasting caused by cachexia, the Ca2+-binding protein calpain induces Ca2+-dependent proteolysis and the breakdown of myofibrillar proteins [93]. The calpains activate ER-bound transcription factor 11 (TCF11)/Nrf1, which activates the 26S proteasome subunit genes [94]. Calpain-1 cleaves TCF11/Nrf1 to generate the active form, and the inhibition of calpain-1 slows down the degradation of TCF11/Nrf1 [94]. Another ER-related protein RNF122 interacts with Ca2+ to modulate the cyclophilin ligand (CAML) to stabilize RNF122, thus inhibiting the ubiquitination of RNF122 [91]. The lectin chaperone calreticulin, which maintains [Ca2+]i homeostasis, regulates the proteasome activity [95]. In calreticulin-deficient cells, the number of ubiquitinated proteins and proteasome activity are increased [95]. In addition, the Ca2+-binding protein S100, which regulates the tumor cell viability [96], interacts with the E3 ubiquitin ligase C-terminus of the Hsc70-interacting protein (CHIP) to inhibit ubiquitination and the proteasome system [97]. The current understanding of Ca2+ signaling and its associated proteins in the UPS is summarized in Table 1.
3. Ubiquitination in Organ Systems
The UPS is expressed in various mammalian tissues, including the pituitary gland [98], liver [99,100], lung [101], kidney [101], skeletal muscle [102], lens [103], and placenta [104]. In this section, we summarize the physiological roles of the UPS with Ca2+-mediated proteins in various organ systems based on experimental evidence.
3.1. The Nervous System
The UPS regulates the nervous system through the modulation of nerve cell activity [105]. The inhibition of voltage-gated sodium channels or gamma-aminobutyric acid (GABA) receptors induces the UPS-mediated degradation of postsynaptic density proteins in rat hippocampal neurons [106]. Keil et al. demonstrated that STIM1 is expressed in hippocampal neurons and is a candidate for synaptic ubiquitinated proteins [49]. Measurement of the Ca2+ influx through SOCE in the presence of MG-132 proteasome inhibitors shows increases of the surface STIM1 [49]. The proteasome localizes to the neuronal plasma membrane to induce neuronal Ca2+ signaling [69]. In addition, inhibition of the UPS induces autophagy, subsequently leading to the degradation of STIM1/STIM2, which causes neurodegenerative diseases, including AD and Parkinson’s disease [50]. Peptides that are generated by the degradation of intracellular proteins via the involvement of proteasomes are delivered into the extracellular matrix and, subsequently, stimulate neuronal signaling through Ca2+ increases [69]. Mutations of the ER membrane-associated ubiquitin ligase RNF170 cause neurodegeneration through the inhibition of IP3-induced Ca2+ signaling [107]. Neuronal activity is also regulated by CaM [89,90,91,108]. The constitutively active CaMKII increases the proteasome activity by the phosphorylation of the proteasome and recruits the proteasome to hippocampal neurons [89,91]. Moreover, CaMKII activation induces proteasome activation to improve spinal abnormalities [90]. Interestingly, in the case of CaV2.2, the voltage-gated Ca2+ channel β-subunits of CaV2.2 protect CaV2.2 from proteasome-induced degradation in sympathetic neurons [57]. The activation of UPS is related to the positive or negative regulation of Ca2+ signaling, according to the location of neuronal tissues based on the current evidence. Thus, the verification of UPS-related Ca2+ signaling in the nervous system is still a challenging issue, and expanding our understanding of the UPS may contribute towards the more effective treatments of neuronal diseases.
3.2. Muscle and Cardiovascular Systems
The UPS regulates muscle atrophy through the degradation of myofibrillar proteins to mediate myogenesis [109]. In addition, the UPS induces the loss of skeletal muscle mass [110] and myocardial remodeling [111]. UPS inhibition causes cardiac dysfunction and heart failure [112]. Since Ca2+ is the main driver of muscle contraction [113], various Ca2+-dependent proteins regulate muscular and vascular functions through the involvement of UPS. Inhibition of the UPS with MG-132 in rat cardiac cells increases the levels of the Ca2+ channels and transporters, including SERCA2, Na+/Ca2+ exchanger (NCX)1, and RyR2 [114]. A UPS malfunction, through mitochondrial stress, oxidative stress, cytotoxic reagents, or infection, causes cardiac dysfunction through protein aggregation, electrophysiological dysfunction, and the accumulation of cardiac remodeling proteins [112]. Activation of the UPS with dexamethasone, which impairs post-injury skeletal muscle regeneration, occurs through an increase in calpain in the myotubes [115]. In addition, a simple increase in [Ca2+]i through the A233187 Ca2+ ionophore increases the proteasome activity via CaMKII- and calpain-dependent mechanisms [92]. SERCA, which is a main component protein of the skeletal muscle, maintains Ca2+ homeostasis in the skeletal muscle [116]. SERCA malfunctions caused by a missense mutation in the ATP2A1 gene, which encodes SERCA isoform 1, induce Chianina cattle congenital pseudomyotonia muscular disorder, which impairs muscle relaxation [117], whereas the inhibition of the UPS with MG-132 results in an increased expression of SERCA1 and increased activity of Ca2+-ATPase [23]. The treatment with MG-132 attenuates carbachol (which induces Ca2+ release from sarcoplasmic reticulum (SR) [118])-induced Ca2+ signaling in SERCA1 mutant-transfected cells, indicating that MG-132 recovers the SERCA1 expression to restore the ER Ca2+ concentration from the cytoplasm [23]. IP3R activates calmodulin to phosphorylate the myosin light chain during muscle contractions [119]. Decreased IP3R expression via H2O2 attenuates the vascular reactivity in rat thoracic aortic rings [44]. In addition, the levels of SR protein RyR2, which regulates excitation–contraction coupling and cardiac cell recovery [120,121], decrease in the heart after ischemia/reperfusion [122,123]. Pedrozo et al. demonstrated that the activation of the UPS through calpain causes the degradation of RyR2 proteins in SR [45]. In addition, CaV1.2 induces Ca2+ influx to stimulate smooth muscle contractions [60] and regulate the arterial blood pressure [124]. CaV1.2 is inhibited by Gal-1, which displaces the β-subunits of CaV1.2, disturbing its protective role against the UPS [64], and its deficiency triggers high blood pressure [125]. Gal-1, therefore, modulates the expression of CaV1.2 through the UPS to maintain the blood pressure [64]. Considering Ca2+ is a major resource for muscle contraction, UPS should be an important process to sustain Ca2+ homeostasis in muscles and cardiovascular tissues. Thus, a wide range of Ca2+-related proteins and Ca2+ signaling interact with UPS in the muscle, and the cardiovascular system provides direct evidence of the interaction between the UPS and Ca2+ signaling and, thus, should be extensively elucidated for future study.
3.3. The Pancreas
The pancreas is one of the exocrine glands and has a role in digestion and glucose metabolism. Although UPS-related experimental evidence is relatively unknown in pancreatic glands, activation of the UPS downregulates IP3R in rat pancreatic islet cells [42]. During the development of pancreatic cells, pancreatic and duodenal homeobox 1 (Pdx1) is a marker of pancreatic progenitor cells [126] and is necessary for β-cell maturation [127]. The proteasome-induced degradation of Pdx1 is protected by the involvement of Ca2+ sensor secretagogin in β cells [128]. Increases in [Ca2+]i in β cells induce the apoptotic pathway through the UPS [68,72]. When [Ca2+]i increases through Ca2+ ionophore A23187, or by the supplementation of Ca2+ in the media in pancreatic β cells, the UPS-mediated degradation of islet-brain 1 and c-Jun N-terminal kinase (JNK) interacting protein 1 (IB1/JIP1) is increased [68]. The IB1/JIP1 are antiapoptotic scaffold proteins and block the JNK pathway [129]. Mutations of IB1/JIP1 induce the activation of the JNK pathway to trigger the apoptosis of β cells and subsequently induce type 2 diabetes [129]. In addition, the increase in [Ca2+]i that occurs through ER stress reduces the proteasome activity in β cells [72]. Impairment of the proteasome activity induces the aggregation of the extracellular human islet amyloid polypeptide (hIAPP), which finally induces β-cell apoptosis [72], suggesting that pancreatic β-cell homeostasis is closely related to Ca2+ signaling and proteasome activity. More recently, nicardipine, a drug to treat high blood pressure that is also known as an L-type Ca2+ channel blocker, blocked the proteasome through CaMKII and increased [Ca2+]i in pancreatic acinar cells [130], suggesting that the treatment of nicardipine should be considered in unwanted pancreatic acinar cell damage. Thus, although direct evidence is rare, verification of the precise mechanism between the UPS and Ca2+ signaling in the pancreas might provide potential strategies for diabetic treatment and pancreatic injury.
3.4. Other Tissues
Several tissues, including reproductive cells, osteoblasts, and mesangial cells, are affected by the relationship between the UPS and Ca2+ signaling. In the meiotic cell cycle, [Ca2+]i affects proteasome activity [65]. Briefly, the treatment with A23187 in metaphase-anaphase transition transiently induces the modulation of proteasome activity, and BAPTA-AM co-treatment sustains the proteasome activity [65].
Inhibition of the UPS by MG-132 blocks the forskolin-mediated decrease of core binding factor α-1 (Cbfa1) [131]. Briefly, core binding factor α-1 (Cbfa1) is a master regulator of osteoblastic differentiation [132,133]. Cbfa1 expression is decreased in forskolin-treated osteoblastic cell lines [131]. The treatment with forskolin increases cyclic adenosine monophosphate (cAMP), which is stimulated by [Ca2+]i and induces Ca2+ signaling [134]. Ca2+-related cAMP thus regulates Cbfa1 expression through the UPS to inhibit osteoblastic differentiation [131].
In addition to the renal system, the treatment of mesangial cells with high glucose attenuates Orai1 expression through ubiquitination of Orai1, whereas MG-132 recovers the expression of Orai1 [135]. A high glucose treatment induces dysregulated SOCE of the mesangial cell through the UPS-mediated degradation of Orai1 [135]. The maintenance of SOCE in the renal system is a critical process against diabetic injury. Above all, although the interaction between the UPS and Ca2+ signaling is multifaceted, their precise modulation according to each specific tissue type requires additional research.
4. Ubiquitination in Cancer
The UPS, which degrades tumor suppressor proteins, contributes to the development and sustaining of the cancerous phenotype [136,137], whereas inhibition of the UPS through the knockdown of UPS-related proteins decreases cancer cell survival [138,139]. Thus, proteasome inhibition has been suggested as a potential target for cancer therapy [140,141]. In this section, we will discuss the relationship between the proteasome and Ca2+ signaling in cancer cells based on the experimental evidence. In triple-negative breast cancer cells, inhibition of the proteasome induces cancer cell death via the involvement of several Ca2+-signaling pathways. The proteasome inhibitor bortezomib (BTZ) induces 5′-adenosine monophosphate (AMP)-activated protein kinase (AMPK) activation by increasing [Ca2+]i and CaMKII subunit β in MDA-MB-231 and MDA-MB-498 cells [142]. Indirubin-3-monoxime (I3M, which enhances apoptosis through oxidative stress [143]) and curcumin are known anticancer components that induce cancer cell apoptosis [143,144]. These compounds induce paraptosis through mitochondrial Ca2+ overload accompanied by proteasome impairment in malignant breast cancer cells [76,145]. Paraptosis is a type of programmed cell death that initiates cytoplasmic vacuolization, which is generated from the ER and mitochondria [76]. I3M induces proteasome impairment-mediated ER stress and the transference of ER Ca2+ into the mitochondria through a mitochondrial Ca2+ uniporter, which causes paraptosis in MDA-MB-231 cells [145]. Similarly, curcumin inhibits proteasome to increase the mitochondrial Ca2+ overload, which is followed by paraptosis in MDA-MB-435S and MDA-MB-231 cells [76]. In human liver carcinoma cells, NNC 55-0396, a T-type Ca2+ channel blocker, inhibits tumor angiogenesis through the UPS-induced degradation of HIF-1 [78].
5. The Crosstalk between the UPS and Autophagy with Ca2+ Signaling
To understand the relationship between the UPS and Ca2+ signaling in the cellular clearance system, autophagy as another clearance system should be also illuminated. Thus, in this section, we describe the crosstalk between the UPS and autophagy in Ca2+ signaling. Several researchers have proposed that the UPS is associated with the autophagic process [5,146,147]. In mammalian cells, there are three types of autophagy, including macroautophagy, chaperone-mediated autophagy, and microautophagy [148]. Although three types of autophagy are distinct in mechanisms, all types of autophagic mechanisms are based on lysosomal protein degradation and recycling [149]. The relationship between the UPS and autophagy has to be further clarified to understand the details of interconnection between the UPS and autophagy. Thus, in this review, we deal with only macroautophagy, which is extensively studied. Macroautophagy proceeds via two components, the autophagosome and the lysosome, which are stimulated by starvation and the mTORC1 complex [150]. The autophagosome recruits target molecules to bind p62 [151,152,153], and the lysosome contains various hydrolases to degrade proteins. Autophagosome and lysosome fusion occurs during autophagy and performs a degradative function [154,155].
Similar to the UPS, Ca2+ signaling is the key signaling modulator of autophagic flux through lysosomal Ca2+ release, which is a critical cellular component of autophagy [156,157,158]. The lysosome is a small cellular compartment that sustains the luminal pH and contains several ions that are essential for lysosomal activity [159]. Most importantly, lysosomal Ca2+ channels play a pivotal role in various cellular physiological functions, as well as lysosomal functions [160]. The regulatory role of Ca2+ in autophagy has been extensively studied [160,161,162,163,164]; however, the relationship between the UPS and lysosomal Ca2+ signaling is rarely studied. The crosstalk of the UPS and autophagy occurs to sustain the protein activity and to supplement proteolysis where needed [5,50]. In particular, p62, which is a major component of autophagy, is associated with the UPS [147]. When the UPS is downregulated, the p62 activation is increased and then competes with Nrf2 for Kelch-like ECH-associated protein 1 (Keap1) [147]. The p62–Keap1 complex triggers the aggregation of ubiquitinated proteins in the UPS [147]. The overexpression of p62 enhances the activity of the UPS to form aggregates of ubiquitinated proteins, whereas the downregulation of p62 inhibits the UPS [147]. Crosstalk between the UPS and autophagy via Ca2+ signaling is, therefore, an essential interaction for maintaining cellular homeostasis.
The ER stress-related regulation of proteolysis has been thoroughly investigated. Treatment with the UPS inhibitor MG-132 increases [Ca2+]i via ER stress in HCT116 colon cancer cells [165] and generates ROS in C6 glioma cells [166] (Figure 1). MG-132 induces cellular vacuolization, leading to autophagosome–lysosome fusion, which is impaired by Ca2+ chelation through BAPTA-AM [165]. Additionally, in hormone receptor-positive breast cancer MCF-7 cells, MG-132 induces autophagy through ER stress and, subsequently, apoptosis [167]. The accumulation of misfolded proteins and perturbed unfolded protein response in the ER causes ER stress and produces ROS, and these misfolded proteins are generally degraded by UPS or autophagy [168]. The downregulation of one side of the pathway induces the supplementary upregulation of the other side, so that, when the UPS is inhibited, the autophagy is increased. Consequently, the inhibition of both the UPS and autophagy induces cell death. A combination of UPS inhibitors, including BTZ, and autophagy inhibitors, including bafilomycin A1 (Baf) or 2-aminoethyldiphenylborinate (2-APB), may therefore be an effective clinical cancer therapeutic strategy [169,170]. The treatment with only Baf (10 nM, 48 h) and only BTZ (10 nM, 48 h) decreases the cellular viability by approximately 20% in U266 myeloma cells [169]. However, a co-treatment with Baf (10 nM) and BTZ (10 nM) for 48 h results in a remarkable decrease in the viability of approximately 90% [169]. The combination of BTZ and 2-APB results in enhanced cell death compared to the treatment with BTZ alone in A549 lung cancer cells [170] (Figure 1). In addition, a co-treatment with BTZ and 2-APB decreases the lung tumor volume and weight in vivo compared to BTZ or 2-APB treatment alone [170]. Malfunction of the proteasome through BTZ increases the Ca2+-related protein activity, including calcineurin, and activates autophagy through the calcineurin-transcription factor EB-p62 pathway in cardiomyocytes (Figure 1) [171,172]. In a recent study, ubiquitinated CaV1.2 was degraded by autophagy through the ubiquitin-binding proteins RFP2 and p62 (Figure 1) [173]. p62 senses ubiquitinated proteins in order to degrade CaV1.2 through autophagy and induces the action potential duration [173]. In thymus cells, the stimulation of TRPV1 via capsaicin reduces the proteasome activity (Figure 1) [174]. However, the induction of autophagy reverts capsaicin-induced UPS inhibition [174]. In addition, the deletion of TRPV1 attenuates both the UPS- and autophagy-related protein levels [174]. In this case, the UPS and autophagy do not contribute to the complementary proteolysis, and TRPV1 obviously modulates both the UPS and autophagy. Thus, the lineage of UPS-autophagy-Ca2+ signaling reveals a convergence and may also be required to overcome its complexity.
6. Conclusions
The studies of the relationship between the UPS and Ca2+ signaling propose the key mechanism to maintain cellular homeostasis as a cellular clearance system. It is well-known that oxidative stress and Ca2+ signaling have a mutual interplay, including increases of [Ca2+]i from the ER and the stimulation of mitochondrial ROS [18,175]. From the point of view that oxidative stress interacts with the UPS, an understanding of the UPS and Ca2+ signaling is needed to comprehend the delicate signaling modulation. Various organ systems have been elucidated with regards to UPS regulation. Briefly, in the nervous system, protein aggregation is considered the hallmark of neurodegeneration, and the key proteins, which are associated with Huntington’s disease (mutant huntingtin), Parkinson’s disease (α-synuclein), and amyotrophic lateral sclerosis (superoxide dismutase), are substrates of the UPS [176]. Additionally, inhibition of the UPS induces inflammatory toxicity (lymphopenia [177]), cardiomyopathies (arrhythmia [178]), the depletion of alloreactive T cells [179], and ischemia–reperfusion injury [180]. Although protein aggregation could be attenuated by activation of the UPS, paradoxically, proteasome inhibitor BTZ has been suggested as a potential drug for cancer therapy [181]. A recent study suggested that proteasome inhibitors could be therapeutic targets for various diseases, including infectious diseases, autoimmune diseases, and neurodegenerative diseases [182]. However, the clinical approaches of proteasome inhibitors should be carefully considered due to their limitations [182]. Especially, BTZ treatment occurs alongside the damage of nerves, including peripheral neuropathy [183,184], and causes a dose limitation of BTZ when treating myeloma patients [185]. Thus, we suggest that the verification of Ca2+ signaling as the checklist of clinical approaches in proteasome modulation might be beneficial to avoid unwanted effects.
Author Contributions
J.H.H. and D.L. contributed to the conception and writing of the manuscript; D.L. drafted the article, collected the referenced articles, and prepared Figure 1; and J.H.H. approved the version of the manuscript and accepted responsibility for all aspects of the work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT; NRF-2022R1A2C1003890 (JHH)) and the Gachon University research fund of 2020 (GCU-202008440008(JHH)).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Breusing, N.; Grune, T. Regulation of proteasome-mediated protein degradation during oxidative stress and aging. Biol. Chem 2008, 389, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Grune, T. The proteasome and its role in the degradation of oxidized proteins. IUBMB Life 2008, 60, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol 2018, 6, 128. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [Green Version]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Heinemeyer, W.; Ramos, P.C.; Dohmen, R.J. The ultimate nanoscale mincer: Assembly, structure and active sites of the 20S proteasome core. Cell Mol. Life Sci. 2004, 61, 1562–1578. [Google Scholar] [CrossRef]
- Paniagua Soriano, G.; De Bruin, G.; Overkleeft, H.S.; Florea, B.I. Toward understanding induction of oxidative stress and apoptosis by proteasome inhibitors. Antioxid. Redox Signal 2014, 21, 2419–2443. [Google Scholar] [CrossRef]
- Bonet-Costa, V.; Pomatto, L.C.D.; Davies, K.J.A. The Proteasome and Oxidative Stress in Alzheimer’s Disease. Antioxid. Redox Sign 2016, 25, 886–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, U.; Anwar, A.; Savage, R.S.; Thornalley, P.J.; Rabbani, N. Protein oxidation, nitration and glycation biomarkers for early-stage diagnosis of osteoarthritis of the knee and typing and progression of arthritic disease. Arthritis. Res. Ther. 2016, 18, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworski, R. Oxidant stress in asthma. Thorax 2000, 55, S51–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Thomas, S.R.; Keaney, J.F., Jr. Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic. Biol. Med. 2003, 35, 117–132. [Google Scholar] [CrossRef]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir Crit Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Nowell, T.R., Jr.; Taylor, A. Removal of oxidatively damaged proteins from lens cells by the ubiquitin-proteasome pathway. Exp. Eye Res. 2001, 73, 229–238. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Masaki, H.; Izutsu, Y.; Yahagi, S.; Okano, Y. Reactive oxygen species in HaCaT keratinocytes after UVB irradiation are triggered by intracellular Ca2+ levels. J. Investig. Dermatol. Symp. Proc. 2009, 14, 50–52. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Szent-Gyorgyi, A.G. Calcium regulation of muscle contraction. Biophys. J. 1975, 15, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, E.; Testoni, S.; Gentile, A.; Cali, T.; Ottolini, D.; Villa, A.; Brini, M.; Betto, R.; Mascarello, F.; Nissen, P.; et al. Inhibition of ubiquitin proteasome system rescues the defective sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1) protein causing Chianina cattle pseudomyotonia. J. Biol. Chem. 2014, 289, 33073–33082. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Ambudkar, I.S. Calcium signalling in salivary gland physiology and dysfunction. J. Physiol. 2016, 594, 2813–2824. [Google Scholar] [CrossRef] [Green Version]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Bio 2018, 19, 697–712. [Google Scholar] [CrossRef] [Green Version]
- Coux, O.; Zieba, B.A.; Meiners, S. The Proteasome System in Health and Disease. Adv. Exp. Med. Biol 2020, 1233, 55–100. [Google Scholar] [CrossRef]
- Campello, L.; Esteve-Rudd, J.; Cuenca, N.; Martin-Nieto, J. The ubiquitin-proteasome system in retinal health and disease. Mol. Neurobiol. 2013, 47, 790–810. [Google Scholar] [CrossRef]
- Hegde, A.N.; Upadhya, S.C. The ubiquitin-proteasome pathway in health and disease of the nervous system. Trends Neurosci. 2007, 30, 587–595. [Google Scholar] [CrossRef]
- Brownlee, C.; Hetherington, A. Introduction to a Virtual Special Issue on calcium signalling in plants. New Phytol. 2011, 192, 786–789. [Google Scholar] [CrossRef]
- Granqvist, E.; Sun, J.; Op den Camp, R.; Pujic, P.; Hill, L.; Normand, P.; Morris, R.J.; Downie, J.A.; Geurts, R.; Oldroyd, G.E. Bacterial-induced calcium oscillations are common to nitrogen-fixing associations of nodulating legumes and nonlegumes. New Phytol. 2015, 207, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Sadanandom, A.; Bailey, M.; Ewan, R.; Lee, J.; Nelis, S. The ubiquitin-proteasome system: Central modifier of plant signalling. New Phytol. 2012, 196, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xu, W.; Wang, J.; Wang, L.; Yao, W.; Yang, Y.; Xu, Y.; Ma, F.; Du, Y.; Wang, Y. The Chinese wild grapevine (Vitis pseudoreticulata) E3 ubiquitin ligase Erysiphe necator-induced RING finger protein 1 (EIRP1) activates plant defense responses by inducing proteolysis of the VpWRKY11 transcription factor. New Phytol. 2013, 200, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Kaandorp, J.A.; Ositelu, O.O.; Beaudry, V.; Knight, A.; Nanfack, Y.F.; Cunningham, K.W. Simulating calcium influx and free calcium concentrations in yeast. Cell Calcium 2009, 45, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Carraro, M.; Bernardi, P. Calcium and reactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium 2016, 60, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.C.; Wu, E.; Sailer, C.; Jando, J.; Styles, E.; Eisenkolb, I.; Kuschel, M.; Bitschar, K.; Wang, X.R.; Huang, L.; et al. Ubiquitin orchestrates proteasome dynamics between proliferation and quiescence in yeast. Mol. Biol. Cell 2017, 28, 2479–2491. [Google Scholar] [CrossRef]
- Hilt, W.; Heinemeyer, W.; Wolf, D.H. Studies on the yeast proteasome uncover its basic structural features and multiple in vivo functions. Enzym. Protein 1993, 47, 189–201. [Google Scholar] [CrossRef]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar] [CrossRef]
- Fedorenko, O.A.; Popugaeva, E.; Enomoto, M.; Stathopulos, P.B.; Ikura, M.; Bezprozvanny, I. Intracellular calcium channels: Inositol-1,4,5-trisphosphate receptors. Eur. J. Pharmacol. 2014, 739, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.P.; Wang, Y.; Sliter, D.A.; Pearce, M.M.; Wojcikiewicz, R.J. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem. 2011, 286, 24426–24433. [Google Scholar] [CrossRef] [Green Version]
- Oberdorf, J.; Webster, J.M.; Zhu, C.C.; Luo, S.G.; Wojcikiewicz, R.J.H. Down-regulation of types I, II and III inositol 1,4,5-trisphosphate receptors is mediated by the ubiquitin/proteasome pathway. Biochem. J. 1999, 339, 453–461. [Google Scholar] [CrossRef]
- Lee, B.; Gai, W.; Laychock, S.G. Proteasomal activation mediates down-regulation of inositol 1,4,5-trisphosphate receptor and calcium mobilization in rat pancreatic islets. Endocrinology 2001, 142, 1744–1751. [Google Scholar] [CrossRef]
- Bhanumathy, C.D.; Nakao, S.K.; Joseph, S.K. Mechanism of proteasomal degradation of inositol trisphosphate receptors in CHO-K1 cells. J. Biol Chem. 2006, 281, 3722–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Garrido, A.; Boyano-Adanez, M.C.; Alique, M.; Calleros, L.; Serrano, I.; Griera, M.; Rodriguez-Puyol, D.; Griendling, K.K.; Rodriguez-Puyol, M. Hydrogen peroxide down-regulates inositol 1,4,5-trisphosphate receptor content through proteasome activation. Free Radic. Bio. Med. 2009, 47, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Pedrozo, Z.; Sanchez, G.; Torrealba, N.; Valenzuela, R.; Fernandez, C.; Hidalgo, C.; Lavandero, S.; Donoso, P. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim. Biophys. Acta 2010, 1802, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 279–305. [Google Scholar] [CrossRef] [PubMed]
- Moller, J.V.; Juul, B.; le Maire, M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta 1996, 1286, 1–51. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Keil, J.M.; Shen, Z.X.; Briggs, S.P.; Patrick, G.N. Regulation of STIM1 and SOCE by the Ubiquitin-Proteasome System (UPS). PLoS ONE 2010, 5, 13465. [Google Scholar] [CrossRef]
- Kuang, X.L.; Liu, Y.; Chang, Y.; Zhou, J.; Zhang, H.; Li, Y.; Qu, J.; Wu, S. Inhibition of store-operated calcium entry by sub-lethal levels of proteasome inhibition is associated with STIM1/STIM2 degradation. Cell Calcium 2016, 59, 172–180. [Google Scholar] [CrossRef]
- Jurkovicova-Tarabova, B.; Lacinova, L. Structure, function and regulation of CaV 2.2 N-type calcium channels. Gen. Physiol. Biophys. 2019, 38, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldo, L.; Sandoval, A.; Garza-Lopez, E.; Felix, R. Involvement of Parkin in the ubiquitin proteasome system-mediated degradation of N-type voltage-gated Ca2+ channels. PLoS ONE 2017, 12, e0185289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marangoudakis, S.; Andrade, A.; Helton, T.D.; Denome, S.; Castiglioni, A.J.; Lipscombe, D. Differential ubiquitination and proteasome regulation of Ca(V)2.2 N-type channel splice isoforms. J. Neurosci. 2012, 32, 10365–10369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, M.A.; Henriquez, D.R.; Grimaldo, L.; Sandoval, A.; Altier, C.; Zamponi, G.W.; Felix, R.; Gonzalez-Billault, C. CaV2.2 channel cell surface expression is regulated by the light chain 1 (LC1) of the microtubule-associated protein B (MAP1B) via UBE2L3-mediated ubiquitination and degradation. Pflugers. Arch. 2014, 466, 2113–2126. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Y.; Zhai, X.; Dai, J.; Jin, G.; Wang, L.; Zhu, L.; Yang, Y.; Liu, J.; Chu, M.; et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat. Genet. 2013, 45, 1499–1503. [Google Scholar] [CrossRef]
- Page, K.M.; Rothwell, S.W.; Dolphin, A.C. The CaVbeta Subunit Protects the I-II Loop of the Voltage-gated Calcium Channel CaV2.2 from Proteasomal Degradation but Not Oligoubiquitination. J. Biol. Chem. 2016, 291, 20402–20416. [Google Scholar] [CrossRef] [Green Version]
- Waithe, D.; Ferron, L.; Page, K.M.; Chaggar, K.; Dolphin, A.C. Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. J. Biol. Chem. 2011, 286, 9598–9611. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Smooth muscle cell calcium activation mechanisms. J. Physiol. 2008, 586, 5047–5061. [Google Scholar] [CrossRef]
- Altier, C.; Garcia-Caballero, A.; Simms, B.; You, H.; Chen, L.; Walcher, J.; Tedford, H.W.; Hermosilla, T.; Zamponi, G.W. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 2011, 14, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, J.W.; Yu, D.; Soon, J.L.; de Kleijn, D.P.; Foo, R.; Liao, P.; Colecraft, H.M.; Soong, T.W. Aberrant Splicing Promotes Proteasomal Degradation of L-type CaV1.2 Calcium Channels by Competitive Binding for CaVbeta Subunits in Cardiac Hypertrophy. Sci. Rep. 2016, 6, 35247. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thio, S.S.; Yang, S.S.; Yu, D.; Yu, C.Y.; Wong, Y.P.; Liao, P.; Li, S.; Soong, T.W. Splice variant specific modulation of CaV1.2 calcium channel by galectin-1 regulates arterial constriction. Circ. Res. 2011, 109, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Li, G.; Wang, J.W.; Chong, S.Y.; Yu, D.; Wang, X.; Soon, J.L.; Liang, M.C.; Wong, Y.P.; Huang, N.; et al. Regulation of Blood Pressure by Targeting CaV1.2-Galectin-1 Protein Interaction. Circulation 2018, 138, 1431–1445. [Google Scholar] [CrossRef]
- Kawahara, H.; Yokosawa, H. Intracellular calcium mobilization regulates the activity of 26 S proteasome during the metaphase-anaphase transition in the ascidian meiotic cell cycle. Dev. Biol. 1994, 166, 623–633. [Google Scholar] [CrossRef]
- Aizawa, H.; Kawahara, H.; Tanaka, K.; Yokosawa, H. Activation of the proteasome during Xenopus egg activation implies a link between proteasome activation and intracellular calcium release. Biochem. Bioph. Res. Co. 1996, 218, 224–228. [Google Scholar] [CrossRef]
- Yazaki, M.; Kashiwagi, K.; Aritake, K.; Urade, Y.; Fujimori, K. Rapid degradation of cyclooxygenase-1 and hematopoietic prostaglandin D synthase through ubiquitin-proteasome system in response to intracellular calcium level. Mol. Biol. Cell 2012, 23, 12–21. [Google Scholar] [CrossRef]
- Allaman-Pillet, N.; Storling, J.; Oberson, A.; Roduit, R.; Negri, S.; Sauser, C.; Nicod, P.; Beckmann, J.S.; Schorderet, D.F.; Mandrup-Poulsen, T.; et al. Calcium- and proteasome-dependent degradation of the JNK scaffold protein islet-brain 1. J. Biol. Chem. 2003, 278, 48720–48726. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, K.V.; Margolis, S.S. A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat. Struct. Mol. Biol. 2017, 24, 419. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.A.; Fan, Y.; Gandhirajan, R.K.; Madesh, M.; Zong, W.X. Hyperactivation of the mammalian degenerin MDEG promotes caspase-8 activation and apoptosis. J. Biol. Chem. 2013, 288, 2952–2963. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.S.; Hong, H.; Kim, C.; Mook-Jung, I. Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci Rep. 2015, 5, 8805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gafner, S.; Lee, S.K.; Cuendet, M.; Barthelemy, S.; Vergnes, L.; Labidalle, S.; Mehta, R.G.; Boone, C.W.; Pezzuto, J.M. Biologic evaluation of curcumin and structural derivatives in cancer chemoprevention model systems. Phytochemistry 2004, 65, 2849–2859. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharm. 2006, 71, 1397–1421. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kim, E.H.; Kwon, T.K.; Park, S.A.; Choi, K.S. Simultaneous mitochondrial Ca2+ overload and proteasomal inhibition are responsible for the induction of paraptosis in malignant breast cancer cells. Cancer Lett. 2012, 324, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, D.; Park, J.Y.; Jung, H.J.; Cho, Y.H.; Kim, H.K.; Han, J.; Choi, K.Y.; Kwon, H.J. NNC 55-0396, a T-type Ca2+ channel inhibitor, inhibits angiogenesis via suppression of hypoxia-inducible factor-1alpha signal transduction. J. Mol. Med. 2015, 93, 499–509. [Google Scholar] [CrossRef]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E.; Runnels, L.W.; Strubing, C. The TRP ion channel family. Nat. Rev. Neurosci. 2001, 2, 387–396. [Google Scholar] [CrossRef]
- Huang, K.F.; Ma, K.H.; Jhap, T.Y.; Liu, P.S.; Chueh, S.H. Ultraviolet B irradiation induced Nrf2 degradation occurs via activation of TRPV1 channels in human dermal fibroblasts. Free Radic. Biol. Med. 2019, 141, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, J.; Qiu, L. Transient receptor potential vanilloid 1 promotes EGFR ubiquitination and modulates EGFR/MAPK signalling in pancreatic cancer cells. Cell Biochem. Funct. 2020, 38, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Organista-Juarez, D.; Carretero-Ortega, J.; Vicente-Fermin, O.; Vazquez-Victorio, G.; Sosa-Garrocho, M.; Vazquez-Prado, J.; Macias-Silva, M.; Reyes-Cruz, G. Calcium-sensing receptor inhibits TGF-beta-signaling by decreasing Smad2 phosphorylation. IUBMB Life 2013, 65, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.C. Calmodulin: An introduction. Can. J. Biochem. Cell Biol. 1983, 61, 906–910. [Google Scholar] [CrossRef]
- Tansey, M.G.; Luby-Phelps, K.; Kamm, K.E.; Stull, J.T. Ca2+-dependent phosphorylation of myosin light chain kinase decreases the Ca2+ sensitivity of light chain phosphorylation within smooth muscle cells. J. Biol. Chem. 1994, 269, 9912–9920. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Okui, Y.; Inaba, M.; Okuno, S.; Yukioka, K.; Miki, T.; Watanabe, Y.; Morii, H. Calcium/calmodulin-mediated action of calcitonin on lipid metabolism in rats. J. Clin. Invest. 1988, 82, 1165–1172. [Google Scholar] [CrossRef]
- Lledo, P.M.; Hjelmstad, G.O.; Mukherji, S.; Soderling, T.R.; Malenka, R.C.; Nicoll, R.A. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 11175–11179. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; Bhattacharya, A.; Sau, A.; Basu, S.; Chakrabarti, S.; Chakrabarti, O. Calmodulin regulates MGRN1-GP78 interaction mediated ubiquitin proteasomal degradation system. FASEB J. 2019, 33, 1927–1945. [Google Scholar] [CrossRef] [Green Version]
- Djakovic, S.N.; Schwarz, L.A.; Barylko, B.; DeMartino, G.N.; Patrick, G.N. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 2009, 284, 26655–26665. [Google Scholar] [CrossRef] [Green Version]
- Izumi, H.; Kawahata, I.; Shinoda, Y.; Helmstetter, F.J.; Fukunaga, K. SAK3 Administration Improves Spine Abnormalities and Cognitive Deficits in App(NL-G-F/NL-G-F) Knock-in Mice by Increasing Proteasome Activity through CaMKII/Rpt6 Signaling. Int. J. Mol. Sci. 2020, 21, 3833. [Google Scholar] [CrossRef]
- Bingol, B.; Wang, C.F.; Arnott, D.; Cheng, D.; Peng, J.; Sheng, M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 2010, 140, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menconi, M.J.; Wei, W.; Yang, H.; Wray, C.J.; Hasselgren, P.O. Treatment of cultured myotubes with the calcium ionophore A23187 increases proteasome activity via a CaMK II-caspase-calpain-dependent mechanism. Surgery 2004, 136, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Reffo, P.; Penna, F.; Autelli, R.; Bonelli, G.; Baccino, F.M. Ca2+-dependent proteolysis in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2134–2146. [Google Scholar] [CrossRef]
- Nowak, K.; Taubert, R.M.; Haberecht, S.; Venz, S.; Kruger, E. Inhibition of calpain-1 stabilizes TCF11/Nrf1 but does not affect its activation in response to proteasome inhibition. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uvarov, A.V.; Mesaeli, N. Enhanced ubiquitin-proteasome activity in calreticulin deficient cells: A compensatory mechanism for cell survival. Biochim. Biophys. Acta 2008, 1783, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Logsdon, C.D. S100P: A novel therapeutic target for cancer. Amino Acids 2011, 41, 893–899. [Google Scholar] [CrossRef]
- Shimamoto, S.; Kubota, Y.; Yamaguchi, F.; Tokumitsu, H.; Kobayashi, R. Ca2+/S100 proteins act as upstream regulators of the chaperone-associated ubiquitin ligase CHIP (C terminus of Hsc70-interacting protein). J. Biol. Chem. 2013, 288, 7158–7168. [Google Scholar] [CrossRef] [Green Version]
- Wilk, S.; Orlowski, M. Cation-sensitive neutral endopeptidase: Isolation and specificity of the bovine pituitary enzyme. J. Neurochem. 1980, 35, 1172–1182. [Google Scholar] [CrossRef]
- Rivett, A.J. Purification of a liver alkaline protease which degrades oxidatively modified glutamine synthetase. Characterization as a high molecular weight cysteine proteinase. J. Biol. Chem. 1985, 260, 12600–12606. [Google Scholar] [CrossRef]
- Tanaka, K.; Ii, K.; Ichihara, A.; Waxman, L.; Goldberg, A.L. A high molecular weight protease in the cytosol of rat liver. I. Purification, enzymological properties, and tissue distribution. J. Biol. Chem. 1986, 261, 15197–15203. [Google Scholar] [CrossRef]
- Zolfaghari, R.; Baker, C.R., Jr.; Canizaro, P.C.; Amirgholami, A.; Behal, F.J. A high-molecular-mass neutral endopeptidase-24.5 from human lung. Biochem. J. 1987, 241, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlmann, B.; Kuehn, L.; Rutschmann, M.; Reinauer, H. Purification and characterization of a multicatalytic high-molecular-mass proteinase from rat skeletal muscle. Biochem. J. 1985, 228, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Harris, H. Purification of neutral lens endopeptidase: Close similarity to a neutral proteinase in pituitary. Proc. Natl. Acad. Sci. USA 1985, 82, 7545–7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, M.; Ishiura, S.; Yamamoto, T.; Okuyama, T.; Furuya, H.; Sugita, H. Purification and characterization of a high-molecular-weight protease, ingensin, from human placenta. J. Biochem. 1986, 99, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Turker, F.; Cook, E.K.; Margolis, S.S. The proteasome and its role in the nervous system. Cell Chem. Biol. 2021, 28, 903–917. [Google Scholar] [CrossRef]
- Ehlers, M.D. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 2003, 6, 231–242. [Google Scholar] [CrossRef]
- Wright, F.A.; Lu, J.P.; Sliter, D.A.; Dupre, N.; Rouleau, G.A.; Wojcikiewicz, R.J. A Point Mutation in the Ubiquitin Ligase RNF170 That Causes Autosomal Dominant Sensory Ataxia Destabilizes the Protein and Impairs Inositol 1,4,5-Trisphosphate Receptor-mediated Ca2+ Signaling. J. Biol. Chem. 2015, 290, 13948–13957. [Google Scholar] [CrossRef] [Green Version]
- Jarome, T.J.; Ferrara, N.C.; Kwapis, J.L.; Helmstetter, F.J. CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol. Learn. Mem. 2016, 128, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Khalil, R. Ubiquitin-Proteasome Pathway and Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 235–248. [Google Scholar] [CrossRef]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef]
- Pagan, J.; Seto, T.; Pagano, M.; Cittadini, A. Role of the ubiquitin proteasome system in the heart. Circ. Res. 2013, 112, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Gilda, J.E.; Gomes, A.V. Proteasome dysfunction in cardiomyopathies. J. Physiol. 2017, 595, 4051–4071. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in muscle contraction. Cold Spring Harb. Perspect Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Bahrudin, U.; Morikawa, K.; Takeuchi, A.; Kurata, Y.; Miake, J.; Mizuta, E.; Adachi, K.; Higaki, K.; Yamamoto, Y.; Shirayoshi, Y.; et al. Impairment of ubiquitin-proteasome system by E334K cMyBPC modifies channel proteins, leading to electrophysiological dysfunction. J. Mol. Biol. 2011, 413, 857–878. [Google Scholar] [CrossRef]
- Wang, L.; Luo, G.J.; Wang, J.J.; Hasselgren, P.O. Dexamethasone stimulates proteasome- and calcium-dependent proteolysis in cultured L6 myotubes. Shock 1998, 10, 298–306. [Google Scholar] [CrossRef]
- Fleischer, S.; Inui, M. Biochemistry and biophysics of excitation-contraction coupling. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 333–364. [Google Scholar] [CrossRef]
- Drogemuller, C.; Drogemuller, M.; Leeb, T.; Mascarello, F.; Testoni, S.; Rossi, M.; Gentile, A.; Damiani, E.; Sacchetto, R. Identification of a missense mutation in the bovine ATP2A1 gene in congenital pseudomyotonia of Chianina cattle: An animal model of human Brody disease. Genomics 2008, 92, 474–477. [Google Scholar] [CrossRef] [Green Version]
- White, C.; McGeown, J.G. Carbachol triggers RyR-dependent Ca2+ release via activation of IP3 receptors in isolated rat gastric myocytes. J. Physiol. 2002, 542, 725–733. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. 2000, 522, 177–185. [Google Scholar] [CrossRef]
- An, J.; Bosnjak, Z.J.; Jiang, M.T. Myocardial protection by isoflurane preconditioning preserves Ca2+ cycling proteins independent of sarcolemmal and mitochondrial KATP channels. Anesth. Analg. 2007, 105, 1207–1213. [Google Scholar] [CrossRef]
- Zucchi, R.; Ronca, F.; Ronca-Testoni, S. Modulation of sarcoplasmic reticulum function: A new strategy in cardioprotection? Pharmacol. Ther. 2001, 89, 47–65. [Google Scholar] [CrossRef]
- Singh, R.B.; Chohan, P.K.; Dhalla, N.S.; Netticadan, T. The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J. Mol. Cell Cardiol. 2004, 37, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Temsah, R.M.; Netticadan, T.; Chapman, D.; Takeda, S.; Mochizuki, S.; Dhalla, N.S. Alterations in sarcoplasmic reticulum function and gene expression in ischemic-reperfused rat heart. Am. J. Physiol. 1999, 277, H584–H594. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Schulla, V.; Welling, A.; Feil, R.; Feil, S.; Wegener, J.W.; Hofmann, F.; Klugbauer, N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003, 22, 6027–6034. [Google Scholar] [CrossRef]
- Freitag, N.; Tirado-Gonzalez, I.; Barrientos, G.; Herse, F.; Thijssen, V.L.; Weedon-Fekjaer, S.M.; Schulz, H.; Wallukat, G.; Klapp, B.F.; Nevers, T.; et al. Interfering with Gal-1-mediated angiogenesis contributes to the pathogenesis of preeclampsia. Proc. Natl. Acad. Sci. USA 2013, 110, 11451–11456. [Google Scholar] [CrossRef] [Green Version]
- Oliver-Krasinski, J.M.; Kasner, M.T.; Yang, J.; Crutchlow, M.F.; Rustgi, A.K.; Kaestner, K.H.; Stoffers, D.A. The diabetes gene Pdx1 regulates the transcriptional network of pancreatic endocrine progenitor cells in mice. J. Clin. Invest. 2009, 119, 1888–1898. [Google Scholar] [CrossRef] [Green Version]
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401. [Google Scholar] [CrossRef]
- Malenczyk, K.; Szodorai, E.; Schnell, R.; Lubec, G.; Szabo, G.; Hokfelt, T.; Harkany, T. Secretagogin protects Pdx1 from proteasomal degradation to control a transcriptional program required for beta cell specification. Mol. Metab. 2018, 14, 108–120. [Google Scholar] [CrossRef]
- Bonny, C.; Oberson, A.; Steinmann, M.; Schorderet, D.F.; Nicod, P.; Waeber, G. IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J. Biol. Chem. 2000, 275, 16466–16472. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Lin, H.M.; Liu, B.G.; Jin, J.F. CaMKII/proteasome/cytosolic calcium/cathepsin B axis was present in tryspin activation induced by nicardipine. Biosci. Rep. 2019, 39, BSR20190516. [Google Scholar] [CrossRef] [Green Version]
- Tintut, Y.; Parhami, F.; Le, V.; Karsenty, G.; Demer, L.L. Inhibition of osteoblast-specific transcription factor Cbfa1 by the cAMP pathway in osteoblastic cells. Ubiquitin/proteasome-dependent regulation. J. Biol. Chem. 1999, 274, 28875–28879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Alasbahi, R.H.; Melzig, M.F. Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie 2012, 67, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zou, S.B.; Chaudhari, S.; Ma, R. Short-term high-glucose treatment decreased abundance of Orai1 protein through posttranslational mechanisms in rat mesangial cells. Am. J. Physiol.-Renal. 2018, 314, F855–F863. [Google Scholar] [CrossRef]
- Ali, A.; Farooqui, S.R.; Rai, J.; Singh, J.; Kumar, V.; Mishra, R.; Banerjea, A.C. HIV-1 Nef promotes ubiquitination and proteasomal degradation of p53 tumor suppressor protein by using E6AP. Biochem. Bioph. Res. Co. 2020, 529, 1038–1044. [Google Scholar] [CrossRef]
- Yue, S.; Chen, Y.; Cheng, S.Y. Hedgehog signaling promotes the degradation of tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 2009, 28, 492–499. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.J.; Chen, X.B. The p73 Tumor Suppressor Is Targeted by Pirh2 RING Finger E3 Ubiquitin Ligase for the Proteasome-dependent Degradation. J. Biol. Chem. 2011, 286, 35388–35395. [Google Scholar] [CrossRef] [Green Version]
- Thacker, G.; Mishra, M.; Sharma, A.; Singh, A.K.; Sanyal, S.; Trivedi, A.K. CDK2 destabilizes tumor suppressor C/EBP alpha expression through ubiquitin-mediated proteasome degradation in acute myeloid leukemia. J. Cell Biochem. 2020, 121, 2839–2850. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Devine, T.; Dai, M.S. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr. Pharm. Des. 2013, 19, 3248–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, R.R.; Dou, Q.P. Proteasome inhibitors induce AMPK activation via CaMKKbeta in human breast cancer cells. Breast Cancer Res. Treat. 2015, 153, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Dilshara, M.G.; Molagoda, I.M.N.; Jayasooriya, R.; Choi, Y.H.; Park, C.; Lee, K.T.; Lee, S.; Kim, G.Y. p53-Mediated Oxidative Stress Enhances Indirubin-3’-Monoxime-Induced Apoptosis in HCT116 Colon Cancer Cells by Upregulating Death Receptor 5 and TNF-Related Apoptosis-Inducing Ligand Expression. Antioxidants 2019, 8, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilshara, M.G.; Neelaka Molagoda, I.M.; Prasad Tharanga Jayasooriya, R.G.; Choi, Y.H.; Park, C.; Kim, G.Y. Indirubin-3’-monoxime induces paraptosis in MDA-MB-231 breast cancer cells by transmitting Ca2+ from endoplasmic reticulum to mitochondria. Arch. Biochem. Biophys. 2021, 698, 108723. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Cuervo, A.M. Selective autophagy: Talking with the UPS. Cell Biochem. Biophys. 2013, 67, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox. Signal 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy. FEBS Lett. 2010, 584, 1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell. Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; He, M.X.; McLeod, I.X.; He, Y.W. Autophagy, a novel pathway to regulate calcium mobilization in T lymphocytes. Front. Immunol. 2013, 4, 179. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Hong, J.H. Nanoparticle-Mediated Therapeutic Application for Modulation of Lysosomal Ion Channels and Functions. Pharmaceutics 2020, 12, 217. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Huang, P.; Dong, X.P. Lysosomal Calcium Channels in Autophagy and Cancer. Cancers 2021, 13, 1299. [Google Scholar] [CrossRef]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 2015, 11, 970–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smaili, S.S.; Pereira, G.J.; Costa, M.M.; Rocha, K.K.; Rodrigues, L.; do Carmo, L.G.; Hirata, H.; Hsu, Y.T. The role of calcium stores in apoptosis and autophagy. Curr. Mol. Med. 2013, 13, 252–265. [Google Scholar] [CrossRef]
- Hu, Y.X.; Han, X.S.; Jing, Q. Ca2+ Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hou, Y.; Ni, H.M.; Ding, W.X. Role of intracellular calcium in proteasome inhibitor-induced endoplasmic reticulum stress, autophagy, and cell death. Pharm. Res. 2013, 30, 2279–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W.H.; Hou, Y.; Meng, F.K.; Wang, X.F.; Luo, Y.N.; Ge, P.F. Proteasome inhibitor MG-132 induces C6 glioma cell apoptosis via oxidative stress. Acta. Pharm. Sin. 2011, 32, 619–625. [Google Scholar] [CrossRef]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulsky, A.V. The intersecting roles of endoplasmic reticulum stress, ubiquitin- proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney. Int. 2013, 84, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Miyazawa, K.; Moriya, S.; Ohtomo, T.; Che, X.F.; Naito, M.; Itoh, M.; Tomoda, A. Combined treatment with bortezomib plus bafilomycin A1 enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: Crosstalk among proteasome, autophagy-lysosome and ER stress. Int. J. Oncol. 2011, 38, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.Q.; Gordillo-Martinez, F.; Law, B.Y.K.; Han, Y.; Wu, A.; Zeng, W.; Lam, W.K.; Ho, C.; Mok, S.W.F.; He, H.Q.; et al. 2-Aminoethoxydiphenylborane sensitizes anti-tumor effect of bortezomib via suppression of calcium-mediated autophagy. Cell Death Dis. 2018, 9, 361. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Wang, X. Proteasome malfunction activates the PPP3/calcineurin-TFEB-SQSTM1/p62 pathway to induce macroautophagy in the heart. Autophagy 2020, 16, 2114–2116. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Li, J.; Parajuli, N.; Tian, Z.; Wu, P.; Lewno, M.T.; Zou, J.; Wang, W.; Bedford, L.; Mayer, R.J.; et al. The Calcineurin-TFEB-p62 Pathway Mediates the Activation of Cardiac Macroautophagy by Proteasomal Malfunction. Circ. Res. 2020, 127, 502–518. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, J.; Gong, Y.; Wang, D.; Wang, X.; Yun, F.; Liu, Z.; Zhang, S.; Li, W.; Zhao, X.; et al. Autophagy exacerbates electrical remodeling in atrial fibrillation by ubiquitin-dependent degradation of L-type calcium channel. Cell Death Dis. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Farfariello, V.; Cardinali, C.; Morelli, M.B.; Marinelli, O.; Nabissi, M.; Santoni, M.; Bonfili, L.; Cecarini, V.; Eleuteri, A.M.; et al. The TRPV1 ion channel regulates thymocyte differentiation by modulating autophagy and proteasome activity. Oncotarget 2017, 8, 90766–90780. [Google Scholar] [CrossRef] [Green Version]
- Feno, S.; Butera, G.; Reane, D.V.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid Med. Cell Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [Green Version]
- Witt, S. Protein Chaperones and Protection from Neurodegenerative Diseases; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Jagannath, S.; Barlogie, B.; Berenson, J.; Siegel, D.; Irwin, D.; Richardson, P.G.; Niesvizky, R.; Alexanian, R.; Limentani, S.A.; Alsina, M.; et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br. J. Haematol. 2004, 127, 165–172. [Google Scholar] [CrossRef]
- Schlossarek, S.; Carrier, L. The ubiquitin-proteasome system in cardiomyopathies. Curr. Opin. Cardiol. 2011, 26, 190–195. [Google Scholar] [CrossRef]
- Blanco, B.; Sanchez-Abarca, L.I.; Caballero-Velazquez, T.; Santamaria, C.; Inoges, S.; Perez-Simon, J.A. Depletion of alloreactive T-cells in vitro using the proteasome inhibitor bortezomib preserves the immune response against pathogens. Leukemia Res. 2011, 35, 1412–1415. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Lundberg, K.C.; Humphries, K.M.; Sadek, H.A.; Szweda, P.A.; Friguet, B.; Szweda, L.I. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem. 2001, 276, 30057–30063. [Google Scholar] [CrossRef] [Green Version]
- Frankland-Searby, S.; Bhaumik, S.R. The 26S proteasome complex: An attractive target for cancer therapy. Biochim Biophys Acta 2012, 1825, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhu, H.; He, R.; Kong, L.; Shao, J.; Zhuang, R.; Xi, J.; Zhang, J. Proteasome, a Promising Therapeutic Target for Multiple Diseases Beyond Cancer. Drug Des. Devel. Ther. 2020, 14, 4327–4342. [Google Scholar] [CrossRef] [PubMed]
- Cengiz Seval, G.; Beksac, M. The safety of bortezomib for the treatment of multiple myeloma. Expert. Opin. Drug Saf. 2018, 17, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Briemberg, H.; Jagannath, S.; Wen, P.Y.; Barlogie, B.; Berenson, J.; Singhal, S.; Siegel, D.S.; Irwin, D.; Schuster, M.; et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J. Clin. Oncol. 2006, 24, 3113–3120. [Google Scholar] [CrossRef]
- Badros, A.; Goloubeva, O.; Dalal, J.S.; Can, I.; Thompson, J.; Rapoport, A.P.; Heyman, M.; Akpek, G.; Fenton, R.G. Neurotoxicity of bortezomib therapy in multiple myeloma: A single-center experience and review of the literature. Cancer 2007, 110, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The crosstalk between the UPS and autophagy in the presence of Ca2+ signaling. The schematic illustration demonstrates the relationship between the UPS and autophagy through proteasome inhibitors in the presence of oxidative or ER stress and the subsequent intracellular Ca2+ increase. The treatment of MG-132, BTZ, and TRPV1 agonist capsaicin inhibits the proteasome activity. The inhibition of the proteasome induces ER stress, which, finally, stimulates calcineurin activity. Calcineurin triggers the dephosphorylation of TFEB to induce the translation associated with autophagy, and BTZ blocks the inhibitory effect of mTOR, which maintains the phosphorylation of TFEB. The inhibition of the proteasome is accompanied by an autophagic flux, which, finally, degrades CaV1.2. Thus, the simultaneous inhibition of the proteasome and autophagy destroys the protein homeostasis to induce cell death. 2-APB, 2-aminoethyldiphenylborinate; Baf, bafilomycin A1; BAPTA, 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetra acetic acid; BTZ, bortezomib; CaV1.2, voltage-gated calcium channel 1.2; TFEB, transcription factor EB; TRPV1, transient receptor potential vanilloid 1; ROS, reactive oxygen species.
Figure 1.
The crosstalk between the UPS and autophagy in the presence of Ca2+ signaling. The schematic illustration demonstrates the relationship between the UPS and autophagy through proteasome inhibitors in the presence of oxidative or ER stress and the subsequent intracellular Ca2+ increase. The treatment of MG-132, BTZ, and TRPV1 agonist capsaicin inhibits the proteasome activity. The inhibition of the proteasome induces ER stress, which, finally, stimulates calcineurin activity. Calcineurin triggers the dephosphorylation of TFEB to induce the translation associated with autophagy, and BTZ blocks the inhibitory effect of mTOR, which maintains the phosphorylation of TFEB. The inhibition of the proteasome is accompanied by an autophagic flux, which, finally, degrades CaV1.2. Thus, the simultaneous inhibition of the proteasome and autophagy destroys the protein homeostasis to induce cell death. 2-APB, 2-aminoethyldiphenylborinate; Baf, bafilomycin A1; BAPTA, 1,2-bis(o-aminophenoxy) ethane-N,N,N′,N′-tetra acetic acid; BTZ, bortezomib; CaV1.2, voltage-gated calcium channel 1.2; TFEB, transcription factor EB; TRPV1, transient receptor potential vanilloid 1; ROS, reactive oxygen species.


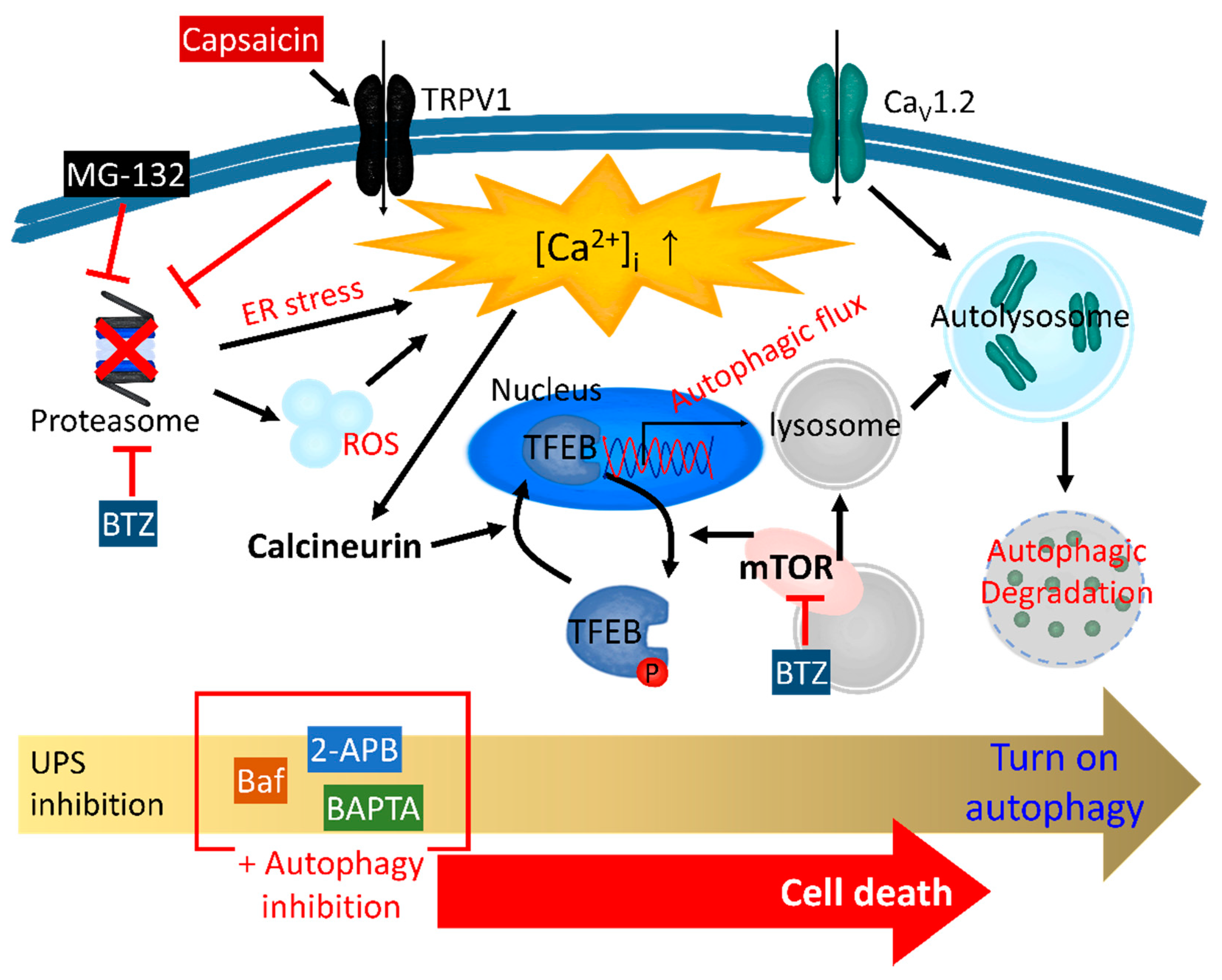{kind=link}
Table 1.
The effect of Ca2+ signaling on the UPS.
| Related Signaling | Effect on UPS | Details | Ref |
|---|---|---|---|
| Mitochondrial Ca2+ release | Inhibition | Curcumin inhibits the UPS to induce paraptosis. | [76] |
| T-type Ca2+ channel | Inhibition | NNC 55-0396 inhibits T-type Ca2+ channels to attenuate cancer angiogenesis. | [78] |
| TRPV1 | Activation | Activation of TRPV1 induces the ubiquitination of Nrf2. | [81] |
| Overexpressed TRPV1 increases the ubiquitination of EGFR to induce the UPS. | [82] | ||
| CaSR | Activation | CaSR maintains Ca2+ homeostasis through the UPS. | [83] |
| CaM | Activation | CaM induces the translocation of GP78 for ER-associated UPS. | [88] |
| CaMKII | Activation | Phosphorylation of Rpt6 through CaMKII enhances the UPS. | [90] |
| Calpain | Activation | Calpain-induced activation of Nrf1 stimulates the 26S proteasome subunit gene. | [94] |
| CAML | Inhibition | CAML stabilizes RNF122. | [91] |
| Calreticulin | Inhibition | Deficiency of calreticulin increases the UPS. | [95] |
| S100 | Inhibition | Inhibition of the E3 ubiquitin ligase. | [97] |
Abbreviations: TRPV1, transient receptor potential vanilloid 1; CaSR, Ca2+-sensing receptor; CaM, calmodulin; CaMKII, Ca2+/calmodulin-dependent protein kinase II; CAML, Ca2+-modulating cyclophilin ligand; UPS, ubiquitin proteasome system; Nrf, nuclear factor erythroid 2-related factor; GP78, glycoprotein 78; RNF122, ring finger protein 122.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, D.; Hong, J.H. Physiological Overview of the Potential Link between the UPS and Ca2+ Signaling. Antioxidants 2022, 11, 997. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050997
AMA Style
Lee D, Hong JH. Physiological Overview of the Potential Link between the UPS and Ca2+ Signaling. Antioxidants. 2022; 11(5):997. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050997
Chicago/Turabian StyleLee, Dongun, and Jeong Hee Hong. 2022. "Physiological Overview of the Potential Link between the UPS and Ca2+ Signaling" Antioxidants 11, no. 5: 997. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11050997
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.