Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress

 ,
,  ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Treatment
2.3. Isometric Tension Recording Experiments
2.4. Detection of Oxidative Burst in Whole Blood and Mitochondrial Reactive Oxygen Species (ROS) Formation in Aorta
2.5. Dot Blot Analysis
2.6. Detection of Mitochondrial Superoxide Formation by mitoSOX HPLC Method and Plate Reader Assay in Isolated Heart Mitochondria
2.7. Detection of Mitochondrial ROS Formation in Isolated Heart Mitochondria
2.8. Statistical Analysis
3. Results
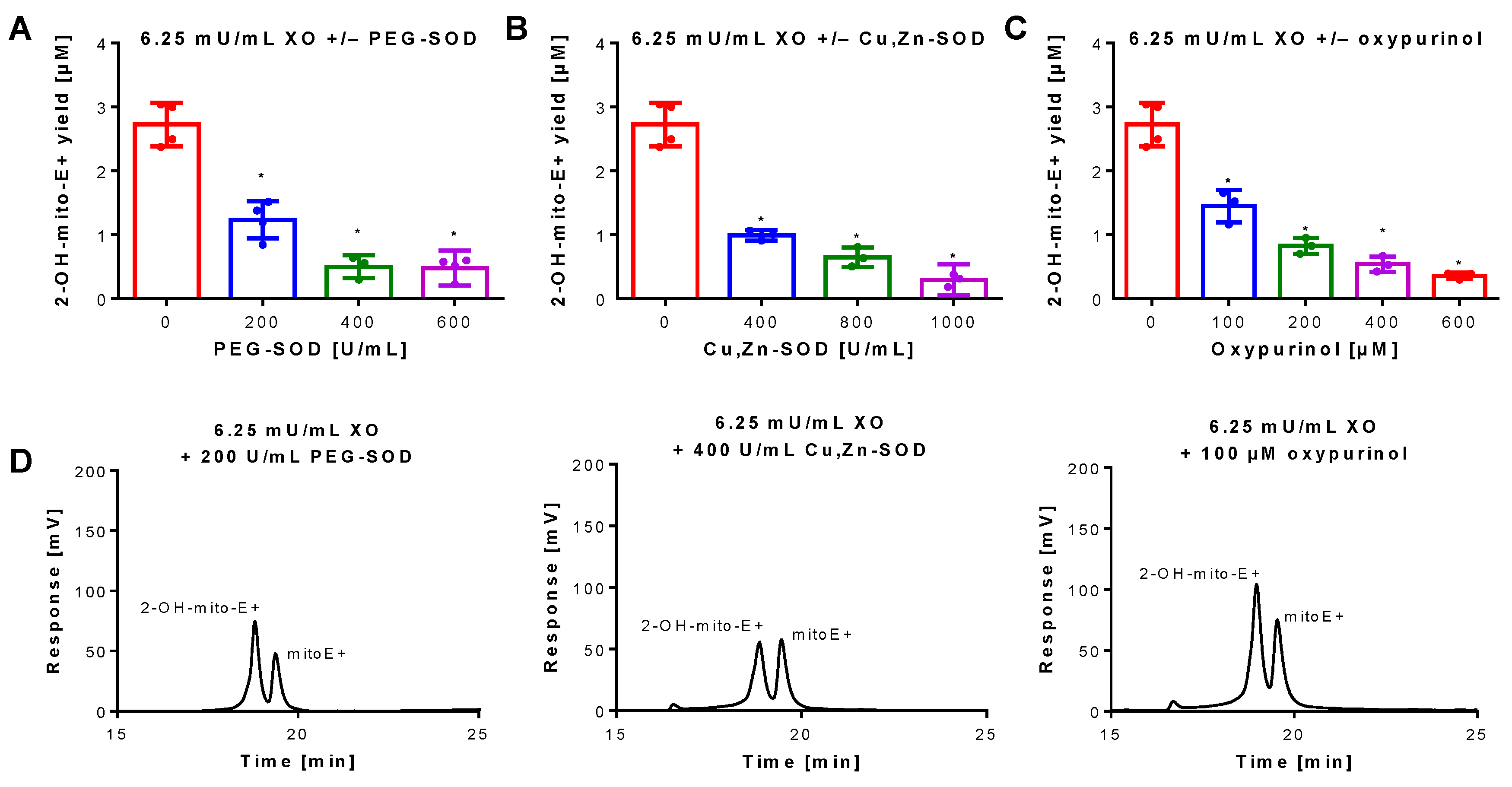
3.1. Detection of Superoxide Generation by Xanthine Oxidase Using mitoSOX HPLC
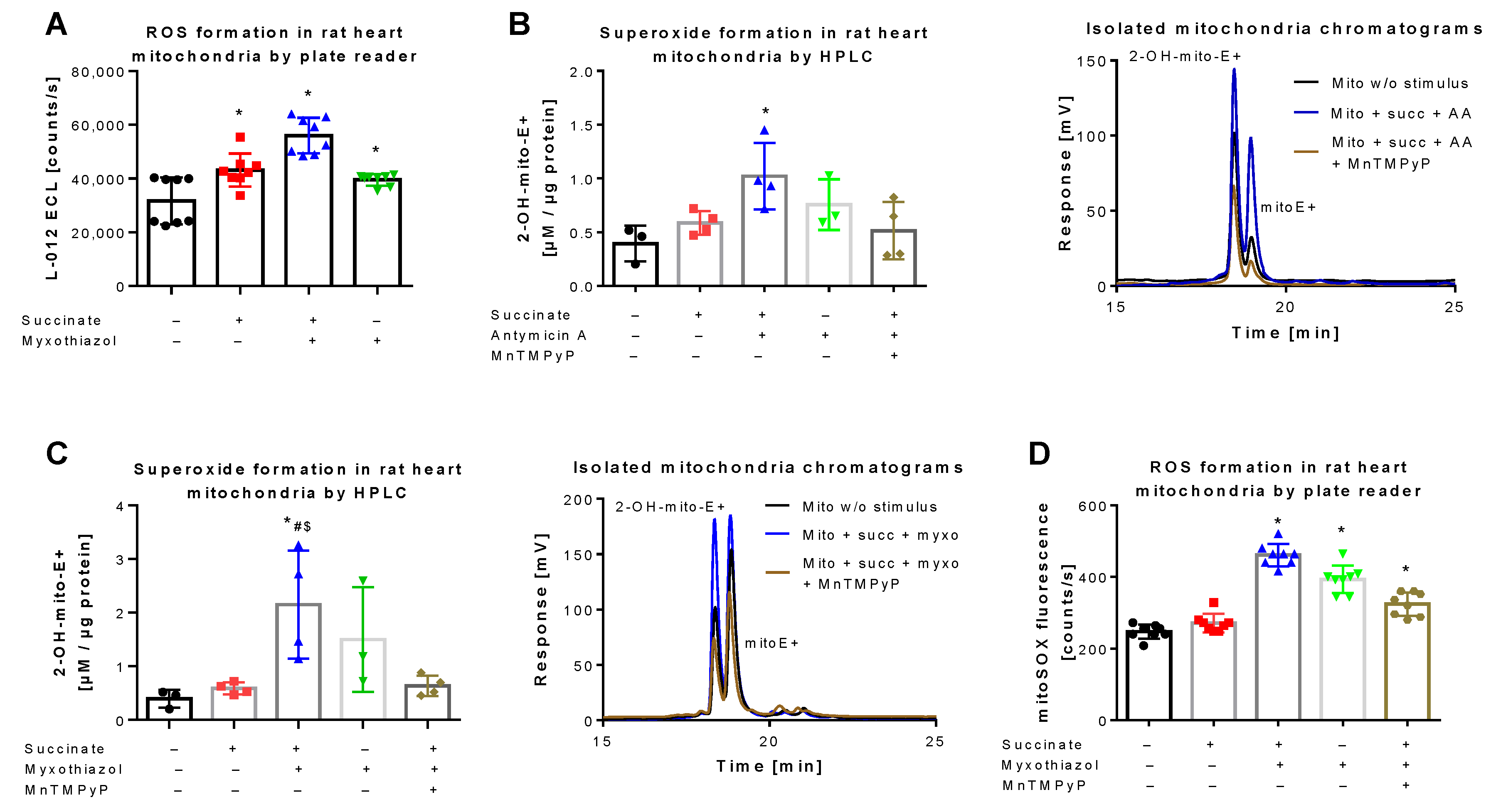
3.2. Detection of Superoxide Generation by Isolated Rat Heart Mitochondria Using mitoSOX HPLC
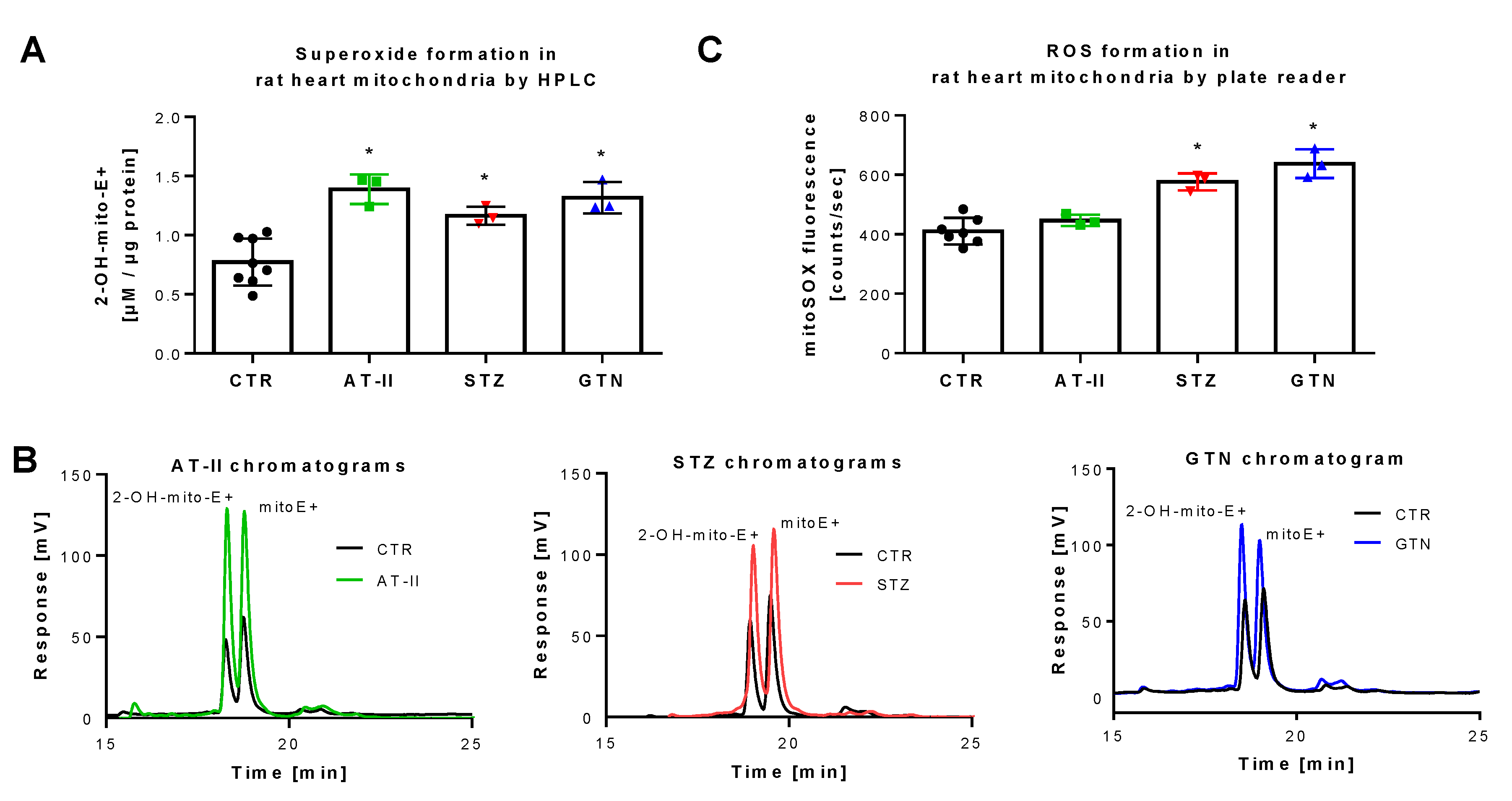
3.3. Detection of Superoxide Generation by Isolated Rat Heart Mitochondria From Rats with Preestablished Oxidative Stress Using mitoSOX HPLC
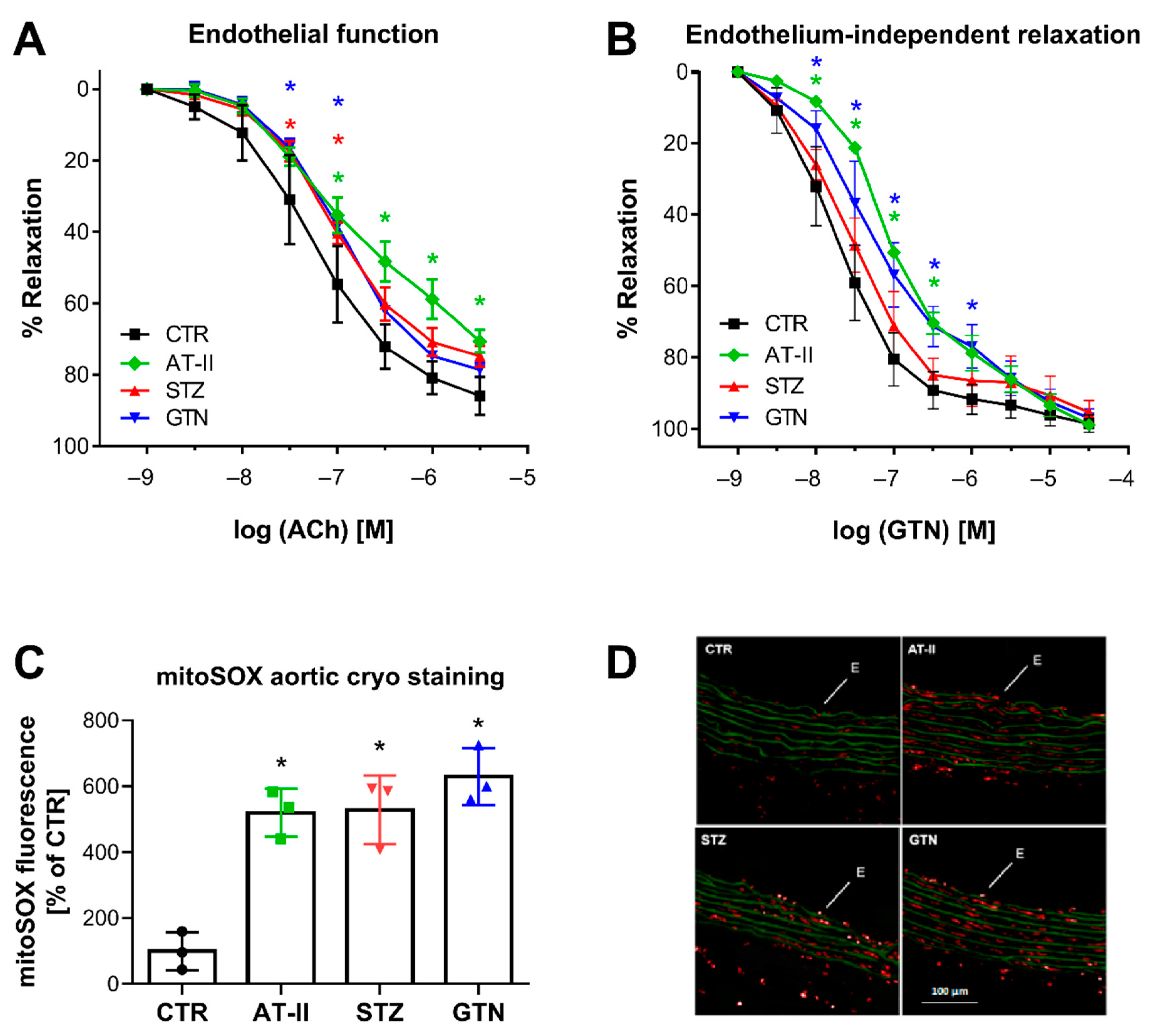
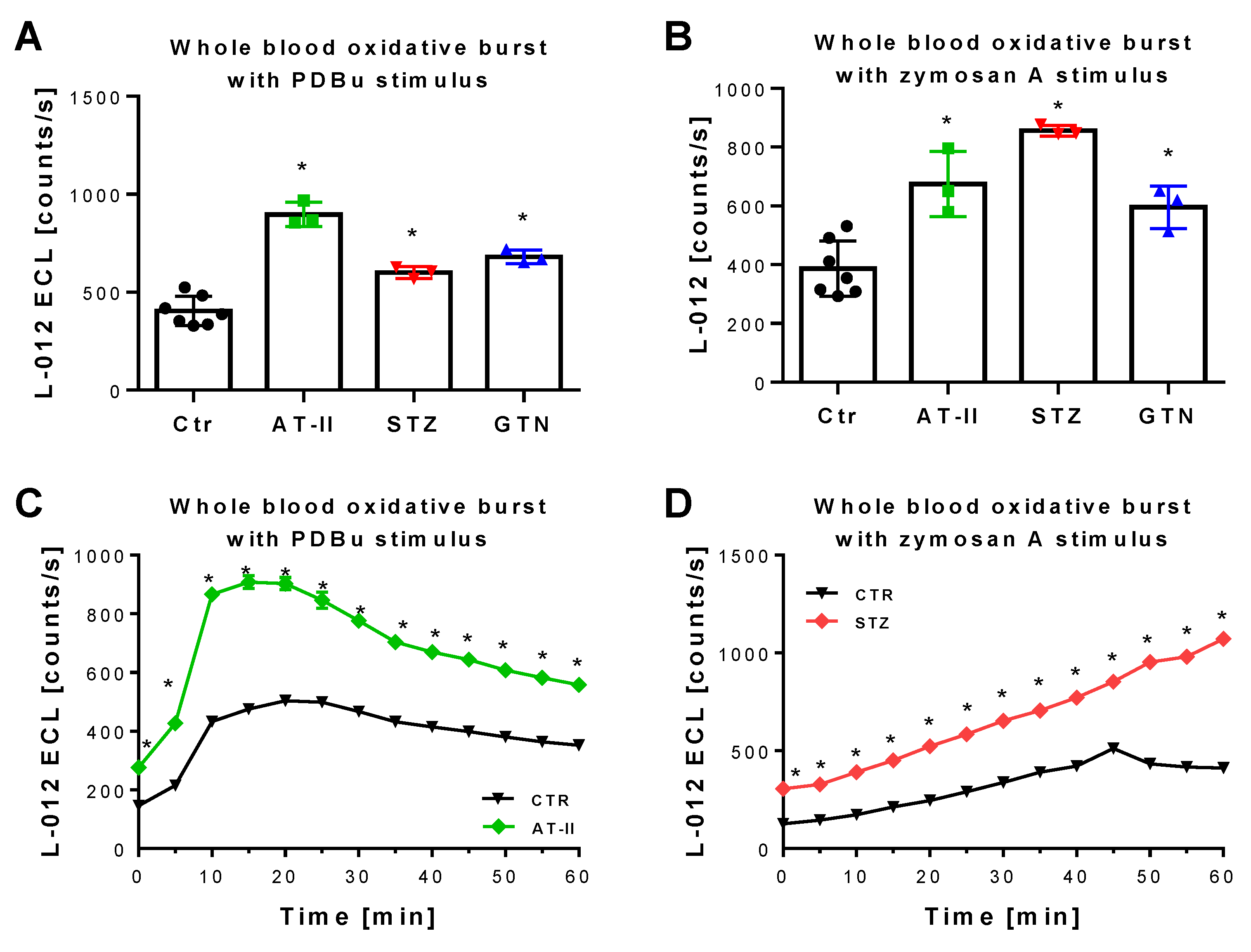
3.4. Comparison of mitoSOX HPLC Results with Vascular Function and Other Oxidative Stress Markers
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part I: Basic mechanisms and in vivo monitoring of ros. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative Stress: Oxidants and Antioxidants; Academic Press: London, UK, 1991. [Google Scholar]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Ullrich, V.; Kissner, R. Redox signaling: Bioinorganic chemistry at its best. J. Inorg. Biochem. 2006, 100, 2079–2086. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Murad, F. Regulation of cytosolic guanylyl cyclase by nitric oxide: The no-cyclic gmp signal transduction system. Adv. Pharmacol 1994, 26, 19–33. [Google Scholar]
- Rossig, L.; Fichtlscherer, B.; Breitschopf, K.; Haendeler, J.; Zeiher, A.M.; Mulsch, A.; Dimmeler, S. Nitric oxide inhibits caspase-3 by s-nitrosation in vivo. J. Biol. Chem. 1999, 274, 6823–6826. [Google Scholar] [CrossRef]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Keele, B.B., Jr.; Fridovich, I. An enzyme-based theory of obligate anaerobiosis: The physiological function of superoxide dismutase. Proc. Natl. Acad. Sci. USA 1971, 68, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Noda, Y.; Tahara, S.; Kaneko, T.; Kinoshita, N.; Hatsuta, H.; Murayama, S.; Barnham, K.J.; Irie, K.; et al. Sod1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of alzheimer disease. J. Biol. Chem. 2011, 286, 44557–44568. [Google Scholar] [CrossRef]
- Fukai, T.; Folz, R.J.; Landmesser, U.; Harrison, D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 2002, 55, 239–249. [Google Scholar] [CrossRef]
- Iida, S.; Chu, Y.; Francis, J.; Weiss, R.M.; Gunnett, C.A.; Faraci, F.M.; Heistad, D.D. Gene transfer of extracellular superoxide dismutase improves endothelial function in rats with heart failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H525–CH532. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ros: A summary of the findings and prospects for the future from the cost action bm1203 (eu-ros). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef]
- Wenzel, P.; Kossmann, S.; Munzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation-immunomodulatory function of mitochondrial and nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with nadph oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Steven, S.; Kroller-Schon, S.; Munzel, T. Taking up the cudgels for the traditional reactive oxygen and nitrogen species detection assays and their use in the cardiovascular system. Redox Biol. 2017, 12, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Schulz, E.; Oelze, M.; Muller, J.; Schuhmacher, S.; Alhamdani, M.S.; Debrezion, J.; Hortmann, M.; Reifenberg, K.; Fleming, I.; et al. At1-receptor blockade by telmisartan upregulates gtp-cyclohydrolase i and protects enos in diabetic rats. Free Radic. Biol. Med. 2008, 45, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Kroller-Schon, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinssius, E.; Agdauletova, S.; et al. The sodium-glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Oelze, M.; Daiber, A.; Brandes, R.P.; Hortmann, M.; Wenzel, P.; Hink, U.; Schulz, E.; Mollnau, H.; von Sandersleben, A.; Kleschyov, A.L.; et al. Nebivolol inhibits superoxide formation by nadph oxidase and endothelial dysfunction in angiotensin ii-treated rats. Hypertension 2006, 48, 677–684. [Google Scholar] [CrossRef]
- Hausding, M.; Jurk, K.; Daub, S.; Kroller-Schon, S.; Stein, J.; Schwenk, M.; Oelze, M.; Mikhed, Y.; Kerahrodi, J.G.; Kossmann, S.; et al. Cd40l contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res. Cardiol. 2013, 108, 386. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Sulyok, S.; Coldewey, M.; Schulz, E.; Treiber, N.; Hink, U.; Mulsch, A.; Scharffetter-Kochanek, K.; Munzel, T. Heterozygous deficiency of manganese superoxide dismutase in mice (mn-sod+/−): A novel approach to assess the role of oxidative stress for the development of nitrate tolerance. Mol. Pharmacol. 2005, 68, 579–588. [Google Scholar] [CrossRef]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.; Muller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First evidence for a crosstalk between mitochondrial and nadph oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1447. [Google Scholar] [CrossRef]
- Xiao, Y.; Meierhofer, D. Are hydroethidine-based probes reliable for reactive oxygen species detection? Antioxid. Redox Signal. 2019, 31, 359–367. [Google Scholar] [CrossRef]
- Zielonka, J.; Srinivasan, S.; Hardy, M.; Ouari, O.; Lopez, M.; Vasquez-Vivar, J.; Avadhani, N.G.; Kalyanaraman, B. Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: Identification of homo- and heterodimers. Free Radic. Biol. Med. 2008, 44, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Schuhmacher, S.; Oelze, M.; Bollmann, F.; Kleinert, H.; Otto, C.; Heeren, T.; Steven, S.; Hausding, M.; Knorr, M.; Pautz, A.; et al. Vascular dysfunction in experimental diabetes is improved by pentaerithrityl tetranitrate but not isosorbide-5-mononitrate therapy. Diabetes 2011, 60, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Knorr, M.; Schuhmacher, S.; Heeren, T.; Otto, C.; Schulz, E.; Reifenberg, K.; Wenzel, P.; Munzel, T.; Daiber, A. Vascular dysfunction in streptozotocin-induced experimental diabetes strictly depends on insulin deficiency. J. Vasc. Res. 2011, 48, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, S.; Wenzel, P.; Schulz, E.; Oelze, M.; Mang, C.; Kamuf, J.; Gori, T.; Jansen, T.; Knorr, M.; Karbach, S.; et al. Pentaerythritol tetranitrate improves angiotensin ii-induced vascular dysfunction via induction of heme oxygenase-1. Hypertension 2010, 55, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Daiber, A.; Oelze, M.; Chen, Z.; August, M.; Wendt, M.; Ullrich, V.; Mulsch, A.; Schulz, E.; Keaney, J.F., Jr.; et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J. Clin. Invest. 2004, 113, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Oelze, M.; Coldewey, M.; Bachschmid, M.; Wenzel, P.; Sydow, K.; Wendt, M.; Kleschyov, A.L.; Stalleicken, D.; Ullrich, V.; et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: A comparison of pentaerythritol tetranitrate with other organic nitrates. Mol. Pharmacol. 2004, 66, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; August, M.; Baldus, S.; Wendt, M.; Oelze, M.; Sydow, K.; Kleschyov, A.L.; Munzel, T. Measurement of nad(p)h oxidase-derived superoxide with the luminol analogue l-012. Free Radic. Biol. Med. 2004, 36, 101–111. [Google Scholar] [CrossRef]
- Kroller-Schon, S.; Daiber, A.; Steven, S.; Oelze, M.; Frenis, K.; Kalinovic, S.; Heimann, A.; Schmidt, F.P.; Pinto, A.; Kvandova, M.; et al. Crucial role for nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur. Heart J. 2018, 39, 3528–3539. [Google Scholar] [CrossRef]
- Oelze, M.; Kroller-Schon, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396. [Google Scholar] [CrossRef]
- Oelze, M.; Knorr, M.; Schell, R.; Kamuf, J.; Pautz, A.; Art, J.; Wenzel, P.; Munzel, T.; Kleinert, H.; Daiber, A. Regulation of human mitochondrial aldehyde dehydrogenase (aldh-2) activity by electrophiles in vitro. J. Biol. Chem. 2011, 286, 8893–8900. [Google Scholar] [CrossRef]
- Wenzel, P.; Schulz, E.; Gori, T.; Ostad, M.A.; Mathner, F.; Schildknecht, S.; Gobel, S.; Oelze, M.; Stalleicken, D.; Warnholtz, A.; et al. Monitoring white blood cell mitochondrial aldehyde dehydrogenase activity: Implications for nitrate therapy in humans. J. Pharmacol. Exp. Ther. 2009, 330, 63–71. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; August, M.; Wendt, M.; Sydow, K.; Wieboldt, H.; Kleschyov, A.L.; Munzel, T. Detection of superoxide and peroxynitrite in model systems and mitochondria by the luminol analogue l-012. Free Radic. Res. 2004, 38, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane nadh/nadph oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin ii-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Steven, S.; Kossmann, S.; Scholz, A.; Daub, S.; Oelze, M.; Xia, N.; Hausding, M.; Mikhed, Y.; Zinssius, E.; et al. Molecular mechanisms of the crosstalk between mitochondria and nadph oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 2014, 20, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.; Warnholtz, A.; et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001, 88, e14–e22. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.C.; Daiber, A.; Kleschyov, A.L.; Mulsch, A.; Sydow, K.; Schulz, E.; Chen, K.; Keaney, J.F., Jr.; Lassegue, B.; Walter, U.; et al. Differential effects of diabetes on the expression of the gp91(phox) homologues nox1 and nox4. Free Radic. Biol. Med. 2005, 39, 381–391. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kroller-Schon, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Godtel-Armbrust, U.; Xia, N.; et al. The sglt2 inhibitor empagliflozin improves the primary diabetic complications in zdf rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Munzel, T.; Sayegh, H.; Freeman, B.A.; Tarpey, M.M.; Harrison, D.G. Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J. Clin. Invest. 1995, 95, 187–194. [Google Scholar] [CrossRef]
- Zielonka, J.; Lambeth, J.D.; Kalyanaraman, B. On the use of l-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of nadph oxidase: A reevaluation. Free Radic. Biol. Med. 2013, 65, 1310–1314. [Google Scholar] [CrossRef]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef]
- Casas, A.I.; Dao, V.T.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive oxygen-related diseases: Therapeutic targets and emerging clinical indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in translational medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Cross talk between mitochondria and nadph oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Munzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ros. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in nlrp3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Swalwell, H.; Latimer, J.; Haywood, R.M.; Birch-Machin, M.A. Investigating the role of melanin in uva/uvb- and hydrogen peroxide-induced cellular and mitochondrial ros production and mitochondrial DNA damage in human melanoma cells. Free Radic. Biol. Med. 2012, 52, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Hasko, G.; Hawkins, B.J.; Madesh, M.; Pacher, P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat. Protoc. 2007, 2, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species (Apex) 2016, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kroller-Schon, S.; Jansen, T.; Schuler, A.; Oelze, M.; Wenzel, P.; Hausding, M.; Kerahrodi, J.G.; Beisele, M.; Lackner, K.J.; Daiber, A.; et al. Peroxisome proliferator-activated receptor gamma, coactivator 1alpha deletion induces angiotensin ii-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.I., 3rd; Han, Z.; Presley, T.; Varadharaj, S.; Zweier, J.L.; Ilangovan, G.; Alevriadou, B.R. Endothelial cell respiration is affected by the oxygen tension during shear exposure: Role of mitochondrial peroxynitrite. Am. J. Physiol. Cell Physiol. 2008, 295, C180–C191. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kholmukhamedov, A.; Lindsey, C.C.; Beeson, C.C.; Jaeschke, H.; Lemasters, J.J. Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: Protection by starch-desferal and minocycline. Free Radic. Biol. Med. 2016, 97, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyanaraman, B.; Dranka, B.P.; Hardy, M.; Michalski, R.; Zielonka, J. Hplc-based monitoring of products formed from hydroethidine-based fluorogenic probes--the ultimate approach for intra- and extracellular superoxide detection. Biochim. Biophys. Acta 2014, 1840, 739–744. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and mitosox-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef]
- Robinson, K.M.; Janes, M.S.; Beckman, J.S. The selective detection of mitochondrial superoxide by live cell imaging. Nat. Protoc. 2008, 3, 941–947. [Google Scholar] [CrossRef]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Dias Duarte Machado, L.G.; Thiebaut, D.; Ouari, O.; et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J. Biol. Chem. 2018, 293, 10363–10380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zou, M.H. Measurement of reactive oxygen species (ROS) and mitochondrial ROS in AMPK knockout mice blood vessels. Methods Mol. Biol. 2018, 1732, 507–517. [Google Scholar] [PubMed]
- Fraccarollo, D.; Galuppo, P.; Motschenbacher, S.; Ruetten, H.; Schafer, A.; Bauersachs, J. Soluble guanylyl cyclase activation improves progressive cardiac remodeling and failure after myocardial infarction. Cardioprotection over ace inhibition. Basic Res. Cardiol. 2014, 109, 421. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinovic, S.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Vujacic-Mirski, K.; Kvandová, M.; Schmal, I.; Al Zuabi, A.; Münzel, T.; Daiber, A. Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress. Antioxidants 2019, 8, 514. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8110514
Kalinovic S, Oelze M, Kröller-Schön S, Steven S, Vujacic-Mirski K, Kvandová M, Schmal I, Al Zuabi A, Münzel T, Daiber A. Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress. Antioxidants. 2019; 8(11):514. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8110514
Chicago/Turabian StyleKalinovic, Sanela, Matthias Oelze, Swenja Kröller-Schön, Sebastian Steven, Ksenija Vujacic-Mirski, Miroslava Kvandová, Isabella Schmal, Ahmad Al Zuabi, Thomas Münzel, and Andreas Daiber. 2019. "Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress" Antioxidants 8, no. 11: 514. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox8110514