Antioxidant Therapies in Traumatic Brain Injury



 , , ,
, , , 
Abstract
:1. Introduction
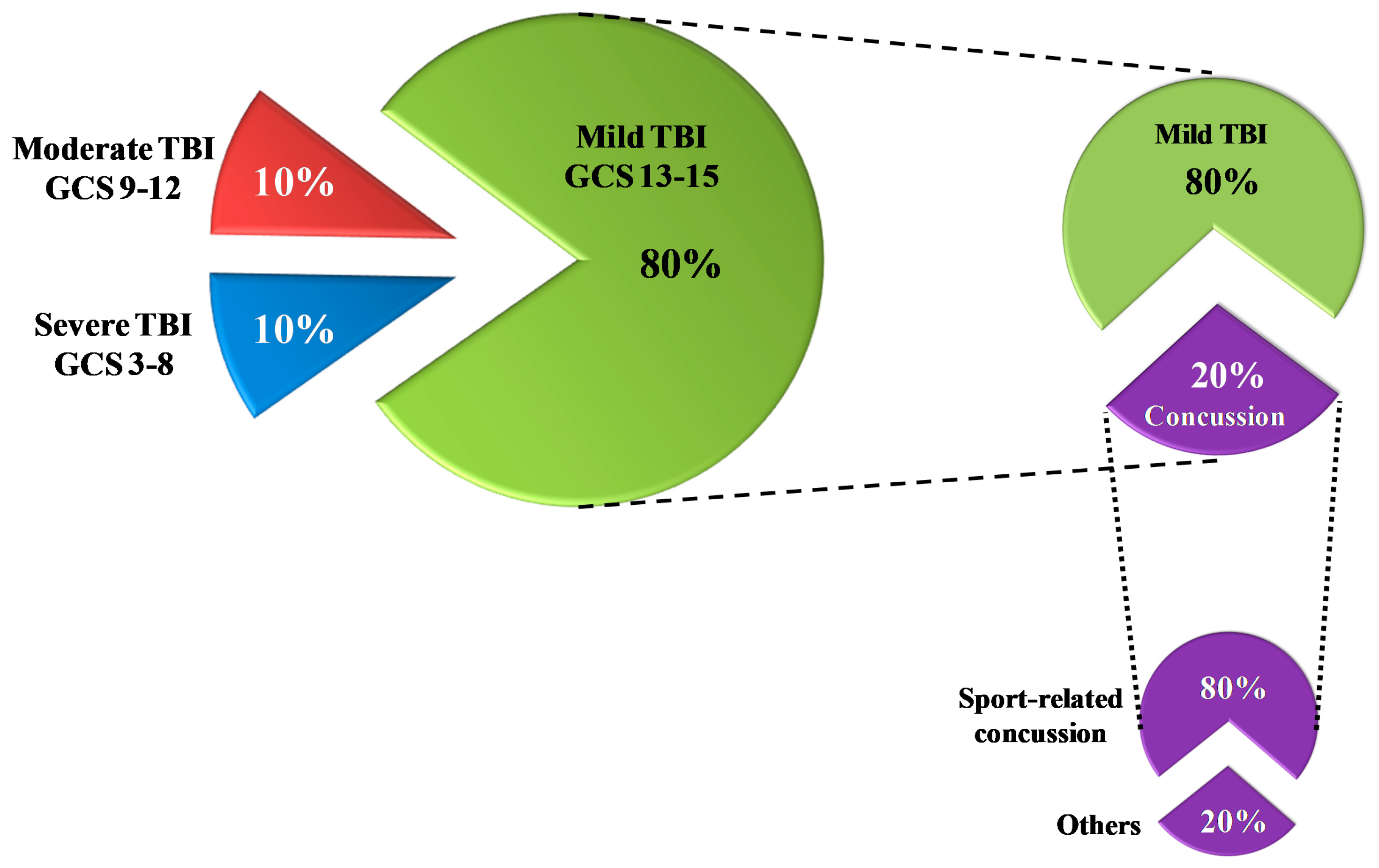
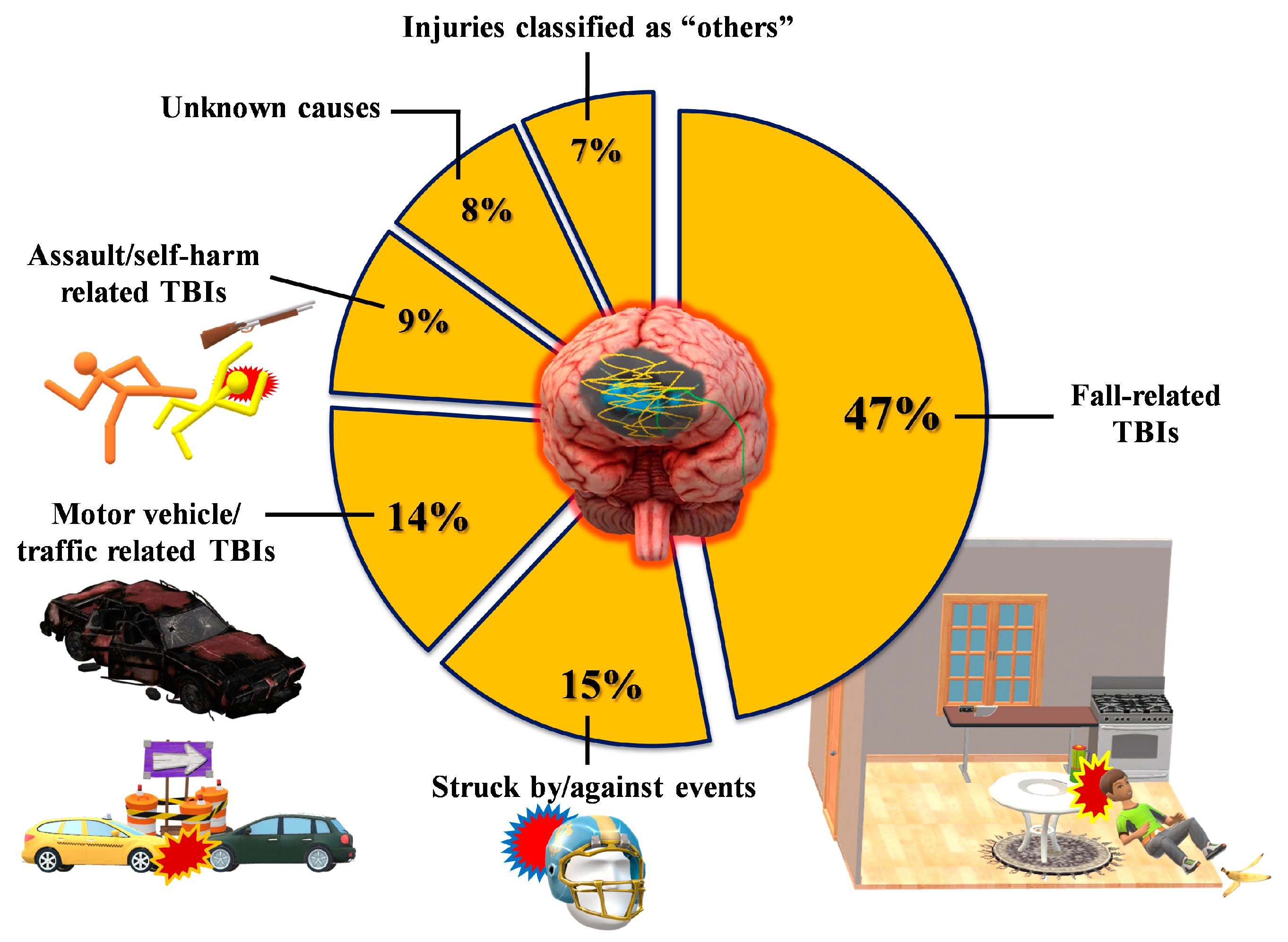
1.1. Definition/Classification
1.2. Triage
1.3. Therapy
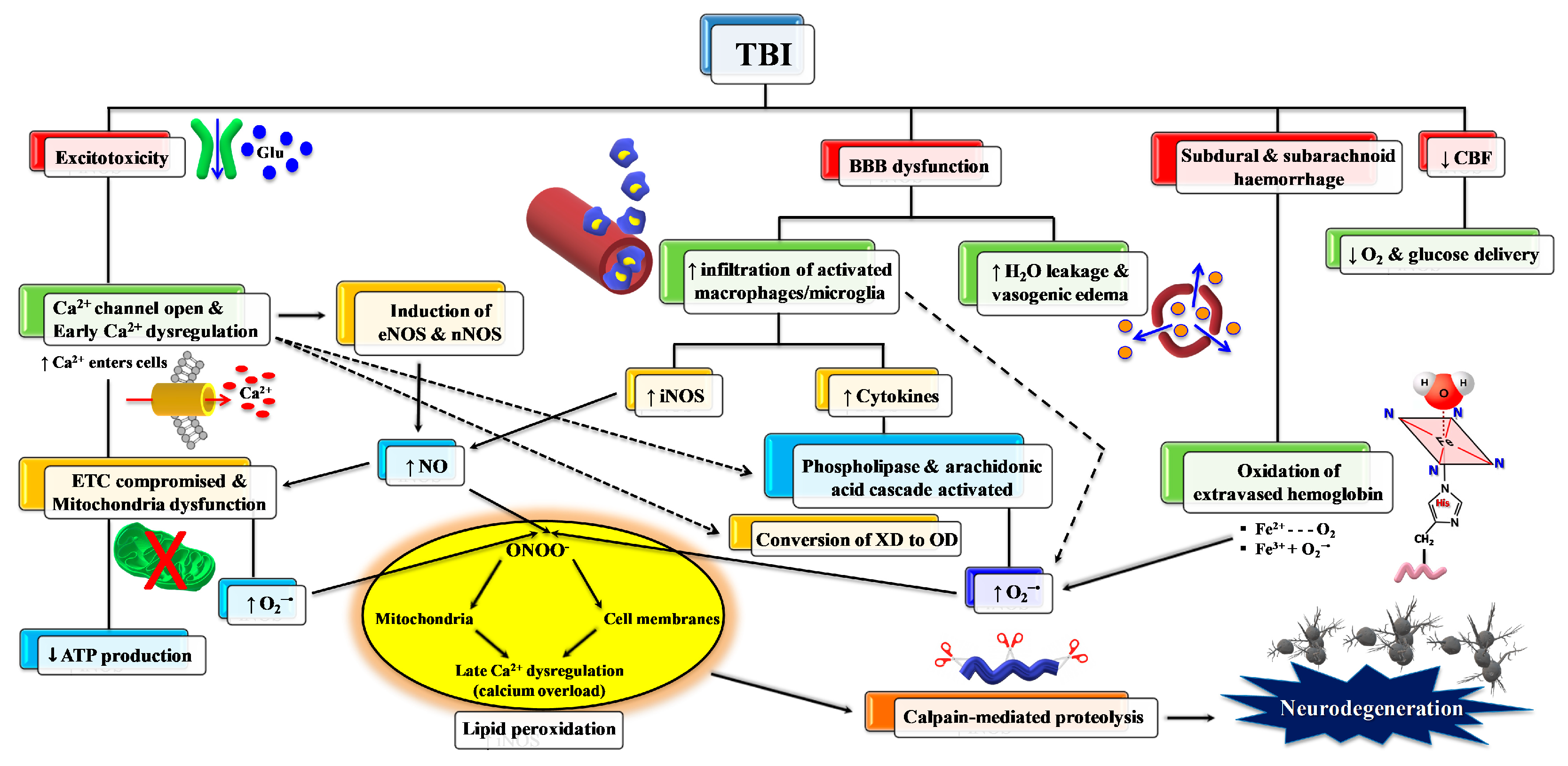
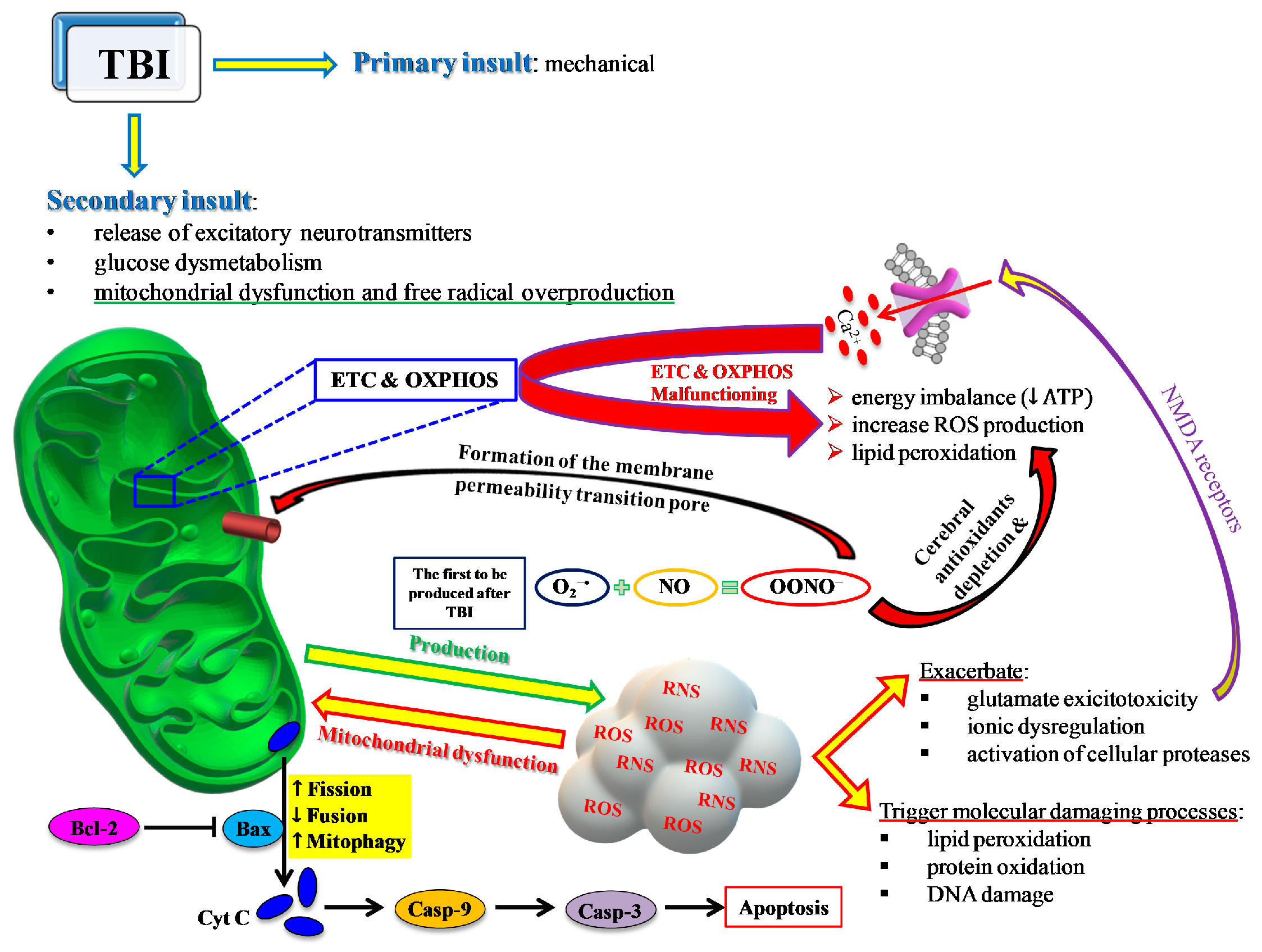
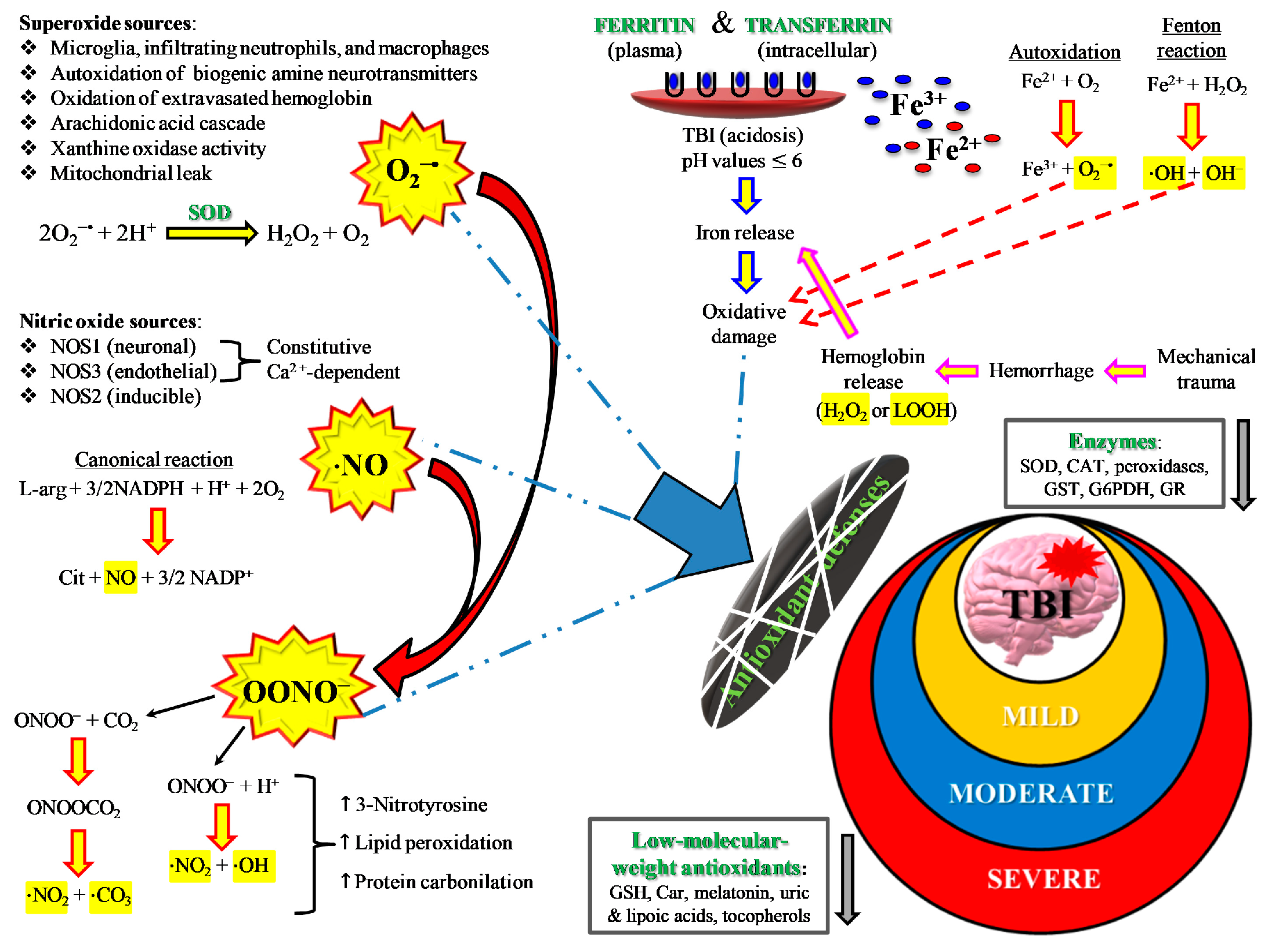
2. TBI and Oxidative/Nitrosative Stress: A Rationale for Antioxidant-Based Therapies
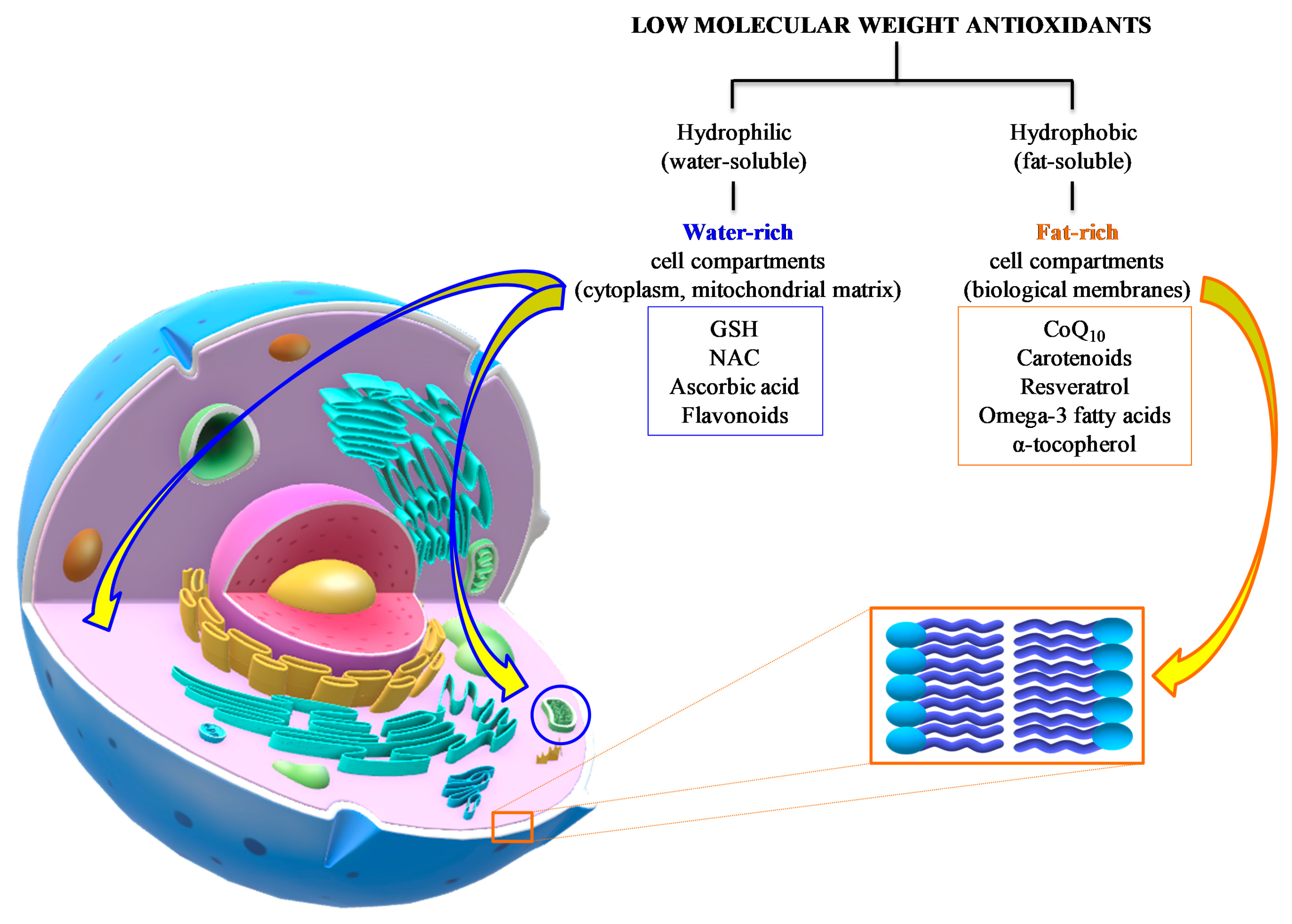
3. Low Molecular Weight Antioxidants in TBI: Summary of Preclinical and Clinical Studies
3.1. Ascorbic Acid (Vitamin C)
3.2. N-Acetyl-Cysteine
3.3. Flavonoids
3.4. Resveratrol
3.5. α-Tocopherol (Vitamin E)
3.6. Coenzyme Q10
3.7. Carotenoids
3.8. Omega-3 Fatty Acids
4. Discussion
Antioxidant Therapies in Sports-Related Concussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Ascorbic Acid |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AQP4 | Aquaporin-4 |
| BBB | Blood–Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CBF | Cerebral Blood Flow |
| CBV | Cerebral Blood Volume |
| CCI | Controlled Cortical Impact |
| COX | Cyclooxygenase |
| CSF | Cerebro-Spinal Fluid |
| CT | Computed Tomography |
| CTE | Chronic Traumatic Encephalopathy |
| DHA | Docosohexaenoic Acid |
| DHE | Dihydroethidium Staining |
| DRP1 | Dynamin-Related Protein 1 |
| EEG | Electroencephalography |
| eNOS | Endothelial Nitric Oxide Synthase |
| EPA | Eicosapentaenoic Acid |
| ETC | Electron Transfer Chain |
| FIS1 | Fission Protein 1, mitochondrial |
| FJB/C | Fluoro Jade B or C staining |
| FST | Forced Swimming Test |
| FPI | Fluid Percussion Injury |
| GAP-43 | Growth-Associated Protein-43 |
| GCS | Glasgow Coma Scale |
| GFAP | Glial Fibrillary Acidic Protein |
| GPx | Glutathione Peroxidases |
| GSH | Reduced Glutathione |
| GSK-3β | Glycogen Synthase Kinase 3 Beta |
| GSNA | Greater Splanchnic Nerve Activity |
| GSNO | Nitrosoglutathione |
| GR | GSH Reductase |
| GSSG | Oxidized Glutathione |
| Hb | Haemoglobin |
| H&E | Hematoxylin and Eosin staining rotocol |
| ICH | Immunohistochemistry |
| ICP | Intracranial Pressure |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| i.m. | Intramuscular |
| i.v. | Intravenous |
| lncRNA Neat1 | Long noncoding RNA nuclear enriched abundant transcript 1 |
| MDA | Malondialdehyde |
| MFN1 or 2 | Mitofusin 1 or 2 |
| MINO | Minocycline |
| mNSS | Modified Neurological Severity Score |
| mTBI | Mild Traumatic Brain Injury |
| MWM | Morris Water Maze |
| NAC | N-Acetylcystein |
| NFL | Neurofilament Light |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NICUs | Neurosurgical Intensive Care Units |
| NKCC | Sodium–potassium–chloride cotransporter |
| NMDA | N-Methyl-D-Aspartate |
| nNOS | Neuronal Nitric Oxide Synthase |
| NO | Nitric oxide |
| NOR | Novel Object Recognition |
| NOS | Nitric Oxide Synthases |
| NOX | NADPH Oxidase |
| NP | Neuropsychological |
| Nrf2-ARE | Nuclear factor E2-related factor 2 Antioxidant Response Elements |
| NSS | Neurological Soft Signs |
| OPA1 | Optic dominant Atrophy 1 |
| OXPHOS | Oxidative Phosphorylation |
| PARK2 | Parkin |
| PAT | Passive Avoidance Task |
| PEG | Polyethylene Glycol |
| PINK1 | mitochondrial serine/threonine-protein kinase 1 |
| PTSD | Posttraumatic Stress Disorder |
| PUFA | Polyunsaturated Fatty Acids |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SIS | Second Impact Syndrome |
| SOD | Superoxide Dismutase |
| SVCT1 or 2 | Sodium dependent vitamin C transporter 1 or 2 |
| SYP | Synaptophysin |
| TAC | Total Antioxidant Capacity |
| TBARS | Thiobarbituric Acid Reactive Substance |
| TBI | Traumatic Brain Injury |
| TEM | Transmission Electron Microscopy |
| TST | Tail Suspension Test |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling assay |
| UCH-L1 | Ubiquitin Carboxy-terminal Hydrolase-L1 |
| VCS | Veterinary Coma Scale |
| VR | Virtual Reality |
| WB | Western Blot |
| WD | Weight Drop |
| XO | Xanthine Oxidase |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Flavonoid | Dosage and Route of Administration | Time Points | Tests and Assays | TBI Model | Main Findings | Ref |
|---|---|---|---|---|---|---|
| Chrysin | 25, 50, or 100 mg/kg orally, started immediately post-injury and continued for up to 3 or 14 days | 1 day before injury; 0, 1, 4 h, and 1, 2, 3, 4, 7, 13, 14, 28 days post-injury | VCS, rotarod test, PAT, biochemical assay, histochemical staining, TUNEL, IHC | WD | Chrysin enhanced post-injury motor function, cognitive status and neuronal loss via reduction of oxidative stress (increased concentrations of SOD, CAT, GPx, GSH, and decreased MDA level) and inhibition of the apoptotic proteins (decreased Bax and increased Bcl-2) in the cerebral cortex and CA3 hippocampal neurons | [133] |
| Wogonin | 1, 2.5, 5 mg/kg. (i.v.) before and after moderate TBI; Alternatively, 2 µL (10 mM) injected into the lateral cerebral ventricle (i.c.v.) before and after moderate TBI | Before injury and 0.5, 1, 2, 4 h post-injury | Arterial pressure, heart rate, baroreflex and GSNA, histochemical staining of hippocampus | FPI | Wogonin administered before or after TBI significantly improved the cardiovascular changes (MAP, HR, and baroreflex) occurring after FPI. | [134] |
| Isoliquiritigenin (ILG) | 20 mg/kg (i.p.) 1 h post-TBI | 1 and 7 days after TBI | Sensorimotor Garcia test, brain water content, BBB permeability assay, histochemical staining, cell viability assay, WB, RT–qPCR, fluorescence immunoassays | CCI | ILG improved neurologic functions by reducing brain edema, BBB permeability, apoptosis (decrease in expression of cleaved caspase 3), and oxidative stress (translocation of Nrf2 into the nucleus with activation of downstream proteins). | [135] |
| Hesperidin | 50 mg/kg orally from day 10 to 24 post-injury | Day 21 and 24 post-injury | Sucrose preference test, FST, suppressed feeding test, TST, biochemical analysis | WD | Hesperidin reduced depression symptoms, levels of IL-1β, TNF-α and MDA, and increased BDNF levels in the hippocampus | [136] |
| Icariin | Oral administration of 3, 10, 30 mg/kg on injury induction and daily for 7 days post-injury | 0, 3, 7, 8 days post-TBI | Rotarod test, balance beam test, Y-maze, WB, IHC, histochemical staining, protein expression | Modified CCI | Icariin improved sensory motor and cognitive functions, and upregulated BDNF, SYP, and PSD-95 after injury. However, no improvement of brain histology and neuronal death were found. | [137] |
| Baicalin | 50, 100, 150 mg/kg (i.p.) 30 min after TBI | 2 h, and 1 and 3 days post-TBI | NSS, brain water content, TUNEL assay, WB Immunostaining, RT-qPCR | WD | Baicalin improved neurological function, brain edema, apoptosis, and oxidative stress via activating the Akt/Nrf2 pathways | [138] |
| Formononetin | Intragastrical administration of 10, 30 mg/kg/die for up to 7 days after TBI | 7 days post-injury | ELISA of tissue and serum cytokines, Cytohistologic stains, RT-qPCR, WB | WD | Formononetin counteracted TBI-induced neuroinflammation by decreasing tissue and serum levels of IL-6 and BDNF, and increasing tissue and serum levels IL-10 | [139] |
| Troxerutin | 1.5 mL/kg (i.p.) for 5 days before TBI | 3 days post-injury | NSS, MRI, ELISA, Nissl, TEM, WB and Immunostaining | WD | Troxerutin improved neurovascular function and integrity post-injury through action on eNOS and NO level, with consequent decrease in peroxynitrite formation | [140] |
| Quercetin | 50 mg/kg (i.p.) at 30 min, 12 h and 24 h post-TBI | 1, 3, 5 days post-injury | Brain water content, NSS, H&E staining and neuron count, IHC, WB | WD | Reduced brain edema, neural apoptosis and improved motor function (inhibition of extracellular signal-regulated kinase 1/2 phosphorylation and activated Akt serine/threonine protein kinase phosphorylation) | [141] |
| Resveratrol | 0.05, 0.1 mg/kg via oral gavage for 10 consecutive days, 7 days after TBI | 7 days post-injury | MWM, WB, TAC, Apoptosis, DHE staining | CCI | Reduced cognitive deficits, ROS generation, and apoptosis after TBI via recovered activation of p38/Nrf2/HO1 signaling pathway | [142] |
| Silymarin | 50 mg/kg via gavage for 20 days before injury | 8–10, 11, 18–20, 21 post-injury | Open field, elevated plus-maze, light-dark box, elevated zero-maze, sucrose preference, FST, TST, TNF-α | WD | Decreased anxiety and depression-like behaviours after injury due to reduced TNF-α levels in the prefrontal cortex and hippocampus | [143] |
| Diosmin | 100 mg/kg (p.o.) for 7 days before TBI | –1 h, 1 h, 1,2,15days post-injury | VCS, passive avoidance memory, BBB permeability, brain edema, ELISA | WD | Protective effects against TBI-induced memory and long-term potentiation impairment through reduction of TNF-α concentration in hippocampus | [144] |
| Catechin | 1, 5, 10, 20 or 30 mg/kg daily via gavage up to 28 days post-TBI | 3, 5, 7, 14, 21, 28 days pot-injury | Brain infarct volume edema, foot-fault test, MWM, BBB permeability, RT-qPCR, WB | CCI | Catechin prevented tight junction disruption and preserved BBB integrity, reducing post-injury inflammatory reaction | [145] |
| 7,8-dihydroxyflavone | 5 mg/kg (i.p) post-TBI either had access to voluntary wheel running for 7 days after injury or were sedentary | 7, 14 days post-injury | Barnes maze, voluntary running wheel exercise, WB, rsfMRI | FPI | 7,8-DHF enhanced the levels of cell energy metabolism (COII, PGC-1α, AMPK) and hippocampal functional connectivity | [146] |
| Pycnogenol | 50, 100 mg/kg (i.p.) 15 min, 3 h, 6 h post-TBI | 2, 5, 7, 12 days post-injury | MWM, FJB, cortical tissue sparing | CCI | No improvement of cognitive ability post-injury in MWM maze task. Pycnogenol suppressed NO through the inhibition of iNOS and also the NF-kB/AP-1 pathway. | [147] |
| Breviscapine | 50 mg/kg (i.v.) post-TBI | 1, 4, 7, 14, 21 days post-injury | NSS, RT-PCR, WB, IHC, Immunostaining, TUNEL | WD | Neurobehavioral function improved after treatment due to GSK3β signaling pathway inhibition | [148] |
| Hydroxysafflor yellow A | 10, 30 mg/kg orally post-TBI | 6 h, 12 h, 24 h post-TBI | Detection of HSYA, SOD, MDA, CAT, GSH/ GSSG | CCI | HSYA reduced oxidative stress by improving the activities of SOD and CAT, the level of GSH, and the GSH/GSSG ratio. Additionally, it decreased the levels of MDA and GSSG | [149] |
| Genistein | 15 mg/kg (i.p.) 30 min and again 24 h after TBI | –1, 1, 2 days post-TBI | Brain oedema, BBB permeability, ICP, VCS, beam-walk task | WD | Genistein inhibited brain edema, BBB permeability, and improved ICP after TBI. It also improved neurobehavioral performance and motor disorder | [150] |
| Epicatechin | 5, 15, 45 mg/kg by gavage at 3 h after TBI and once daily for 3 days or 15 mg/kg EC at 3 h after TBI and then once daily for 7 days | 1, 2, 3, 7, 14, 21, 28 days post-TBI | Neurologic deficit score, forelimb placing test, wire-hanging test, rotarod test, TST, FST, sucrose preference test, IHC, brain water content, Hb, WB, Immunostaining | CCI | EC significantly reduced lesion volume, edema, and cell death and improved neurologic function on days 3 and 28. Cognitive performance and depression-like behaviors were also improved by activating the Nrf2 pathway, inhibiting heme oxygenase-1 protein expression, and reducing iron deposition | [151] |
| Procyanidins | 100 mg/kg (i.v.) PC within 30 min post-TBI | 24 h, and 11, 12, 13, 14 days post-injury | MWM, MDA, GSH, SOD, ELISA, WB | CCI | Procyanidins improved cognitive performance by reducing the level of MDA, increasing GSH and activity of SOD, elevating the levels of BDNF, phosphorylation-cAMP-response element-binding protein (pCREB), total CREB, and cyclic AMP (cAMP) | [152] |
| Proanthocyanidin | Not mentioned | 72 h post-injury | Cerebral water content, TBARS, nitrite and nitrate, Thiols | Cold injury | Proanthocyanidin attenuated oxidative and nitrosative stress and decreased brain edema | [153] |
| (–)-epigallocatechin gallate (EGCG) | 0.1% (w/v) EGCG pre- and post-TBI. Solution was prepared by dissolving the drug in drinking water | 1, 3, and 7 days post-TBI | MWM, IHC, immunostaining for ssDNA and NeuN, lipid peroxidation | CCI | EGCG treatments improved cognitive impairment through inhibiting free radical-mediated neuronal degeneration and apoptotic cell death around the area damaged by TBI. | [154] |
| Puerarin | 200 mg/kg (i.p.) before injury | 24 h post-injury | WB, MDA, GSH, Naþ-Kþ-ATPase activity, Myeloperoxidase activity, FJC | WD | Puerarin ameliorated oxidative neurodegeneration after TBI through the activation of PI3K-Akt pathway | [155] |
| Luteolin | In vivo 10, 30, 50 (i.p.) mg/kg post-TBI In vitro 5, 10, and 25 µM post-TBI | 1, 3, 7 days post-TBI | Grip test, brain water content, MDA, GPx activity, TUNEL, IHC, WB, nuclear extraction and electrophoresis mobility shift assay, RT-qPCR, cell viability | WD | Luteolin enhanced the translocation of Nrf2 to the nucleus both in vivo and in vitro, upregulation of heme oxygenase 1 (HO1) and NAD(P)H:quinone oxidoreductase 1 (NQO1). Luteolin neuroprotective effects are possibly mediated by the activation of the Nrf2–ARE pathway | [156] |
| Naringin | 100 mg/kg orally 7 days before and 7 days after the TBI | 7 days post-injury | NSS, brain water content, serum and tissue biochemical analysis, WB | WD | Naringin improved behavioral dysfunction by attenuating the increases in MDA and NO; enhancing the activation of SOD; decreasing the over-activation of iNOS; down-regulating the overexpression of IL-1b; and reducing the brain edema. | [157] |
| Baicalein | 30 mg/kg (i.p.) immediately following injury or daily for 4 dayspost-injury | Before injury, 1, 4, 7, 14, 21 days after injury | Rotarod test, y test, mNSS, beam walk test, RT-PCR, WB, IHC, ELISA, FJB | CCI | Baicalein attenuated the contusion’s site expression of TNF-α, IL-1β and IL-6 mRNA and cytokine protein. | [158] |
| Dosage and Route of Administration | Time Points | Tests and Assays | TBI Model | Main Findings | Ref |
|---|---|---|---|---|---|
| DHA: 500 nmol/kg (i.v.) 30 min post-injury | 7, 28 days post-injury | mNSS, MRI, ICH | CCI | Reduced lesion size, microglia, and astrocytic reactivity, decreased the accumulation of beta-amyloid precursor protein (APP) at 7 days post-TBI. Reduced the neurofilament light (NFL) levels in plasma at 28 days. | [159] |
| DHA: 7.5 mg/100 mL (i.p.) 30 min after TBI | 1, 3, 7, 14 days post-injury | Brain nitrates and nitrites (NOx), NOR, markers of microglial activation, astrocyte marker, mRNA of inflammation-related genes | CCI | Decreased oxidative stress at day 1 and pro-inflammatory microglial activation at day 3. Decreased oxidative stress, histologic damage, and mRNA markers of microglial pro-inflammatory activation. Improved short term cognitive function. | [160] |
| NPD 1:50 ng intra-lesionally, immediately following TBI | 1,3 days post-injury | FJB, TUNEL, Immuno-staining and lesion size analyses | PBI | NPD1 decreased the lesion area at 72 h | [161] |
| DHA: 370, 550, 740 mg/kg/day, intragastric administration 30 min after TBI. | 1, 4, 7, 14, 21 days post-injury | NSS, MWM TUNEL, Nrf2-ARE pathway-related genes | FPI | Improved neuromotor and cognitive functions, increased anti-apoptotic protein expression, SOD and GPx activity, translocation of Nrf2 to the nucleus. Increased the expression of the downstream factors NAD(P)H:quinone oxidoreductase (NQO-1) and HO-1. Neuroprotective potentially mediated through activating the Nrf2- ARE pathway. | [162] |
| DHA: 16 mg/kg (i.p) at 30 min, 1, 3, 5 days after TBI | 1, 2, 3, 4, 5 days post-injury | NSS, beam walking test and rotarod test, qRT-PCR, WB, TUNEL, IHC | CCI | Improved neurological and cognitive functions. Decreased apoptosis, TLR4 expression, and the expression of inflammatory mediator NF-Kappa B. | [163] |
| DHA: 370, 740 mg/kg/day (i.v.) 30 min after TBI and daily for 15 days | 2, 7 and 15 days post-injury | Beam-walking, MWM, RT-qPCR | FPI | Protection against motor deficits. Inhibition of caspase-3 upregulating the Bcl-2:Bax ratio. | [164] |
| Resveratrol 50 mg/L via drinking water; Omega3 fatty acids and prebiotics were administered via powdered food (100 g of prebiotic, 300 g of DHA, and 600 g of standard diet per 10 kg of food) for 43 days before TBI. | 43–60 days | Neurological test battery, expression of Aqp4, Gfap, Igf1, Nfl, Sirt1, and Tau genes | Focal closed- head | Treatment altered the behavioral performance and prevented injury-related deficits in the longer-term behavior measures, Decreased expression of Aqp4, Gfap, Igf1, Nfl, and Sirt1in the prefrontal cortex. | [165] |
| DHA: 16 mg/kg (i.p.) at 5 min, 3 to 21 days, after TBI | 3, 7, 21 days post-injury | Activation of microglia or macrophages, inflammatory response, neurons expression of the endoplasmic reticulum (ER) stress marker CHOP | CCI | DHA administration reduced neuronal ER stress and subsequent association with microglial or macrophage polarization after TBI. Potential amelioration of TBI-induced cellular pathology | [166] |
| DHA: 0.1% diet. 1 d before TBI and for 50 days after TBI | 1, 2, 3, 12, 28, 41, 47, 50 days post-injury | Inflammatory cytokines, nitrates, and nitrites, MWM, T2-weighted MRI, diffusion tensor imaging, histological exams | CCI | Decreased cognitive impairment, oxidative stress, and white matter injury in adult rats | [167] |
| ω-3 PUFA: 2 mL/kg (i.p.) 30 min after TBI and daily for 7 days | 1, 3, 7 days post-injury | mNSS, brain water content, NISSl staining, microglial activation, immunofluorescent staining, WB, TLR4/NF-κB | Feeney DM TBI model | ω-3 PUFA supplementation inhibited TBI-induced microglial activation and the subsequent inflammatory response by regulating HMGB1 nuclear translocation and secretion and also HMGB1-mediated activation of the TLR4/NF-κB signaling pathway, leading to neuroprotective effects. | [168] |
| DHA: 16 mg/kg (i.p.) 15 min after the injury and daily for 3 or 7 days. | 3, 7 days post-injury. | RT–qPCR, immunoblotting, immunostaining, DTI, MWM | CCI | DHA administration restored hippocampal lysosomal biogenesis and function, demonstrating its therapeutic potential. | [169] |
| Diet containing 10% corn oil with a fatty acid profile high in ω-6 PUFAs and low in ω-3 PUFAs for 3 days before injury | 3, 6, 12 h | Two-photon laser scanning microscopy (2PLSM), parenchymal cell death and reactive oxidative species (ROS), (GSH), neuroprotectin D1 (NPD1) | Closed-head | Neuroprotection by decreasing cell death, ROS formation, and preserving GSH and NPD1 expression. | [170] |
| DHA (1.2%) enriched diets and/or curcumin (500 ppm) for 2 weeks post-injury | 1, 2, 3, 4, 5 days and 2 weeks | Cognitive tests, WB, markers of lipid peroxidation | FPI | DHA alone or in combination improved cognitive functions, decreased oxidative stress and damage to membrane phospholipids. Curcumin complemented the action of DHA on TBI pathology | [171] |
| DHA = 1.2% of diet coupled with exercise for 12 days | 12 days | Expression of acyl-CoA oxidase 1 (Acox1), 17β-hydroxysteroid dehydrogenase type 4 (17β-HSD4), calcium-independent phospholipases A2 (iPLA2), syntaxin-3 (STX-3), and BDNF | FPI | DHA activity was synergistic with exercise. The effects were evident on restoration of membrane homeostasis after TBI, thus supporting synaptic plasticity and cognition. | [172] |
| Control diet: unhydrogenated soybean oil (70 g/kg). Deficient diet: safflower oil (66.5 g/kg) and soybean oil (3.5 g/kg). | 1, 7, 14, 21, and 28 days after TBI | Ccl2, Gfap, and Mmp 9 mRNA levels, MMP-2 and -9 enzymatic activities, lesion volume | CCI | Decreased brain DHA content in TBI rats fed with deficient diet contributed to poorer sensorimotor outcomes after TBI through a mechanism involving modulation of Timp1 expression. | [173] |
| DHA: 3, 12, 40 mg/kg diet for 30 days prior TBI | 1 week after injury | WMW, IHC | WD | Dietary supplementation with DHA increased DHA serum levels, reduced TBI-associated tissue damage (axonal injury counts, markers for cellular injury and apoptosis), and improved memory assessment by the water maze testing. | [174] |
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. InTBIR Participants and Investigators. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- World Health Oraganization. Neurological Disorders: Public Health Challenges. 2006. Available online: https://www.who.int/mental_health/neurology/chapter_3_b_neuro_disorders_public_h_challenges.pdf?ua=1 (accessed on 19 July 2019).
- Carroll, L.J.; Cassidy, J.D.; Holm, L.; Kraus, J.; Coronado, V.G. WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. Methodological issues and research recommendations for mild traumatic brain injury: The WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J. Rehabil. Med. 2004, 43, 113–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Gardner, A.J.; Zafonte, R. Neuroepidemiology of traumatic brain injury. Handb. Clin. Neurol. 2016, 138, 207–223. [Google Scholar] [PubMed]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Web-based Injury Statistics Query and Reporting System (WISQARS) (2003). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. Available online: www.cdc.gov/injury/wisqars (accessed on 19 July 2019).
- Greenberg, M.S. Handbook of Neurosurgery, 8th ed.; Thieme: New York, NY, USA, 2016; pp. 825–830. [Google Scholar]
- Kolias, A.G.; Guilfoyle, M.R.; Helmy, A.; Allanson, J.; Hutchinson, P.J. Traumatic brain injury in adults. Pract. Neurol. 2013, 13, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Kochanek, P.M.; Dixon, C.E.; Mondello, S.; Wang, K.K.K.; Lafrenaye, A.; Bramlett, H.M.; Dietrich, W.D.; Hayes, R.L.; Shear, D.A.; Gilsdorf, J.S.; et al. Multi-Center Pre-clinical Consortia to Enhance Translation of Therapies and Biomarkers for Traumatic Brain Injury: Operation Brain Trauma Therapy and Beyond. Front. Neurol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Kim, S.; Mortera, M.; Hu, X.; Krishnan, S.; Hoffecker, L.; Herrold, A.; Terhorst, L.; King, L.; Machtinger, J.; Zumsteg, J.M.; et al. Overview of pharmacological interventions after traumatic brain injuries: Impact on selected outcomes. Brain Inj. 2019, 33, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Fujisawa, H. The role of glutamate antagonists for the treatment of CNS injury. J. Neurotrauma 1992, 9 (Suppl. 2), S443–S462. [Google Scholar]
- Tymianski, M.; Tator, C.H. Normal and abnormal calcium homeostasis in neurons: A basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 1996, 38, 1176–1195. [Google Scholar] [PubMed]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell. Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef]
- Cristofori, L.; Tavazzi, B.; Gambin, R.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Fazzina, G.; Lazzarino, G. Biochemical analysis of the cerebrospinal fluid: Evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: A case report. Clin. Biochem. 2005, 38, 97–100. [Google Scholar] [CrossRef]
- Hall, E.D.; Springer, J.E. Neuroprotection and acute spinal cord injury: A reappraisal. NeuroRx 2004, 1, 80–100. [Google Scholar] [CrossRef]
- Ozsuer, H.; Gorgulu, A.; Kiris, T.; Cobanoglu, S. The effects of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurg. Rev. 2005, 28, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, H.; Fang, J.; Dai, W.; Zhou, J.; Wang, X.; Zhou, M. SS-31 Provides Neuroprotection by Reversing Mitochondrial Dysfunction After Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 4783602. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Narabayashi, H.; Sata, T.; Takeshige, K. Kinetics of superoxide formation by respiratory chain NADH- dehydrogenase of bovine heart mitochondria. J. Biochem. 1983, 94, 1301–1306. [Google Scholar] [CrossRef]
- De Campos, R.P.; Siegel, J.M.; Fresta, C.G.; Caruso, G.; da Silva, J.A.; Lunte, S.M. Indirect detection of superoxide in RAW 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal. Bioanal. Chem. 2015, 407, 7003–7012. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, A.G.; Alshboul, O.A. Pathophysiology of traumatic brain injury. Neurosciences 2013, 18, 222–234. [Google Scholar]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Fenoy, A.; Bredt, D.S.; Noble, L.J. Expression of nitric oxide synthase in the cerebral microvasculature after traumatic brain injury in the rat. Brain Res. 1997, 751, 336–338. [Google Scholar] [CrossRef]
- Rao, V.L.; Dogan, A.; Bowen, K.K.; Dempsey, R.J. Traumatic injury to rat brain upregulates neuronal nitric oxide synthase expression and L-[3H]nitroarginine binding. J. Neurotrauma 1999, 16, 865–877. [Google Scholar] [CrossRef]
- Gahm, C.; Holmin, S.; Mathiesen, T. Temporal profiles and cellular sources of three nitric oxide synthase isoforms in the brain after experimental contusion. Neurosurgery 2000, 46, 169–177. [Google Scholar] [CrossRef]
- Cherian, L.; Goodman, J.C.; Robertson, C.S. Brain nitric oxide changes after controlled cortical impact injury in rats. J. Neurophysiol. 2000, 83, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, R.; Lui, H.; Cherian, L.; Goodman, J.C.; O’Brien, W.E.; Contant, C.F.; Robertson, C.S. The role of endothelial nitric oxide synthase in the cerebral hemodynamics after controlled cortical impact injury in mice. J. Neurotrauma 2003, 20, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Siegel, J.M.; Wijesinghe, M.B.; Lunte, S.M. Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Anal. Bioanal. Chem. 2017, 409, 4529–4538. [Google Scholar] [CrossRef]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Andrus, P.K.; Oostveen, J.A.; Fleck, T.J.; Gurney, M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J. Neurosci. Res. 1998, 53, 66–77. [Google Scholar] [CrossRef]
- Castilho, R.F.; Kowaltowski, A.J.; Meinicke, A.R.; Bechara, E.J.; Vercesi, A.E. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic. Biol. Med. 1995, 18, 479–486. [Google Scholar] [CrossRef]
- Jacquard, C.; Trioulier, Y.; Cosker, F.; Escartin, C.; Bizat, N.; Hantraye, P.; Cancela, J.M.; Bonvento, G.; Brouillet, E. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB J. 2006, 20, 1021–1023. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Dhammu, T.S.; Matsuda, F.; Annamalai, B.; Dhindsa, T.S.; Singh, I.; Singh, A.K. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016, 1630, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Lipton, S.A. ‘SNO’-Storms Compromise Protein Activity and Mitochondrial Metabolism in Neurodegenerative Disorders. Trends Endocrinol. Metab. 2017, 28, 879–892. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Petzold, A.; Gasperini, C.; Ruggieri, S.; Quartuccio, M.E.; Lazzarino, G.; Di Stasio, E.; Tavazzi, B. Serum Compounds of Energy Metabolism Impairment Are Related to Disability, Disease Course and Neuroimaging in Multiple Sclerosis. Mol. Neurobiol. 2017, 54, 7520–7533. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of nitric oxide synthases in Parkinson’s disease: A review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Listorti, I.; Muzii, L.; Amorini, A.M.; Longo, S.; Di Stasio, E.; Caruso, G.; D’Urso, S.; Puglia, I.; Pisani, G.; et al. Low-molecular weight compounds in human seminal plasma as potential biomarkers of male infertility. Hum. Reprod. 2018, 33, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Listorti, I.; Bilotta, G.; Capozzolo, T.; Amorini, A.M.; Longo, S.; Caruso, G.; Lazzarino, G.; Tavazzi, B.; Bilotta, P. Water- and Fat-Soluble Antioxidants in Human Seminal Plasma and Serum of Fertile Males. Antioxidants 2019, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Perez-Gregorio, R.; Simal-Gandara, J. A Critical Review of Bioactive Food Components, and of their Functional Mechanisms, Biological Effects and Health Outcomes. Curr. Pharm. Des. 2017, 23, 2731–2741. [Google Scholar] [CrossRef]
- Hasadsri, L.; Wang, B.H.; Lee, J.V.; Erdman, J.W.; Llano, D.A.; Barbey, A.K.; Wszalek, T.; Sharrock, M.F.; Wang, H.J. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J. Neurotrauma 2013, 30, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Rigg, J.L.; Elovic, E.P.; Greenwald, B.D. A review of the effectiveness of antioxidant therapy to reduce neuronal damage in acute traumatic brain injury. J. Head Trauma Rehabil. 2005, 20, 389–391. [Google Scholar] [CrossRef]
- Vonder Haar, C.; Peterson, T.C.; Martens, K.M.; Hoane, M.R. Vitamins and nutrients as primary treatments in experimental brain injury: Clinical implications for nutraceutical therapies. Brain Res. 2016, 1640, 114–129. [Google Scholar] [CrossRef] [Green Version]
- Granger, M.; Eck, P. Dietary Vitamin C in Human Health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar]
- Castiglione, D.; Platania, A.; Conti, A.; Falla, M.; D’Urso, M.; Marranzano, M. Dietary Micronutrient and Mineral Intake in the Mediterranean Healthy Eating, Ageing, and Lifestyle (MEAL) Study. Antioxidants 2018, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Burzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clemencon, B.; Burrier, R.; Hediger, M.A. The sodium-dependent ascorbic acid transporter family SLC23. Mol. Asp. Med. 2013, 34, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2008, 34, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; D’Urso, S.; Longo, S.; Vagnozzi, R.; Signoretti, S.; Clementi, E.; Giardina, B.; et al. Neuroglobin expression and oxidant/antioxidant balance after graded traumatic brain injury in the rat. Free Radic. Biol. Med. 2014, 69, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Parker, W.H.; Qu, Z.C.; May, J.M. Ascorbic acid transport in brain microvascular pericytes. Biochem. Biophys. Res. Commun. 2015, 458, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Angulo, C.; Castro, M.A.; Rivas, C.I.; Segretain, D.; Maldonado, R.; Yanez, A.J.; Slebe, J.C.; Vera, J.C.; Concha, I.I. Molecular identification and functional characterization of the vitamin C transporters expressed by Sertoli cells. J. Cell Physiol. 2008, 217, 708–716. [Google Scholar] [CrossRef]
- Awasthi, D.; Church, D.F.; Torbati, D.; Carey, M.E.; Pryor, W.A. Oxidative stress following traumatic brain injury in rats. Surg. Neurol. 1997, 47, 575–581. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; Sokolova, T.V.; Ritov, V.B.; Quinn, P.J.; Rose, M.; Kochanek, P.; Graham, S.H.; Kagan, V.E. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000, 75, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, G.M.; Saidu, Y.; Bilbis, L.S.; Muhammad, S.A.; Jinjir, N.; Shehu, B.B. Effects of alpha-tocopherol and ascorbic acid in the severity and management of traumatic brain injury in albino rats. J. Neurosci. Rural Pract. 2013, 4, 292–297. [Google Scholar]
- Razmkon, A.; Sadidi, A.; Sherafat-Kazemzadeh, E.; Mehrafshan, A.; Jamali, M.; Malekpour, B.; Saghafinia, M. Administration of vitamin C and vitamin E in severe head injury: A randomized double-blind controlled trial. Clin. Neurosurg. 2011, 58, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.E.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as Anti-inflammatory Drug in Different Models of Canonical and Cell Autonomous Inflammation Through NFκB Inhibition. Front. Immunol. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steullet, P.; Neijt, H.C.; Cuenod, M.; Do, K.Q. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: Relevance to schizophrenia. Neuroscience 2006, 137, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Varga, V.; Jenei, Z.; Janaky, R.; Saransaari, P.; Oja, S.S. Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem. Res. 1997, 22, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Oja, S.S.; Janaky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.; Ruppel, R.A.; Adelson, P.D.; Graham, S.H.; Janesko, K.; Clark, R.S.; Kochanek, P.M. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr. Res. 2002, 51, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Hergenroeder, G.W.; Jeter, C.B.; Choi, H.A.; Kobori, N.; Moore, A.N. Traumatic Brain Injury Alters Methionine Metabolism: Implications for Pathophysiology. Front. Syst. Neurosci. 2016, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Koza, L.; Linseman, D.A. Glutathione precursors shield the brain from trauma. Neural Regen. Res. 2019, 14, 1701–1702. [Google Scholar] [PubMed]
- Reed, T.T.; Owen, J.; Pierce, W.M.; Sebastian, A.; Sullivan, P.G.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: Insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J. Neurosci. Res. 2009, 87, 408–417. [Google Scholar] [PubMed]
- Hicdonmez, T.; Kanter, M.; Tiryaki, M.; Parsak, T.; Cobanoglu, S. Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem. Res. 2006, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Sangobowale, M.; Nikulina, E.; Bergold, P.J. Minocycline plus N-acetylcysteine protect oligodendrocytes when first dosed 12 h after closed head injury in mice. Neurosci. Lett. 2018, 682, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Senol, N.; Naziroglu, M.; Yuruker, V. N-acetylcysteine and selenium modulate oxidative stress, antioxidant vitamin and cytokine values in traumatic brain injury-induced rats. Neurochem. Res. 2014, 39, 685–692. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Feliciano, A.S. Flavonoids: From Structure to Health Issues. Molecules 2017, 22, 477. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Masuelli, L.; Tresoldi, I.; Sacchetti, P.; Modesti, A.; Galvano, F.; Bei, R. The effects of dietary flavonoids on the regulation of redox inflammatory networks. Front. Biosci. (Landmark Ed.) 2012, 17, 2396–2418. [Google Scholar] [CrossRef] [Green Version]
- Theadom, A.; Mahon, S.; Barker-Collo, S.; McPherson, K.; Rush, E.; Vandal, A.C.; Feigin, V.L. Enzogenol for cognitive functioning in traumatic brain injury: A pilot placebo-controlled RCT. Eur. J. Neurol. 2013, 20, 1135–1144. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Ren, J. From French Paradox to cancer treatment: Anti-cancer activities and mechanisms of resveratrol. Anticancer Agents Med. Chem. 2014, 14, 806–825. [Google Scholar] [CrossRef]
- Tsai, H.Y.; Ho, C.T.; Chen, Y.K. Biological actions and molecular effects of resveratrol, pterostilbene, and 3′-hydroxypterostilbene. J. Food Drug Anal. 2017, 25, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.H.; Yang, L.Y.; Shih, C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatson, J.W.; Liu, M.M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Minei, J.P. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J. Trauma Acute Care Surg. 2013, 74, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Irias-Mata, A.; Stuetz, W.; Sus, N.; Hammann, S.; Gralla, K.; Cordero-Solano, A.; Vetter, W.; Frank, J. Tocopherols, Tocomonoenols, and Tocotrienols in Oils of Costa Rican Palm Fruits: A Comparison between Six Varieties and Chemical versus Mechanical Extraction. J. Agric. Food Chem. 2017, 65, 7476–7482. [Google Scholar] [CrossRef] [PubMed]
- Comitato, R.; Ambra, R.; Virgili, F. Tocotrienols: A Family of Molecules with Specific Biological Activities. Antioxidants 2017, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inci, S.; Ozcan, O.E.; Kilinc, K. Time-level relationship for lipid peroxidation and the protective effect of alpha-tocopherol in experimental mild and severe brain injury. Neurosurgery 1998, 43, 330–335. [Google Scholar] [CrossRef]
- Yang, J.; Han, Y.; Ye, W.; Liu, F.; Zhuang, K.; Wu, G. Alpha tocopherol treatment reduces the expression of Nogo-A and NgR in rat brain after traumatic brain injury. J. Surg. Res. 2013, 182, e69–e77. [Google Scholar] [CrossRef]
- Clifton, G.L.; Lyeth, B.G.; Jenkins, L.W.; Taft, W.C.; DeLorenzo, R.J.; Hayes, R.L. Effect of D, alpha-tocopheryl succinate and polyethylene glycol on performance tests after fluid percussion brain injury. J. Neurotrauma 1989, 6, 71–81. [Google Scholar] [CrossRef]
- Aiguo, W.; Zhe, Y.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Artuch, R.; Salviati, L.; Jackson, S.; Hirano, M.; Navas, P. Coenzyme Q10 deficiencies in neuromuscular diseases. Adv. Exp. Med. Biol. 2009, 652, 117–128. [Google Scholar]
- Jorat, M.V.; Tabrizi, R.; Kolahdooz, F.; Akbari, M.; Salami, M.; Heydari, S.T.; Asemi, Z. The effects of coenzyme Q10 supplementation on biomarkers of inflammation and oxidative stress in among coronary artery disease: A systematic review and meta-analysis of randomized controlled trials. Inflammopharmacology 2019, 27, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, M.; Unal, M.M.; Gul, S.; Acikgoz, S.; Kandemir, N.; Hanci, V.; Edebali, N.; Acikgoz, B. Effect of coenzyme Q10 on ischemia and neuronal damage in an experimental traumatic brain-injury model in rats. BMC Neurosci. 2011, 12, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.M. The Antioxidant Role of Non-mitochondrial CoQ10: Mystery Solved! Cell Metab. 2020, 31, 13–15. [Google Scholar] [CrossRef]
- Pierce, J.D.; Shen, Q.; Peltzer, J.; Thimmesch, A.; Hiebert, J.B. A pilot study exploring the effects of ubiquinol on brain genomics after traumatic brain injury. Nurs. Outlook 2017, 65, S44–S52. [Google Scholar] [CrossRef]
- Pierce, J.D.; Gupte, R.; Thimmesch, A.; Shen, Q.; Hiebert, J.B.; Brooks, W.M.; Clancy, R.L.; Diaz, F.J.; Harris, J.L. Ubiquinol treatment for TBI in male rats: Effects on mitochondrial integrity, injury severity, and neurometabolism. J. Neurosci. Res. 2018, 96, 1080–1092. [Google Scholar] [CrossRef]
- Ross, A.C.; Caballero, B.H.; Cousins, R.J.; Tucker, K.L.; Ziegler, T.R. Modern Nutrition in Health and Disease, 11th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2012. [Google Scholar]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent Advances in Studies on the Therapeutic Potential of Dietary Carotenoids in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, Z.; Cui, H.; Wang, Y.; Zhong, C. Astaxanthin protects astrocytes against trauma-induced apoptosis through inhibition of NKCC1 expression via the NF-kappaB signaling pathway. BMC Neurosci. 2017, 18, 42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cui, Z.; Cui, H.; Cao, Y.; Zhong, C.; Wang, Y. Astaxanthin alleviates cerebral edema by modulating NKCC1 and AQP4 expression after traumatic brain injury in mice. BMC Neurosci. 2016, 17, 60. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Peng, D.; Zhang, Y.; Zhang, J.; Wang, Y.; Gao, Y.; Lu, N.; Tang, P. Astaxanthin improves cognitive performance in mice following mild traumatic brain injury. Brain Res. 2017, 1659, 88–95. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Jiang, L.; Huang, Z.; Zhang, H.; Cheng, C.; Liu, H.; He, J.; Wu, J.; Darwazeh, R.; Wu, Y.; et al. The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain Behav. Immun. 2017, 65, 183–194. [Google Scholar] [CrossRef]
- Zhong, J.; Cheng, C.; Liu, H.; Huang, Z.; Wu, Y.; Teng, Z.; He, J.; Zhang, H.; Wu, J.; Cao, F.; et al. Bexarotene protects against traumatic brain injury in mice partially through apolipoprotein E. Neuroscience 2017, 343, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, L.; Rao, W.; Su, N.; Hui, H.; Wang, L.; Peng, C.; Tu, Y.; Zhang, S.; Fei, Z. Neuroprotective effects of crocin against traumatic brain injury in mice: Involvement of notch signaling pathway. Neurosci. Lett. 2015, 591, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Yu, X.; Chen, M.; Chen, J.; Xu, J. Lutein protects against severe traumatic brain injury through antiinflammation and antioxidative effects via ICAM1/Nrf2. Mol. Med. Rep. 2017, 16, 4235–4240. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, L.; Hansen, H.S.; Jorgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nishi, D.; Tanima, Y.; Itakura, M.; Kojima, M.; Hamazaki, K.; Noguchi, H.; Hamazaki, T. Serum pro-BDNF/BDNF as a treatment biomarker for response to docosahexaenoic acid in traumatized people vulnerable to developing psychological distress: A randomized controlled trial. Transl. Psychiatry 2015, 5, e596. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, H.; Nishi, D.; Matsumura, K.; Hamazaki, K.; Hamazaki, T.; Matsuoka, Y.J. Limited effect of omega-3 fatty acids on the quality of life in survivors of traumatic injury: A randomized, placebo-controlled trial. Prostaglandins Leukot. Essent. Fatty Acids 2017, 127, 1–5. [Google Scholar] [CrossRef]
- Gross, B.W.; Gillio, M.; Rinehart, C.D.; Lynch, C.A.; Rogers, F.B. Omega-3 Fatty Acid Supplementation and Warfarin: A Lethal Combination in Traumatic Brain Injury. J. Trauma Nurs. 2017, 24, 15–18. [Google Scholar] [CrossRef]
- McCrory, P.; Feddermann-Demont, N.; Dvorak, J.; Cassidy, J.D.; McIntosh, A.; Vos, P.E.; Echemendia, R.J.; Meeuwisse, W.; Tarnutzer, A.A. What is the definition of sports-related concussion: A systematic review. Br. J. Sports Med. 2017, 51, 877–887. [Google Scholar] [CrossRef]
- McCrory, P.; Meeuwisse, W.; Dvorak, J.; Aubry, M.; Bailes, J.; Broglio, S.; Cantu, R.C.; Cassidy, D.; Echemendia, R.J.; Castellani, R.J.; et al. Consensus statement on concussion in sport-the 5(th) international conference on concussion in sport held in Berlin, October 2016. Br. J. Sports Med. 2017, 51, 838–847. [Google Scholar] [PubMed] [Green Version]
- Signoretti, S.; Tavazzi, B.; Lazzarino, G.; Vagnozzi, R. The Pathophysiology of Concussive Brain Injury. In Concussion and Traumatic Encephalopathy: Causes, Diagnosis and Management; Victoroff, J., Bigler, E.D., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 138–152. [Google Scholar]
- Giza, C.C.; Hovda, D.A. The Neurometabolic Cascade of Concussion. J. Athl. Train. 2001, 36, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75 (Suppl. 4), S24–S33. [Google Scholar] [CrossRef] [Green Version]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta 2016, 1862, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment—Part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Signoretti, S.; Floris, R.; Marziali, S.; Manara, M.; Amorini, A.M.; Belli, A.; Di Pietro, V.; D’Urso, S.; Pastore, F.S.; et al. Decrease in N-acetylaspartate following concussion may be coupled to decrease in creatine. J. Head Trauma Rehabil. 2013, 28, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Vagnozzi, R.; Signoretti, S.; Tavazzi, B.; Floris, R.; Ludovici, A.; Marziali, S.; Tarascio, G.; Amorini, A.M.; Di Pietro, V.; Delfini, R.; et al. Temporal window of metabolic brain vulnerability to concussion: A pilot 1H-magnetic resonance spectroscopic study in concussed athletes—Part III. Neurosurgery 2008, 62, 1286–1295. [Google Scholar] [CrossRef]
- Tavazzi, B.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Oxidative and nitrosative stresses—Part II. Neurosurgery 2007, 61, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Hoffer, M.E.; Balaban, C.; Slade, M.D.; Tsao, J.W.; Hoffer, B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: A double-blind, placebo controlled study. PLoS ONE 2013, 8, e54163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, J.M.; Jones, M.T.; Kirk, K.M.; Gable, D.A.; Repshas, J.T.; Johnson, T.A.; Andreasson, U.; Norgren, N.; Blennow, K.; Zetterberg, H. Effect of Docosahexaenoic Acid on a Biomarker of Head Trauma in American Football. Med. Sci. Sports Exerc. 2016, 48, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Finelli, K.; Bai, X.; Arnett, P.; Bream, T.; Seidenberg, P.; Lynch, S.; Johnson, B.; Slobounov, S. Effect of Enzogenol® Supplementation on Cognitive, Executive, and Vestibular/Balance Functioning in Chronic Phase of Concussion. Dev. Neuropsychol. 2017, 42, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Marmarou, A.; Tavazzi, B.; Signoretti, S.; Di Pierro, D.; del Bolgia, F.; Amorini, A.M.; Fazzina, G.; Sherkat, S.; Lazzarino, G. Changes of cerebral energy metabolism and lipid peroxidation in rats leading to mitochondrial dysfunction after diffuse brain injury. J. Neurotrauma 1999, 16, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Sullivan, P.G.; Deng, Y.; Mbye, L.H.; Hall, E.D. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy. J. Cereb. Blood Flow Metab. 2006, 26, 1407–1418. [Google Scholar] [CrossRef] [Green Version]
- Cristofori, L.; Tavazzi, B.; Gambin, R.; Vagnozzi, R.; Vivenza, C.; Amorini, A.M.; Di Pierro, D.; Fazzina, G.; Lazzarino, G. Early onset of lipid peroxidation after human traumatic brain injury: A fatal limitation for the free radical scavenger pharmacological therapy? J. Investig. Med. 2001, 49, 450–458. [Google Scholar] [CrossRef]
- Rashno, M.; Sarkaki, A.; Farbood, Y.; Rashno, M.; Khorsandi, L.; Naseri, M.K.G.; Dianat, M. Therapeutic effects of chrysin in a rat model of traumatic brain injury: A behavioral, biochemical, and histological study. Life Sci. 2019, 228, 285–294. [Google Scholar] [CrossRef]
- Umemoto, Y.; Patel, A.; Huynh, T.; Chitravanshi, V.C. Wogonin attenuates the deleterious effects of traumatic brain injury in anesthetized Wistar rats. Eur. J. Pharmacol. 2019, 848, 121–130. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, L.L.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Shi, Y.J.; He, Z.L.; Liu, L.; Jiang, C.J.; Hou, R.N.; et al. Isoliquiritigenin Provides Protection and Attenuates Oxidative Stress-Induced Injuries via the Nrf2-ARE Signaling Pathway After Traumatic Brain Injury. Neurochem. Res. 2018, 43, 2435–2445. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Abbasi, M.M.; Salari, A.A. Hesperidin attenuates depression-related symptoms in mice with mild traumatic brain injury. Life Sci. 2018, 213, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Bae, J.; Lee, J.S.; Bang, Y.; Lee, B.J.; Park, J.W.; Lee, K.; Cho, J.H.; Bu, Y. Icariin Improves Functional Behavior in a Mouse Model of Traumatic Brain Injury and Promotes Synaptic Plasticity Markers. Planta Med. 2019, 85, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M. Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway. Drug Des. Dev. Ther. 2018, 12, 2497–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zeng, G.; Zheng, X.; Wang, W.; Ling, Y.; Tang, H.; Zhang, J. Neuroprotective effect of formononetin against TBI in rats via suppressing inflammatory reaction in cortical neurons. Biomed. Pharmacother. 2018, 106, 349–354. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, Y.; Zeng, J.; Li, D.; Huang, Y. Troxerutin cerebroprotein hydrolysate injection ameliorates neurovascular injury induced by traumatic brain injury—Via endothelial nitric oxide synthase pathway regulation. Int. J. Neurosci. 2018, 128, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, Z.; Chen, Y.; Li, Z.; Tian, Y.; Liu, Z.; Liu, B.; Song, J. Quercetin protects rat cortical neurons against traumatic brain injury. Mol. Med. Rep. 2018, 17, 7859–7865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Qiu, W.; Xiao, G.; Cheng, J.; Zhang, N. Resveratrol Attenuates Cognitive Deficits of Traumatic Brain Injury by Activating p38 Signaling in the Brain. Med. Sci. Monit. 2018, 24, 1097–1103. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Abbasi, M.M.; Salari, A.A. Anxiolytic- and antidepressant-like effects of Silymarin compared to diazepam and fluoxetine in a mouse model of mild traumatic brain injury. Toxicol. Appl. Pharmacol. 2018, 338, 159–173. [Google Scholar] [CrossRef]
- Mirshekar, M.A.; Fanaei, H.; Keikhaei, F.; Javan, F.S. Diosmin improved cognitive deficit and amplified brain electrical activity in the rat model of traumatic brain injury. Biomed. Pharmacother. 2017, 93, 1220–1229. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, J.; Cai, Y.; Huang, J.; You, L. Catechin attenuates traumatic brain injury-induced blood-brain barrier damage and improves longer-term neurological outcomes in rats. Exp. Physiol. 2017, 102, 1269–1277. [Google Scholar] [CrossRef]
- Krishna, G.; Agrawal, R.; Zhuang, Y.; Ying, Z.; Paydar, A.; Harris, N.G.; Royes, L.F.; Gomez-Pinilla, F. 7,8-Dihydroxyflavone facilitates the action exercise to restore plasticity and functionality: Implications for early brain trauma recovery. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Roberts, K.N. Cognitive assessment of pycnogenol therapy following traumatic brain injury. Neurosci. Lett. 2016, 634, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Xia, Q.J.; Dong, X.J.; Hu, Y.; Chen, Z.W.; Chen, K.; Wang, K.H.; Liu, J.; Wang, T.H. Neuroprotective effect of breviscapine on traumatic brain injury in rats associated with the inhibition of GSK3beta signaling pathway. Brain Res. 2017, 1660, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Peng, W.; Xia, Z.; Gan, P.; Huang, W.; Shi, Y.; Fan, R. Hydroxysaf fl or yellow A exerts antioxidant effects in a rat model of traumatic brain injury. Mol. Med. Rep. 2016, 14, 3690–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltani, Z.; Khaksari, M.; Jafari, E.; Iranpour, M.; Shahrokhi, N. Is genistein neuroprotective in traumatic brain injury? Physiol. Behav. 2015, 152, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Wang, W.; Li, Q.; Han, X.; Xing, J.; Qi, C.; Lan, X.; Wan, J.; Potts, A.; Guan, F.; et al. Cerebroprotection of flavanol (-)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic. Biol. Med. 2016, 92, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Hao, S.; Zhu, Z.; Zhang, H.; Wu, W.; Xu, F.; Liu, B. Procyanidins protects against oxidative damage and cognitive deficits after traumatic brain injury. Brain Inj. 2015, 29, 86–92. [Google Scholar] [CrossRef]
- Erdem, Y.; Tekiner, A.; Erkoc, Y.S.; Yilmaz, M.B.; Celik, H.; Yildirim, A.E.; Tekiner, A.S.; Bayar, M.A. Antiedema effects of proanthocyanidin on experimental traumatic brain edema. Turk. Neurosurg. 2015, 25, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective effect of (-)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. J. Neural Transm. 2013, 120, 767–783. [Google Scholar] [CrossRef]
- Wang, J.W.; Wang, H.D.; Cong, Z.X.; Zhou, X.M.; Xu, J.G.; Jia, Y.; Ding, Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J. Surg. Res. 2014, 186, 328–337. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.J.; Wang, L.Y.; Wei, Z.X.; Qu, W.S. Continual naringin treatment benefits the recovery of traumatic brain injury in rats through reducing oxidative and inflammatory alterations. Neurochem. Res. 2014, 39, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Hsu, C.W.; Huang, W.H.; Wang, J.Y. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thau-Zuchman, O.; Ingram, R.; Harvey, G.G.; Cooke, T.; Palmas, F.; Pallier, P.N.; Brook, J.; Priestley, J.V.; Dalli, J.; Lopez-Tremoleda, J.; et al. A single injection of docosahexaenoic acid induces a pro-resolving lipid mediator profile in the injured tissue and a long-lasting reduction in neurological deficit after traumatic brain injury in mice. J. Neurotrauma 2020, 37, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.E.; Requena, D.F.; Casper, T.C.; Velhorst, A.K.; Lolofie, A.; McFarlane, K.E.; Otto, T.E.; Terry, C.; Gensel, J.C. Docosahexaenoic acid decreased neuroinflammation in rat pups after controlled cortical impact. Exp. Neurol. 2019, 320, 112971. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.W.V.; Davidsson, J.; Lidin, E.; Angeria, M.; Risling, M.; Gunther, M. Brain tissue saving effects by single-dose intralesional administration of Neuroprotectin D1 on experimental focal penetrating brain injury in rats. J. Clin. Neurosci. 2019, 64, 227–233. [Google Scholar] [CrossRef]
- Zhu, W.; Ding, Y.; Kong, W.; Li, T.; Chen, H. Docosahexaenoic Acid (DHA) Provides Neuroprotection in Traumatic Brain Injury Models via Activating Nrf2-ARE Signaling. Inflammation 2018, 41, 1182–1193. [Google Scholar] [CrossRef]
- Tang, R.; Lin, Y.M.; Liu, H.X.; Wang, E.S. Neuroprotective effect of docosahexaenoic acid in rat traumatic brain injury model via regulation of TLR4/NF-Kappa B signaling pathway. Int. J. Biochem. Cell Biol. 2018, 99, 64–71. [Google Scholar] [CrossRef]
- Zhu, W.; Chi, N.; Zou, P.; Chen, H.; Tang, G.; Zhao, W. Effect of docosahexaenoic acid on traumatic brain injury in rats. Exp. Ther. Med. 2017, 14, 4411–4416. [Google Scholar] [CrossRef]
- Salberg, S.; Yamakawa, G.; Christensen, J.; Kolb, B.; Mychasiuk, R. Assessment of a nutritional supplement containing resveratrol, prebiotic fiber, and omega-3 fatty acids for the prevention and treatment of mild traumatic brain injury in rats. Neuroscience 2017, 365, 146–157. [Google Scholar] [CrossRef]
- Harvey, L.D.; Yin, Y.; Attarwala, I.Y.; Begum, G.; Deng, J.; Yan, H.Q.; Dixon, C.E.; Sun, D. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, M.E.; Requena, D.F.; Abdullah, O.M.; Casper, T.C.; Beachy, J.; Malleske, D.; Pauly, J.R. Dietary Docosahexaenoic Acid Improves Cognitive Function, Tissue Sparing, and Magnetic Resonance Imaging Indices of Edema and White Matter Injury in the Immature Rat after Traumatic Brain Injury. J. Neurotrauma 2016, 33, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, E.; Sun, G.; Yan, H.Q.; Foley, L.M.; Andrzejczuk, L.A.; Attarwala, I.Y.; Hitchens, T.K.; Kiselyov, K.; Dixon, C.E.; et al. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Yang, Z.; Luo, C.; Zeng, H.; Li, P.; Kang, J.X.; Wan, J.B.; He, C.; Su, H. Enriched Endogenous Omega-3 Fatty Acids in Mice Ameliorate Parenchymal Cell Death After Traumatic Brain Injury. Mol. Neurobiol. 2017, 54, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary strategy to repair plasma membrane after brain trauma: Implications for plasticity and cognition. Neurorehabil. Neural Repair 2014, 28, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience 2013, 248, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Russell, K.L.; Berman, N.E.; Levant, B. Low brain DHA content worsens sensorimotor outcomes after TBI and decreases TBI-induced Timp1 expression in juvenile rats. Prostaglandins Leukot. Essent. Fatty Acids 2013, 89, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.D.; Hadley, K.; Bailes, J.E. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 2011, 68, 474–481. [Google Scholar] [CrossRef] [Green Version]






| Score | Eye Opening (E) | Verbal Response (V) | Motor Response(M) |
|---|---|---|---|
| 1 | No eye-opening | No verbal response | No response |
| 2 | Eye-opening to pain | Incoherent | Extension to pain |
| 3 | Eye-opening to speech | Inappropriate words | Flexion to pain |
| 4 | Spontaneous eye-opening | Confused conversation | Withdrawal to pain |
| 5 | Oriented | Localizes to pain | |
| 6 | Follows commands |
| Class | Treatment Drugs | Mechanism of Action |
|---|---|---|
| Osmotic therapy | Mannitol Hypertonic saline | Decrease brain edema, improve cerebral blood flow and blood rheology |
| Antiepileptic drugs | Phenytoin, Phenobarbital, Carbamazepine, Valproate, Levetiracetam | Prevent seizures, especially during the first week after injury thus preventing rise in ICP |
| Sedative agents | Barbiturate: Pentobarbital Benzodiazepine: Midazolam | Reduce neuronal activity, metabolic brain requirements and ICP |
| Pharmacological paralysis | Succinylcholine, Atracurium, Rocuronium | Prevents high intra-thoracic pressure during mechanical ventilation which is transmitted intra-cranially |
| Opioid analgesics | Morphine, Fentanyl, Alfentanil | Pain control via their action on neuronal opioid receptors |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants 2020, 9, 260. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9030260
Di Pietro V, Yakoub KM, Caruso G, Lazzarino G, Signoretti S, Barbey AK, Tavazzi B, Lazzarino G, Belli A, Amorini AM. Antioxidant Therapies in Traumatic Brain Injury. Antioxidants. 2020; 9(3):260. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9030260
Chicago/Turabian StyleDi Pietro, Valentina, Kamal M. Yakoub, Giuseppe Caruso, Giacomo Lazzarino, Stefano Signoretti, Aron K. Barbey, Barbara Tavazzi, Giuseppe Lazzarino, Antonio Belli, and Angela Maria Amorini. 2020. "Antioxidant Therapies in Traumatic Brain Injury" Antioxidants 9, no. 3: 260. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox9030260