
Integrating Immunologic Signaling Networks: The JAK/STAT Pathway in Colitis and Colitis-Associated Cancer

Abstract
:1. Introduction
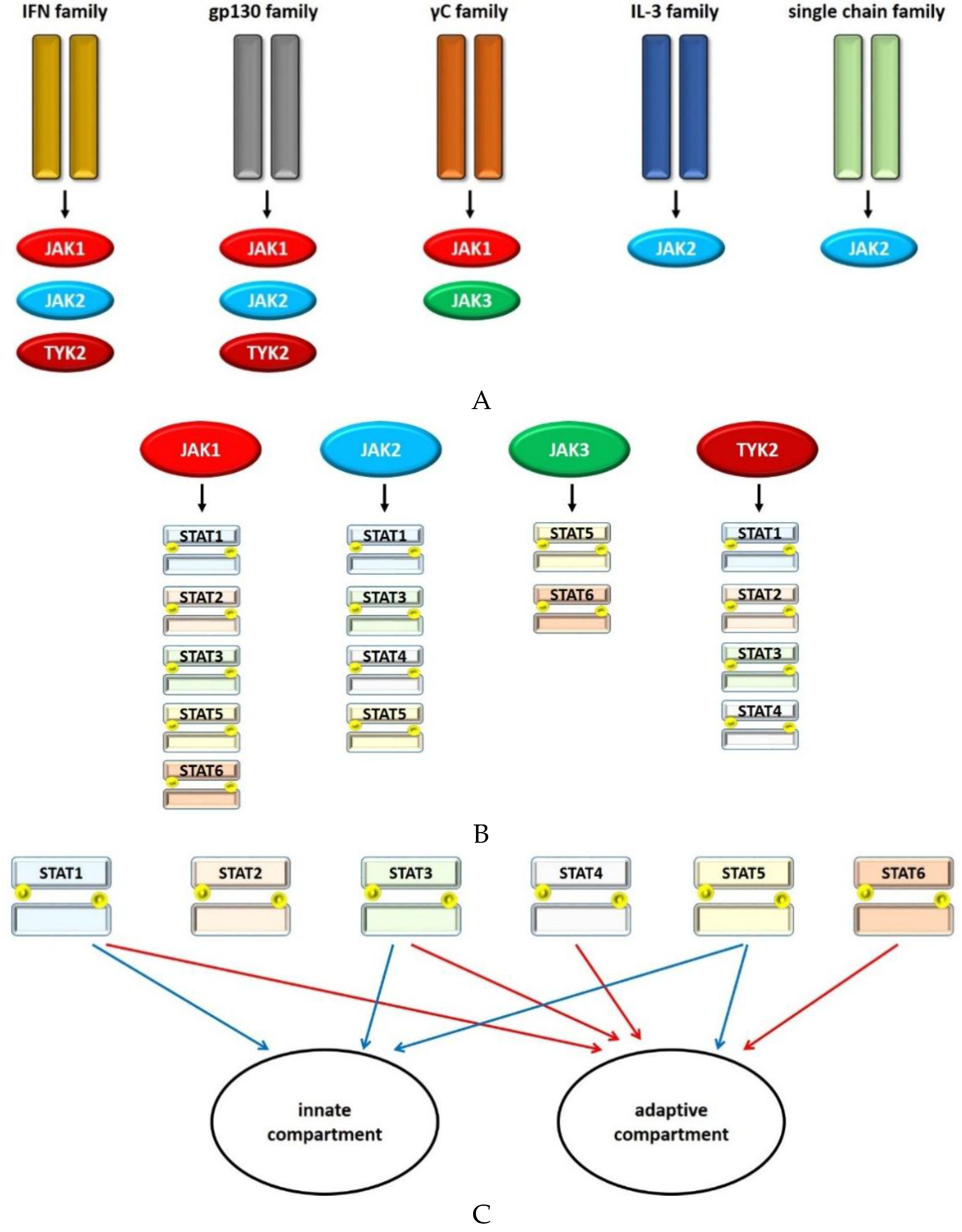
2. Principles of Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) Signaling
2.1. JAKs
2.2. STAT1
2.3. STAT2
2.4. STAT3
2.5. STAT4
2.6. STAT5
2.7. STAT6
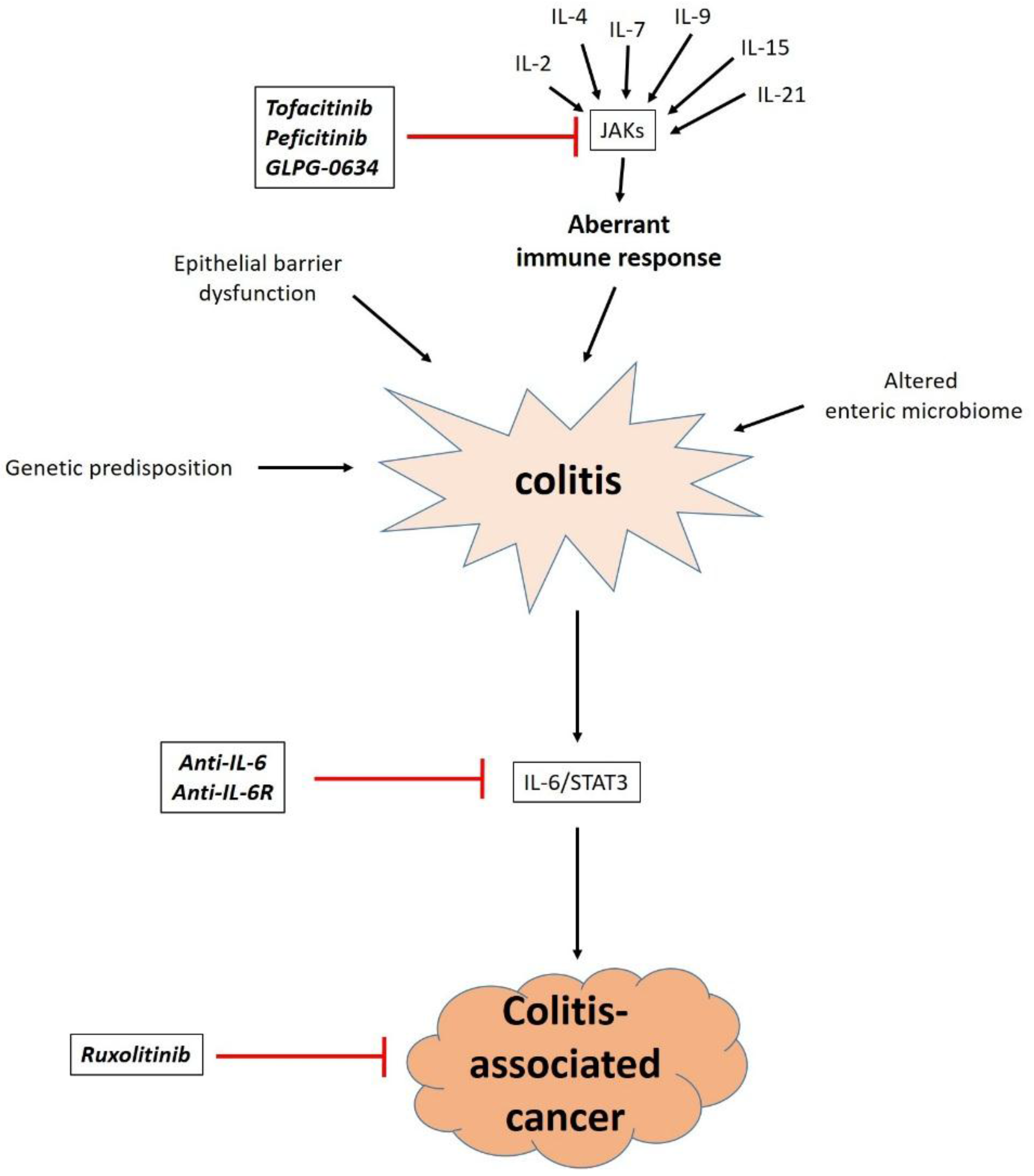
3. Therapeutic Implications
4. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
Abbreviations
| CAC | colitis-associated cancer |
| CCL | chemokine (C-C motif) ligand |
| CD | Crohn’s disease |
| CRC | colorectal carcinoma |
| DC | dendritic cell |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| IBD | inflammatory bowel disease |
| IEC | intestinal epithelial cell |
| IFN | interferon |
| IL | interleukin |
| JAK | janus kinase |
| LPMC | lamina propria mononuclear cell |
| R | receptor |
| SOCS | suppressor of cytokine signaling |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor β |
| TNF-α | tumor necrosis factor α |
| Th cell | T helper cell |
| Treg cell | regulatory T cell |
| UC | ulcerative colitis |
| VEGF | vascular endothelial growth factor |
References
- Danese, S.; Fiocchi, C. Ulcerative Colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. The Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel diseases: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Klag, T.; Stange, E.F.; Wehkamp, J. Defective antibacterial barrier in inflammatory bowel disease. Dig. Dis. Basel Switz. 2013, 31, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-F.; Li, Y.; Zhao, Q.-H.; Wang, Z.-Y.; Hu, S.; Yang, C.-Q.; Ye, K.; Li, L. Association of STAT4 rs7574865 polymorphism with susceptibility to inflammatory bowel disease: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Salzman, N.H.; Porter, E.; Nuding, S.; Weichenthal, M.; Petras, R.E.; Shen, B.; Schaeffeler, E.; Schwab, M.; Linzmeier, R.; et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 18129–18134. [Google Scholar] [CrossRef] [PubMed]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.; Dignass, A.U. Epithelial restitution and wound healing in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 348–353. [Google Scholar] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.-A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L.; Huang, J.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Inhibition of interferon gamma signaling by the short chain fatty acid butyrate. Mol. Cancer Res. 2003, 1, 855–862. [Google Scholar] [PubMed]
- Compare, D.; Nardone, G. Contribution of gut microbiota to colonic and extracolonic cancer development. Dig. Dis. Basel Switz. 2011, 29, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Herszényi, L.; Barabás, L.; Miheller, P.; Tulassay, Z. Colorectal cancer in patients with inflammatory bowel disease: The true impact of the risk. Dig. Dis. Basel Switz. 2015, 33, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Zografos, G.C.; Pikoulis, E. Cytokine networks in animal models of colitis-associated cancer. Anticancer Res. 2015, 35, 19–24. [Google Scholar] [PubMed]
- Waldner, M.J.; Neurath, M.F. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin. Immunol. 2014, 26, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E.T.; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Darnell, J.E. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [PubMed]
- O’Shea, J.J.; Pesu, M.; Borie, D.C.; Changelian, P.S. A new modality for immunosuppression: Targeting the JAK/STAT pathway. Nat. Rev. Drug Discov. 2004, 3, 555–564. [Google Scholar] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Zhang, S.; Fukuda, S.; Lee, Y.; Hangoc, G.; Cooper, S.; Spolski, R.; Leonard, W.J.; Broxmeyer, H.E. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J. Exp. Med. 2000, 192, 719–728. [Google Scholar] [CrossRef]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; Xia, W.; Gu, J.; Ishikawa, H.; Gutman, D.; Barber, G.; Qin, Z.; Jiang, Z. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; Johnson, E.M.; Schreiber, R.D. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; Grosveld, G.; Ihle, J.N. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395. [Google Scholar] [PubMed]
- Neubauer, H.; Cumano, A.; Müller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef]
- Aringer, M.; Cheng, A.; Nelson, J.W.; Chen, M.; Sudarshan, C.; Zhou, Y.J.; O’Shea, J.J. Janus kinases and their role in growth and disease. Life Sci. 1999, 64, 2173–2186. [Google Scholar] [CrossRef]
- Baird, A.M.; Thomis, D.C.; Berg, L.J. T cell development and activation in Jak3-deficient mice. J. Leukoc. Biol. 1998, 63, 669–677. [Google Scholar] [PubMed]
- Shimoda, K.; Kato, K.; Aoki, K.; Matsuda, T.; Miyamoto, A.; Shibamori, M.; Yamashita, M.; Numata, A.; Takase, K.; Kobayashi, S.; et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity 2000, 13, 561–571. [Google Scholar] [CrossRef]
- Shimoda, K.; Tsutsui, H.; Aoki, K.; Kato, K.; Matsuda, T.; Numata, A.; Takase, K.; Yamamoto, T.; Nukina, H.; Hoshino, T.; et al. Partial impairment of interleukin-12 (IL-12) and IL-18 signaling in Tyk2-deficient mice. Blood 2002, 99, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Kamesaki, K.; Numata, A.; Aoki, K.; Matsuda, T.; Oritani, K.; Tamiya, S.; Kato, K.; Takase, K.; Imamura, R.; et al. Cutting edge: tyk2 is required for the induction and nuclear translocation of Daxx which regulates IFN-alpha-induced suppression of B lymphocyte formation. J. Immunol. 2002, 169, 4707–4711. [Google Scholar] [CrossRef] [PubMed]
- Hainzl, E.; Stockinger, S.; Rauch, I.; Heider, S.; Berry, D.; Lassnig, C.; Schwab, C.; Rosebrock, F.; Milinovich, G.; Schlederer, M.; et al. Intestinal epithelial cell tyrosine kinase 2 transduces IL-22 signals to protect from acute colitis. J. Immunol. 2015, 195, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Akimoto, T.; Muromoto, R.; Yokoyama, M.; Ohshiro, Y.; Sekine, Y.; Maeda, H.; Shimoda, K.; Oritani, K.; Matsuda, T. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J. Immunol. 2011, 187, 181–189. [Google Scholar] [PubMed]
- Murata, Y.; Yamashita, A.; Saito, T.; Sugamura, K.; Hamuro, J. The conversion of redox status of peritoneal macrophages during pathological progression of spontaneous inflammatory bowel disease in Janus family tyrosine kinase 3(−/−) and IL-2 receptor gamma(−/−) mice. Int. Immunol. 2002, 14, 627–636. [Google Scholar] [PubMed]
- Mishra, J.; Verma, R.K.; Alpini, G.; Meng, F.; Kumar, N. Role of Janus kinase 3 in mucosal differentiation and predisposition to colitis. J. Biol. Chem. 2013, 288, 31795–31806. [Google Scholar] [CrossRef] [PubMed]
- Seavey, M.; Lu, L.; Stump, K.; Wallace, N.; Hockeimer, W.; O’Kane, T.; Ruggeri, B.; Dobrzanski, P. Therapeutic efficacy of CEP-33779, a novel selective JAK2 inhibitor, in a mouse model of colitis-induced colorectal cancer. Mol. Cancer Ther. 2012, 11, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [PubMed]
- Schreiber, S.; Rosenstiel, P.; Hampe, J.; Nikolaus, S.; Groessner, B.; Schottelius, A.; Kühbacher, T.; Hämling, J.; Fölsch, U.R.; Seegert, D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 2002, 51, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Weigmann, B.; Bartsch, B.; Kiesslich, R.; Strand, D.; Galle, P.R.; Lehr, H.A.; Schmidt, J.; Neurath, M.F. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol. 2005, 100, 64–72. [Google Scholar] [PubMed]
- Wu, F.; Dassopoulos, T.; Cope, L.; Maitra, A.; Brant, S.R.; Harris, M.L.; Bayless, T.M.; Parmigiani, G.; Chakravarti, S. Genome-wide gene expression differences in Crohn’s disease and ulcerative colitis from endoscopic pinch biopsies: insights into distinctive pathogenesis. Inflamm. Bowel Dis. 2007, 13, 807–821. [Google Scholar] [PubMed]
- Bandyopadhyay, S.K.; de la Motte, C.A.; Kessler, S.P.; Hascall, V.C.; Hill, D.R.; Strong, S.A. Hyaluronan-mediated leukocyte adhesion and dextran sulfate sodium-induced colitis are attenuated in the absence of signal transducer and activator of transcription 1. Am. J. Pathol. 2008, 173, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, W.; Wu, L.; Gu, Y.; Gu, L.; Xu, S.; Wu, X.; Shen, Y.; Ke, Y.; Tan, R.; et al. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis. J. Immunol. 2012, 189, 3497–3507. [Google Scholar] [PubMed]
- Azuma, Y.-T.; Matsuo, Y.; Kuwamura, M.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Nakajima, H.; Karow, M.; Takeuchi, T. Interleukin-19 protects mice from innate-mediated colonic inflammation. Inflamm. Bowel Dis. 2010, 16, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Nishimoto, S.; Muto, G.; Sekiya, T.; Tamiya, T.; Kimura, A.; Morita, R.; Asakawa, M.; Chinen, T.; Yoshimura, A. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J. Exp. Med. 2011, 208, 2055–2067. [Google Scholar] [PubMed]
- Lucas, S.; Ghilardi, N.; Li, J.; de Sauvage, F.J. IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 15047–15052. [Google Scholar] [PubMed]
- Diegelmann, J.; Olszak, T.; Göke, B.; Blumberg, R.S.; Brand, S. A novel role for interleukin-27 (IL-27) as mediator of intestinal epithelial barrier protection mediated via differential signal transducer and activator of transcription (STAT) protein signaling and induction of antibacterial and anti-inflammatory proteins. J. Biol. Chem. 2012, 287, 286–298. [Google Scholar] [PubMed]
- Li, C.; Xi, Y.; Li, S.; Zhao, Q.; Cheng, W.; Wang, Z.; Zhong, J.; Niu, X.; Chen, G. Berberine ameliorates TNBS induced colitis by inhibiting inflammatory responses and Th1/Th17 differentiation. Mol. Immunol. 2015, 67, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Qian, C.; Guo, W.; Luo, Q.; Xu, Q.; Sun, Y. Inhibition of Th1/Th17 responses via suppression of STAT1 and STAT3 activation contributes to the amelioration of murine experimental colitis by a natural flavonoid glucoside icariin. Biochem. Pharmacol. 2013, 85, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Owusu, B.Y.; Augenlicht, L.; Klampfer, L. The Role of STAT1 for Crosstalk between Fibroblasts and Colon Cancer Cells. Front. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L.; Huang, J.; Corner, G.; Mariadason, J.; Arango, D.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Oncogenic Ki-ras inhibits the expression of interferon-responsive genes through inhibition of STAT1 and STAT2 expression. J. Biol. Chem. 2003, 278, 46278–46287. [Google Scholar] [PubMed]
- Xu, X.; Fu, X.Y.; Plate, J.; Chong, A.S. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: Requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar] [PubMed]
- Hanada, T.; Kobayashi, T.; Chinen, T.; Saeki, K.; Takaki, H.; Koga, K.; Minoda, Y.; Sanada, T.; Yoshioka, T.; Mimata, H.; et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 2006, 203, 1391–1397. [Google Scholar] [PubMed]
- Kim, H.S.; Lee, M.-S. Essential role of STAT1 in caspase-independent cell death of activated macrophages through the p38 mitogen-activated protein kinase/STAT1/reactive oxygen species pathway. Mol. Cell. Biol. 2005, 25, 6821–6833. [Google Scholar] [PubMed]
- Liddle, F.J.; Frank, D.A. STAT1 expression is not required for polyp formation in Min mice. Mol. Carcinog. 2008, 47, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Tobelaim, W.S.; Beaurivage, C.; Champagne, A.; Pomerleau, V.; Simoneau, A.; Chababi, W.; Yeganeh, M.; Thibault, P.; Klinck, R.; Carrier, J.C.; et al. Tumour-promoting role of SOCS1 in colorectal cancer cells. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Steen, H.C.; Gamero, A.M. STAT2 phosphorylation and signaling. JAK-STAT 2013, 2, e25790. [Google Scholar] [CrossRef] [PubMed]
- Di Trolio, R.; Simeone, E.; Di Lorenzo, G.; Buonerba, C.; Ascierto, P.A. The use of interferon in melanoma patients: A systematic review. Cytokine Growth Factor Rev. 2015, 26, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Peterson, P.W.; Rawlins, J.T.; Dalley, B.K.; Yang, Q.; Albertsen, H.; Jones, D.A. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc. Natl. Acad. Sci. USA 1999, 96, 14007–14012. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Musso, A.; Dentelli, P.; Carlino, A.; Chiusa, L.; Repici, A.; Sturm, A.; Fiocchi, C.; Rizzetto, M.; Pegoraro, L.; Sategna-Guidetti, C.; et al. Signal transducers and activators of transcription 3 signaling pathway: An essential mediator of inflammatory bowel disease and other forms of intestinal inflammation. Inflamm. Bowel Dis. 2005, 11, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 193, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Bai, A.; Hu, P.; Chen, J.; Song, X.; Chen, W.; Peng, W.; Zeng, Z.; Gao, X. Blockade of STAT3 by antisense oligonucleotide in TNBS-induced murine colitis. Int. J. Colorectal Dis. 2007, 22, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sosnowska, D.; Bonkowski, E.L.; Denson, L.A. Growth hormone inhibits signal transducer and activator of transcription 3 activation and reduces disease activity in murine colitis. Gastroenterology 2005, 129, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.-W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Liu, L.; Ji, X.; Gao, Y.; Chen, X.; Liu, Y.; Liu, Y.; Zhao, X.; Li, Y.; Li, Y.; et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat. Immunol. 2015, 16, 1263–1273. [Google Scholar] [PubMed]
- Zundler, S.; Neurath, M.F. Immunopathogenesis of inflammatory bowel diseases: Functional role of T cells and T cell homing. Clin. Exp. Rheumatol. 2015, 33, S19–S28. [Google Scholar] [PubMed]
- Mudter, J.; Amoussina, L.; Schenk, M.; Yu, J.; Brüstle, A.; Weigmann, B.; Atreya, R.; Wirtz, S.; Becker, C.; Hoffman, A.; et al. The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J. Clin. Investig. 2008, 118, 2415–2426. [Google Scholar] [PubMed]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORγ-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Shioya, M.; Nishida, A.; Bamba, S.; Tsujikawa, T.; Kim-Mitsuyama, S.; Fujiyama, Y. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J. Immunol. 2009, 183, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.L.; Montero, M.; Ropeleski, M.J.; Bergstrom, K.S.B.; Ma, C.; Ghosh, S.; Merkens, H.; Huang, J.; Månsson, L.E.; Sham, H.P.; et al. Interleukin-11 reduces TLR4-induced colitis in TLR2-deficient mice and restores intestinal STAT3 signaling. Gastroenterology 2010, 139, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K. Role of STAT3 in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 5110–5114. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Förster, I.; Akira, S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef]
- Alonzi, T.; Newton, I.P.; Bryce, P.J.; Di Carlo, E.; Lattanzio, G.; Tripodi, M.; Musiani, P.; Poli, V. Induced somatic inactivation of STAT3 in mice triggers the development of a fulminant form of enterocolitis. Cytokine 2004, 26, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Zhang, S.S.M.; Wang, T.; Zhang, Z.; Hesslein, D.G.T.; Yin, Z.; Kano, A.; Iwamoto, Y.; Li, E.; Craft, J.E.; et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc. Natl. Acad. Sci. USA 2003, 100, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Ueda, Y.; Sawa, Y.; Jeon, S.G.; Ma, J.S.; Okumura, R.; Kubo, A.; Ishii, M.; Okazaki, T.; Murakami, M.; et al. Intestinal CX3C chemokine receptor 1(high) (CX3CR1(high)) myeloid cells prevent T-cell-dependent colitis. Proc. Natl. Acad. Sci. USA 2012, 109, 5010–5015. [Google Scholar] [PubMed]
- Olszak, T.; Neves, J.F.; Dowds, C.M.; Baker, K.; Glickman, J.; Davidson, N.O.; Lin, C.-S.; Jobin, C.; Brand, S.; Sotlar, K.; et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature 2014, 509, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-Mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [PubMed]
- Du, W.; Hong, J.; Wang, Y.-C.; Zhang, Y.-J.; Wang, P.; Su, W.-Y.; Lin, Y.-W.; Lu, R.; Zou, W.-P.; Xiong, H.; et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J. Cell. Mol. Med. 2012, 16, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [PubMed]
- Waldner, M.J.; Wirtz, S.; Jefremow, A.; Warntjen, M.; Neufert, C.; Atreya, R.; Becker, C.; Weigmann, B.; Vieth, M.; Rose-John, S.; et al. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J. Exp. Med. 2010, 207, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Stolfi, C.; Rizzo, A.; Franzè, E.; Rotondi, A.; Fantini, M.C.; Sarra, M.; Caruso, R.; Monteleone, I.; Sileri, P.; Franceschilli, L.; et al. Involvement of interleukin-21 in the regulation of colitis-associated colon cancer. J. Exp. Med. 2011, 208, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Wang, H.; Deng, L.; Hou, J.; Shi, R.; Yao, M.; Gao, Y.; Yao, A.; Wang, X.; Yu, L.; et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 2013. [Google Scholar] [CrossRef]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Neurath, M.F. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015, 26, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Monteleone, I.; Fina, D.; Vavassori, P.; del Vecchio Blanco, G.; Caruso, R.; Tersigni, R.; Alessandroni, L.; Biancone, L.; Naccari, G.C.; et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology 2005, 128, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Shah, S.; Comiskey, M.; de Jong, Y.P.; Wang, B.; Mizoguchi, E.; Bhan, A.K.; Terhorst, C. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J. Exp. Med. 1998, 187, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Finotto, S.; Kanzler, S.; Lohse, A.W.; Blessing, M.; Lehr, H.A.; Galle, P.R.; Neurath, M.F. Cutting Edge: Chronic intestinal inflammation in STAT-4 transgenic mice: Characterization of disease and adoptive transfer by TNF- Plus IFN-γ-producing CD4+ T cells that respond to bacterial antigens. J. Immunol. 1999, 162, 1884–1888. [Google Scholar] [PubMed]
- O’Malley, J.T.; Eri, R.D.; Stritesky, G.L.; Mathur, A.N.; Chang, H.-C.; Hogenesch, H.; Srinivasan, M.; Kaplan, M.H. STAT4 isoforms differentially regulate Th1 cytokine production and the severity of inflammatory bowel disease. J. Immunol. 2008, 181, 5062–5070. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yang, Y.; Qiu, G.; Lal, G.; Yin, N.; Wu, Z.; Bromberg, J.S.; Ding, Y. Stat4 is critical for the balance between Th17 cells and regulatory T cells in colitis. J. Immunol. 2011, 186, 6597–6606. [Google Scholar] [CrossRef] [PubMed]
- Harbour, S.N.; Maynard, C.L.; Zindl, C.L.; Schoeb, T.R.; Weaver, C.T. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 2015, 112, 7061–7066. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Seiderer, J.; Nagy, M.; Fries, C.; Beigel, F.; Weidinger, M.; Pfennig, S.; Klein, W.; Epplen, J.T.; Lohse, P.; et al. Evidence for STAT4 as a common autoimmune gene: rs7574865 is associated with colonic Crohn’s disease and early disease onset. PLoS ONE 2010, 5, e10373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Gallo, L.M.; Palomino-Morales, R.J.; Gómez-García, M.; Cardeña, C.; Rodrigo, L.; Nieto, A.; Alcain, G.; Cueto, I.; López-Nevot, M.A.; Martin, J. STAT4 gene influences genetic predisposition to ulcerative colitis but not Crohn’s disease in the Spanish population: a replication study. Hum. Immunol. 2010, 71, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.H.; Zheng, C.Q.; Yang, X.Z.; Zhang, W.J. Increased expression and activation of IL-12-induced Stat4 signaling in the mucosa of ulcerative colitis patients. Cell. Immunol. 2007, 248, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Ohtsuka, Y.; Ikuse, T.; Baba, Y.; Yamakawa, Y.; Aoyagi, Y.; Fujii, T.; Kudo, T.; Nagata, S.; Shimizu, T. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2010, 52, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Glosson-Byers, N.L.; Sehra, S.; Kaplan, M.H. STAT4 is required for IL-23 responsiveness in Th17 memory cells and NKT cells. JAK-STAT 2014, 3, e955393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Okamoto, S.; Hisamatsu, T.; Kamada, N.; Chinen, H.; Saito, R.; Kitazume, M.T.; Nakazawa, A.; Sugita, A.; Koganei, K.; et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 2008, 57, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, R.; Miller, L.; Yao, W.; Gupta, S.; Steiner, S.; Kaplan, M.H. Altered STAT4 isoform expression in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Uemura, A.; Takehara, T.; Miyagi, T.; Suzuki, T.; Tatsumi, T.; Ohkawa, K.; Kanto, T.; Hiramatsu, N.; Hayashi, N. Natural killer cell is a major producer of interferon gamma that is critical for the IL-12-induced anti-tumor effect in mice. Cancer Immunol. Immunother. CII 2010, 59, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Klupp, F.; Diers, J.; Kahlert, C.; Neumann, L.; Halama, N.; Franz, C.; Schmidt, T.; Lasitschka, F.; Warth, A.; Weitz, J.; et al. Expressional STAT3/STAT5 ratio is an independent prognostic marker in colon carcinoma. Ann. Surg. Oncol. 2015, 22, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- DiFedele, L.M.; He, J.; Bonkowski, E.L.; Han, X.; Held, M.A.; Bohan, A.; Menon, R.K.; Denson, L.A. Tumor necrosis factor alpha blockade restores growth hormone signaling in murine colitis. Gastroenterology 2005, 128, 1278–1291. [Google Scholar] [PubMed]
- Han, X.; Benight, N.; Osuntokun, B.; Loesch, K.; Frank, S.J.; Denson, L.A. Tumour necrosis factor alpha blockade induces an anti-inflammatory growth hormone signalling pathway in experimental colitis. Gut 2007, 56, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Osuntokun, B.; Benight, N.; Loesch, K.; Frank, S.J.; Denson, L.A. Signal transducer and activator of transcription 5b promotes mucosal tolerance in pediatric Crohn’s disease and murine colitis. Am. J. Pathol. 2006, 169, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ren, X.; Jurickova, I.; Groschwitz, K.; Pasternak, B.A.; Xu, H.; Wilson, T.A.; Hogan, S.P.; Denson, L.A. Regulation of intestinal barrier function by signal transducer and activator of transcription 5b. Gut 2009, 58, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.; Nivarthi, H.; Mayhew, C.N.; Lo, Y.-H.; Noah, T.K.; Vallance, J.; Rülicke, T.; Müller, M.; Jegga, A.G.; Tang, W.; et al. Activated STAT5 confers resistance to intestinal injury by increasing intestinal stem cell proliferation and regeneration. Stem Cell Rep. 2015, 4, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A.; Tato, C.M.; Davidson, T.S.; Kanno, Y.; Chen, Z.; Yao, Z.; Blank, R.B.; Meylan, F.; Siegel, R.; Hennighausen, L.; et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007, 26, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Kanno, Y.; Kerenyi, M.; Stephens, G.; Durant, L.; Watford, W.T.; Laurence, A.; Robinson, G.W.; Shevach, E.M.; Moriggl, R.; et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood 2007, 109, 4368–4375. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Becker, C.; Tubbe, I.; Nikolaev, A.; Lehr, H.A.; Galle, P.; Neurath, M.F. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 2006, 55, 671–680. [Google Scholar] [PubMed]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.-K.; Oh, J.; Li, P.; West, E.E.; Wong, E.A.; Andraski, A.B.; Spolski, R.; Yu, Z.-X.; He, J.; Kelsall, B.L.; et al. The cytokines IL-21 and GM-CSF have opposing regulatory roles in the apoptosis of conventional dendritic cells. Immunity 2013, 38, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Yang, F.; Zhou, Y.; Yang, H.; Low, P.; Kemeny, D.; Tan, P.; Moh, A.; Kaplan, M.; Zhang, Y.; et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014, 24, 1387–1402. [Google Scholar] [PubMed]
- Mao, Y.; Li, Z.; Lou, C.; Zhang, Y. Expression of phosphorylated Stat5 predicts expression of cyclin D1 and correlates with poor prognosis of colonic adenocarcinoma. Int. J. Colorectal Dis. 2011, 26, 29–35. [Google Scholar] [PubMed]
- Wolf, M.J.; Hoos, A.; Bauer, J.; Boettcher, S.; Knust, M.; Weber, A.; Simonavicius, N.; Schneider, C.; Lang, M.; Stürzl, M.; et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 2012, 22, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Su, W.-Y.; Liang, Q.-C.; Zhang, Z.-G.; Chen, H.-M.; Du, W.; Chen, Y.-X.; Fang, J.-Y. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dutta, P.; Tsurumi, A.; Li, J.; Wang, J.; Land, H.; Li, W.X. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Chaturvedi, R.; Washington, M.K.; Kuhnhein, L.A.; Moore, P.D.; Coggeshall, S.S.; McDonough, E.M.; Weitkamp, J.-H.; Singh, A.B.; Coburn, L.A.; et al. STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin-2 and Th2-inducing cytokines. J. Immunol. 2013, 190, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Van Kampen, C.; Gauldie, J.; Collins, S.M. Proinflammatory properties of IL-4 in the intestinal microenvironment. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G111–G117. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.-C.; et al. Interleukin 4 inhibits TGF-β-induced-Foxp3+T cells and generates, in combination with TGF-β, Foxp3-effector T cells that produce interleukins 9 and 10. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Frey, M.R.; Washington, M.K.; Chaturvedi, R.; Kuhnhein, L.A.; Matta, P.; Revetta, F.L.; Wilson, K.T.; Polk, D.B. STAT6 activation in ulcerative colitis: A new target for prevention of IL-13-induced colon epithelial cell dysfunction. Inflamm. Bowel Dis. 2011, 17, 2224–2234. [Google Scholar] [PubMed]
- Cosín-Roger, J.; Ortiz-Masiá, D.; Calatayud, S.; Hernández, C.; Esplugues, J.V.; Barrachina, M.D. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deuring, J.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C.J. STAT1, STAT6 and adenosine 3’,5’-cyclic monophosphate (cAMP) signaling drive SOCS3 expression in inactive ulcerative colitis. Mol. Med. Camb. Mass 2012, 18, 1412–1419. [Google Scholar] [PubMed]
- Klein, W.; Tromm, A.; Folwaczny, C.; Hagedorn, M.; Duerig, N.; Epplen, J.; Schmiegel, W.; Griga, T. The G2964A polymorphism of the STAT6 gene in inflammatory bowel disease. Dig. Liver Dis. 2005, 37, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Wick, E.C.; LeBlanc, R.E.; Ortega, G.; Robinson, C.; Platz, E.; Pardoll, D.M.; Iacobuzio-Donahue, C.; Sears, C.L. Shift from pStat6 to pStat3 predominance is associated with inflammatory bowel disease-associated dysplasia. Inflamm. Bowel Dis. 2012, 18, 1267–1274. [Google Scholar] [PubMed]
- Zhang, M.; Zhou, Y.; Xie, C.; Zhou, F.; Chen, Y.; Han, G.; Zhang, W.J. STAT6 specific shRNA inhibits proliferation and induces apoptosis in colon cancer HT-29 cells. Cancer Lett. 2006, 243, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.B.; Iovino, F.; Lombardo, Y.; Eterno, V.; Höger, T.; Dieli, F.; Stassi, G.; Todaro, M. Survivin is regulated by interleukin-4 in colon cancer stem cells. J. Cell. Physiol. 2010, 225, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wang, N.; Wei, X.; Xie, G.; Li, J.; Liang, H. Interleukin-12 inhibits the survival of human colon cancer stem cells in vitro and their tumor initiating capacity in mice. Cancer Lett. 2012, 322, 92–97. [Google Scholar] [PubMed]
- Sandborn, W.J.; Gasink, C.; Gao, L.-L.; Blank, M.A.; Johanns, J.; Guzzo, C.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Rutgeerts, P.; et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W. Tofacitinib, an oral janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Wang, W.; Niezychowski, W.; Study A3921043 Investigators. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin. Gastroenterol. Hepatol. 2014. [Google Scholar] [CrossRef]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2015. [Google Scholar] [CrossRef]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Billmeier, U.; Rath, T.; Mudter, J.; Vieth, M.; Neumann, H.; Neurath, M. First case report of exacerbated ulcerative colitis after anti-interleukin-6R salvage therapy. World J. Gastroenterol. 2015, 21, 12963–12969. [Google Scholar] [CrossRef] [PubMed]
- McLean, M.H.; Neurath, M.F.; Durum, S.K. Targeting Interleukins for the Treatment of Inflammatory Bowel Disease-What Lies Beyond Anti-TNF Therapy? Inflamm. Bowel Dis. 2014, 20, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.; Kiladjian, J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cell type | STAT | Effects and functions in IBD |
|---|---|---|
| Lymphocytes | STAT1 | Inhibition → amelioration of colitis Important for Th1 function, but impedes Th2 and Treg function |
| STAT3 | IL-6-mediated resistance to apoptosis Inhibition → amelioration of colitis drives Th17 differentiation promotes IL-10-dependent Treg function | |
| STAT4 | drives Th1 differentiation, suppresses Treg function Deletion → protection from colitis hyperactivation → spontaneous colitis | |
| STAT5 | drives Treg differentiation, limits Th17 differentiation | |
| STAT6 | drives Th2 and Th9 differentiation, reduces Treg induction Deficiency → protection from colitis, less Th2 cytokines | |
| Macrophages | STAT1 | IL-19-dependent reduction of pro-inflammatory cytokine production |
| STAT3 | Inactivation → chronic colitis, Th1 response, reduced IL-10 signaling | |
| STAT6 | Polarization of M2-like macrophages | |
| Intestinal epithelial cells | STAT1 | activates antibacterial/anti-inflammatory IDO-1 |
| STAT3 | IL-22-dependent promotion of goblet cells, mucus layer and IEC restitution IL-24-mediated mucus production IL-11-promoted maintenance of mucosal integrity | |
| STAT5 | Deficiency → increased colitis susceptibility, enhanced IEC apoptosis Critical for regeneration of crypt epithelium | |
| STAT6 | IL-13-dependent apoptosis |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zundler, S.; Neurath, M.F. Integrating Immunologic Signaling Networks: The JAK/STAT Pathway in Colitis and Colitis-Associated Cancer. Vaccines 2016, 4, 5. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4010005
Zundler S, Neurath MF. Integrating Immunologic Signaling Networks: The JAK/STAT Pathway in Colitis and Colitis-Associated Cancer. Vaccines. 2016; 4(1):5. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4010005
Chicago/Turabian StyleZundler, Sebastian, and Markus F. Neurath. 2016. "Integrating Immunologic Signaling Networks: The JAK/STAT Pathway in Colitis and Colitis-Associated Cancer" Vaccines 4, no. 1: 5. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4010005