
Comprehensive Characterization of Reference Standard Lots of HIV-1 Subtype C Gp120 Proteins for Clinical Trials in Southern African Regions

 and
and 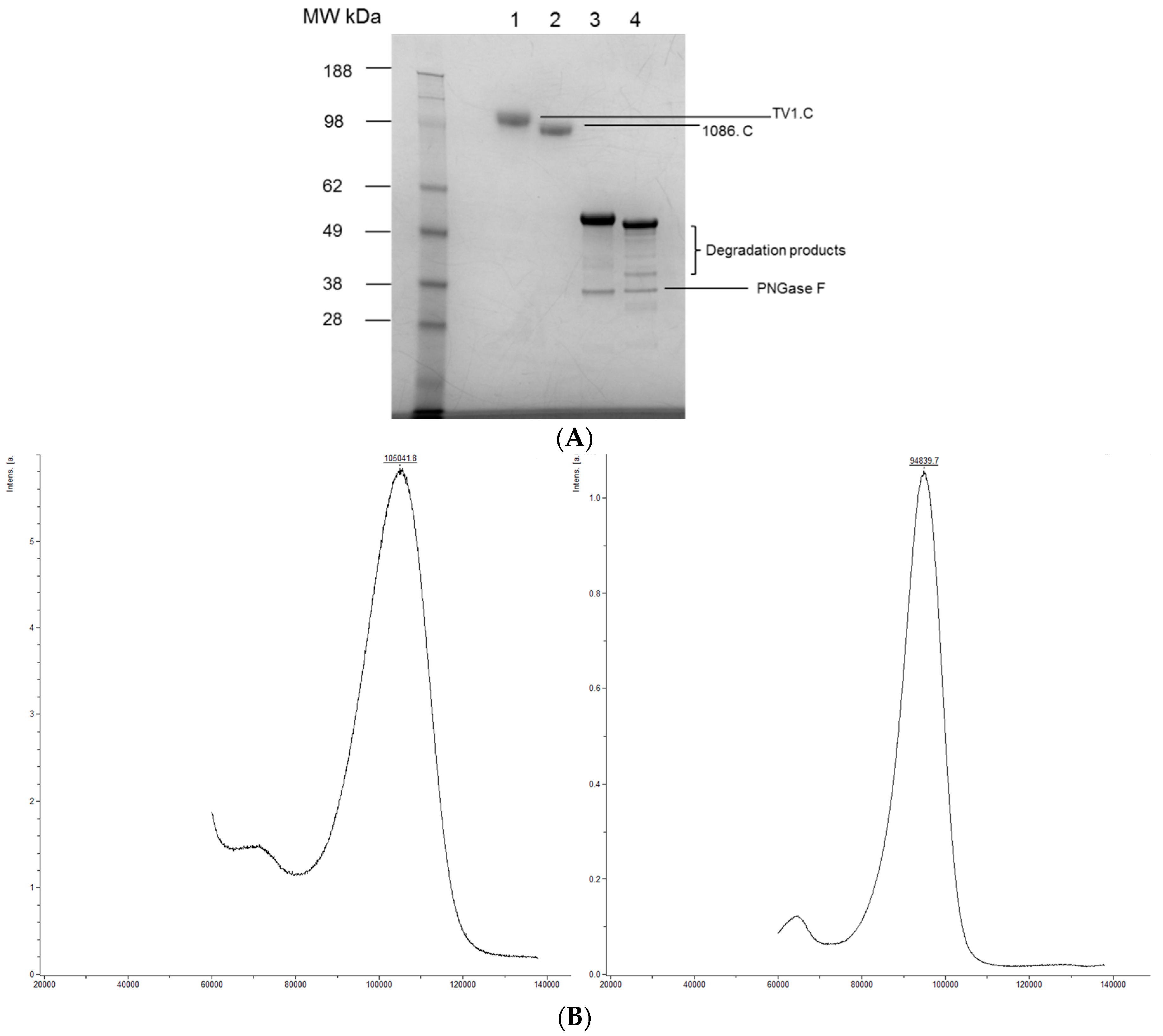{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Production of Reference Lots
2.2. Intact Molecular Weight Determination
2.3. Immunogenicity Assessment
2.4. Peptide Mapping and Isoelectric Focusing (IEF) Gel Electrophoresis
2.5. Differential Scanning Calorimetry (DSC) and Circular Dichroism (CD)
2.6. O-Linked Glycosylation Site Mapping and Identification
2.7. N-Linked Glycosylation Characterization
2.8. Disulfide Bond Mapping
3. Results and Discussion
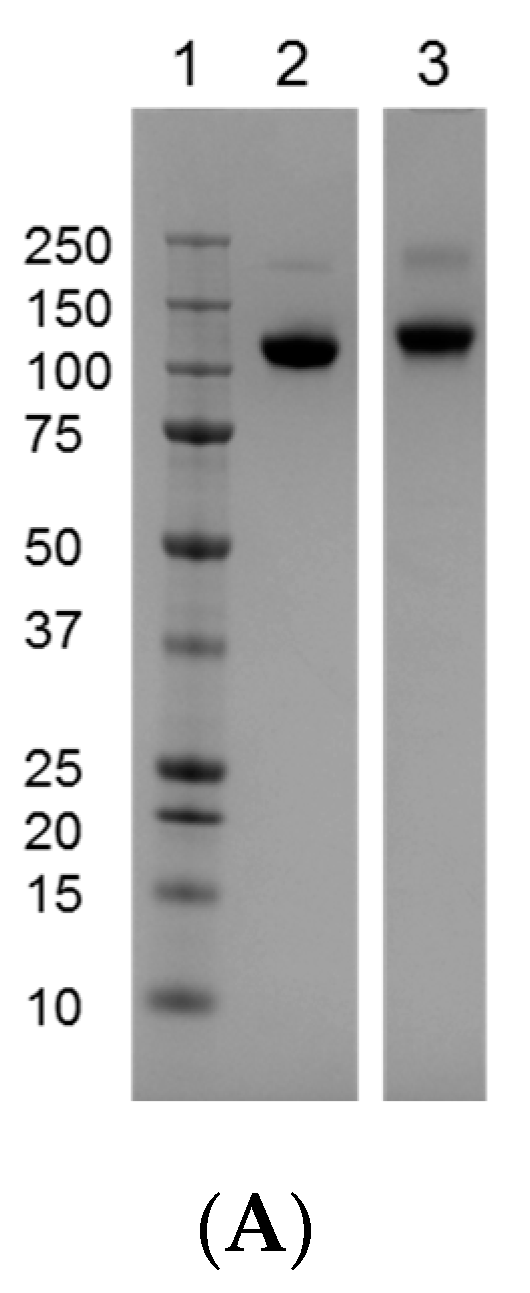
3.1. Comparison of the Reference Materials to the Clinical Trial Material (CTM)
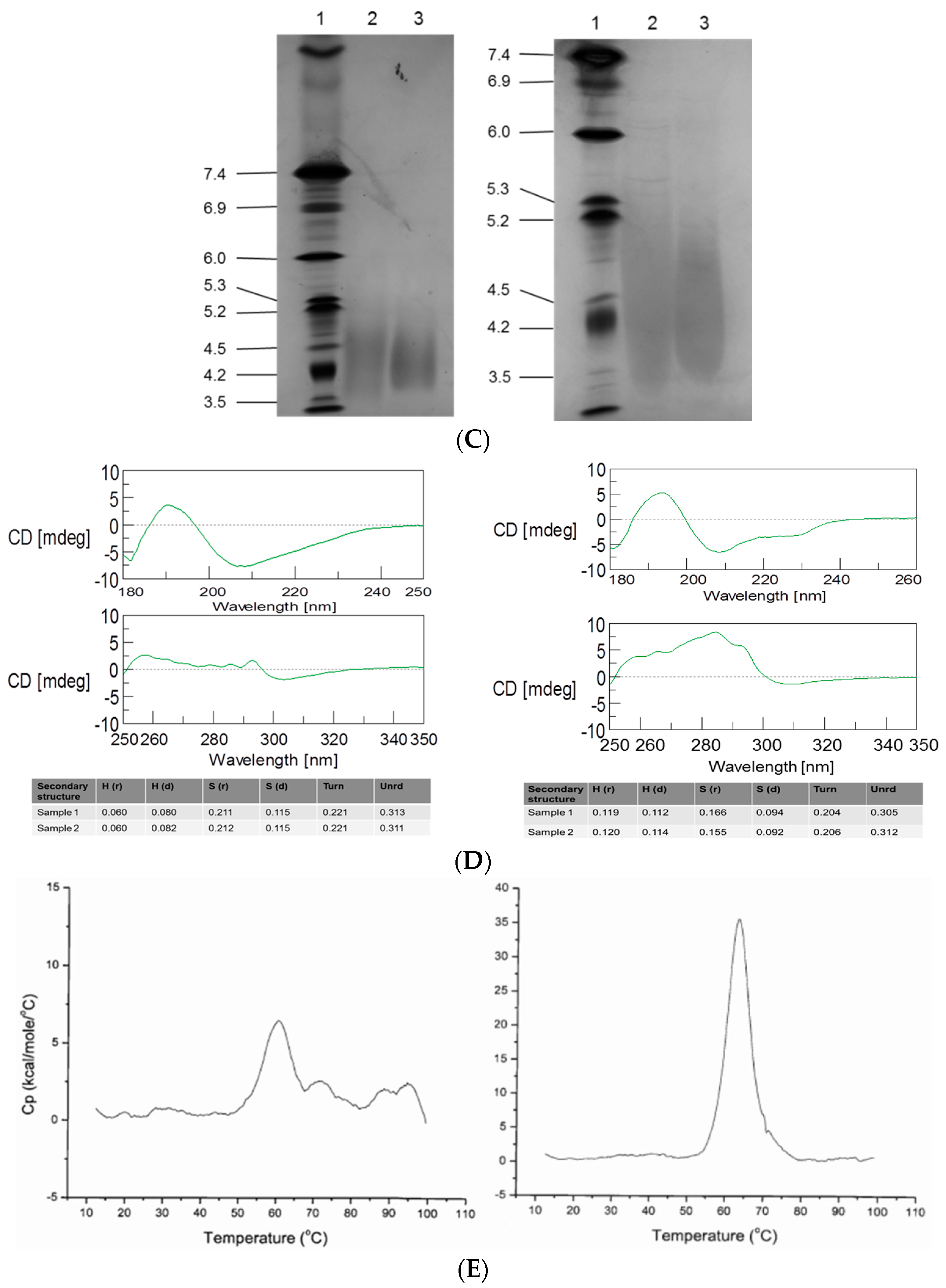
3.2. Intact MW, Charge Heterogeneity, Higher Structure, and Melting Point
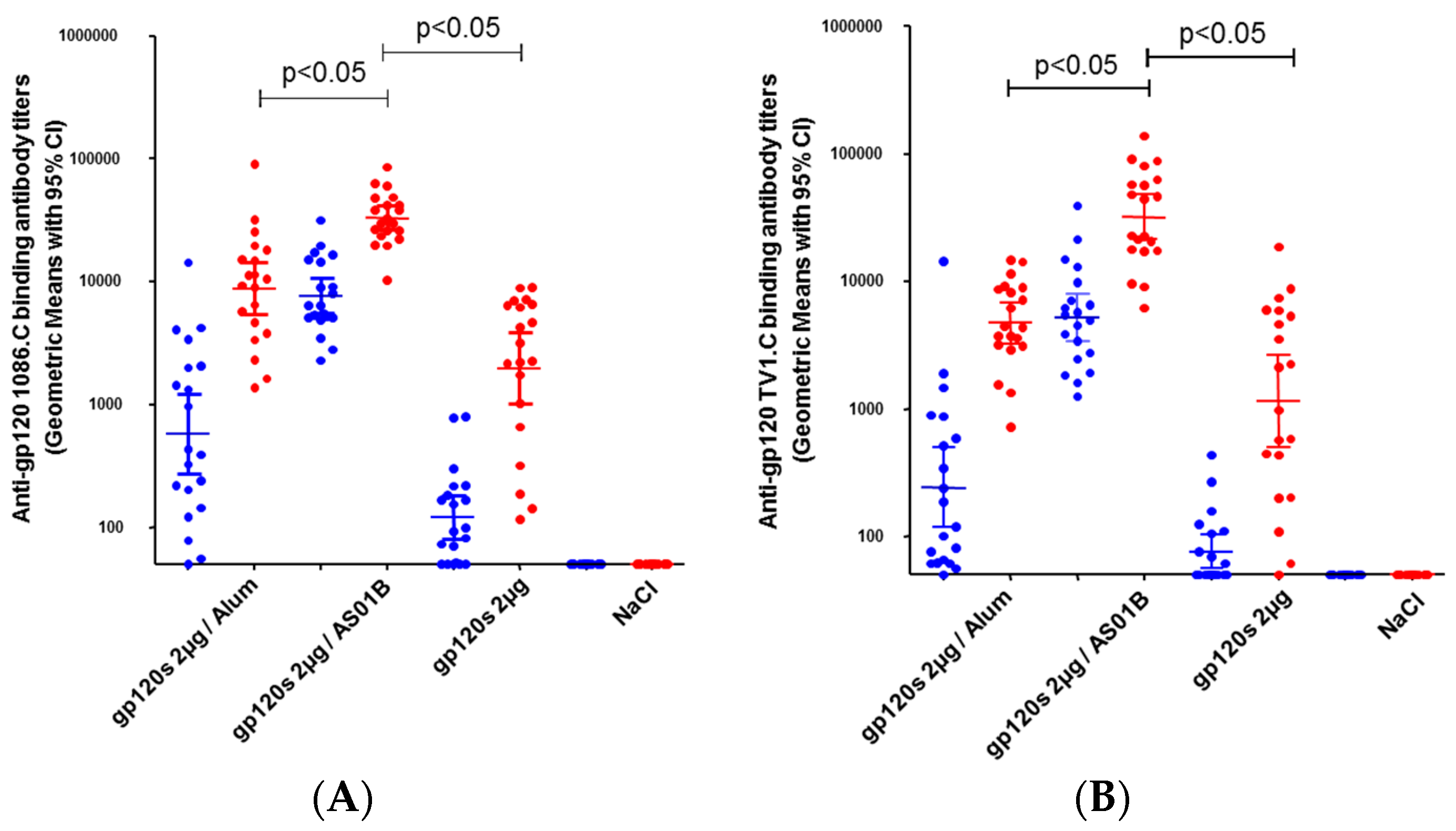
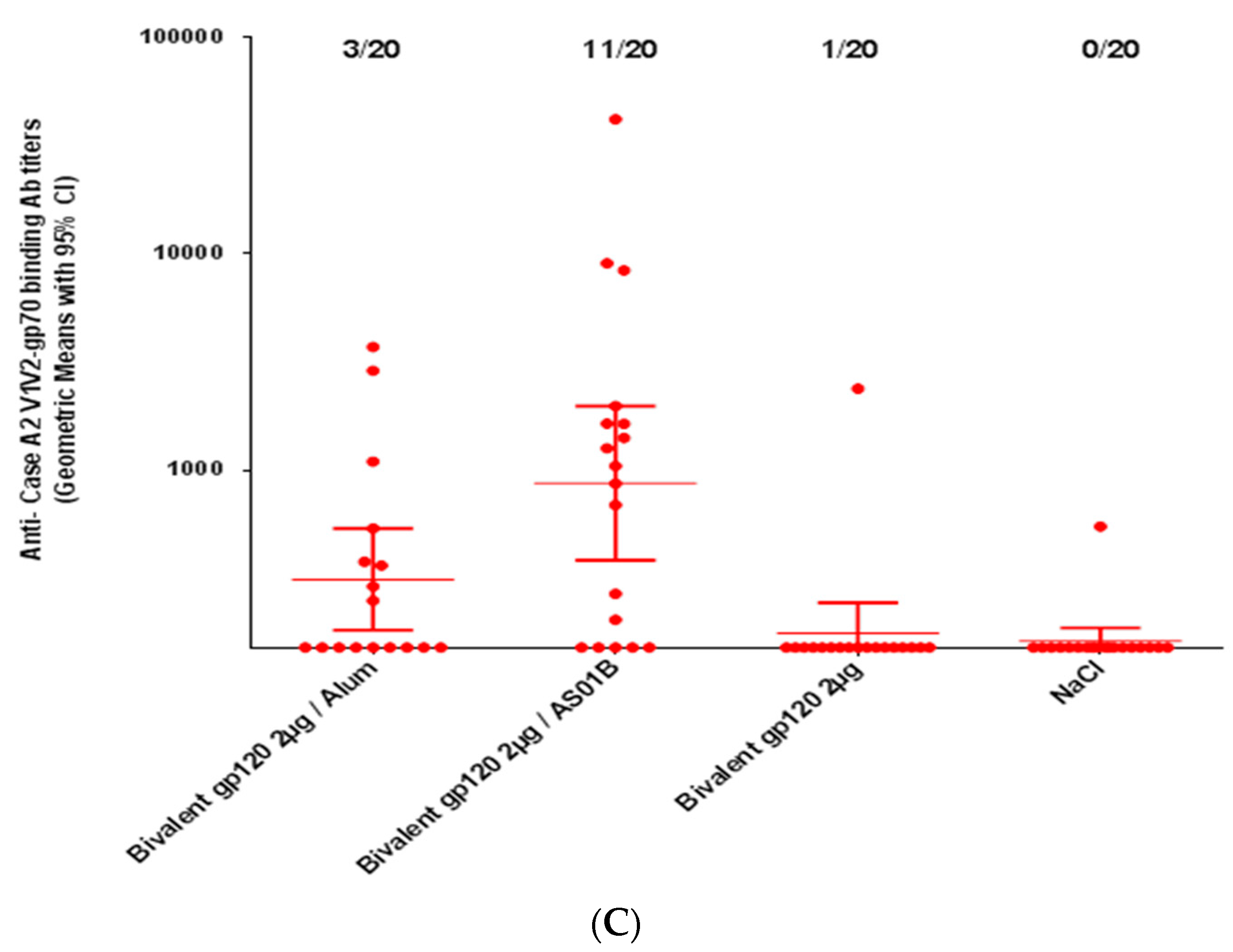
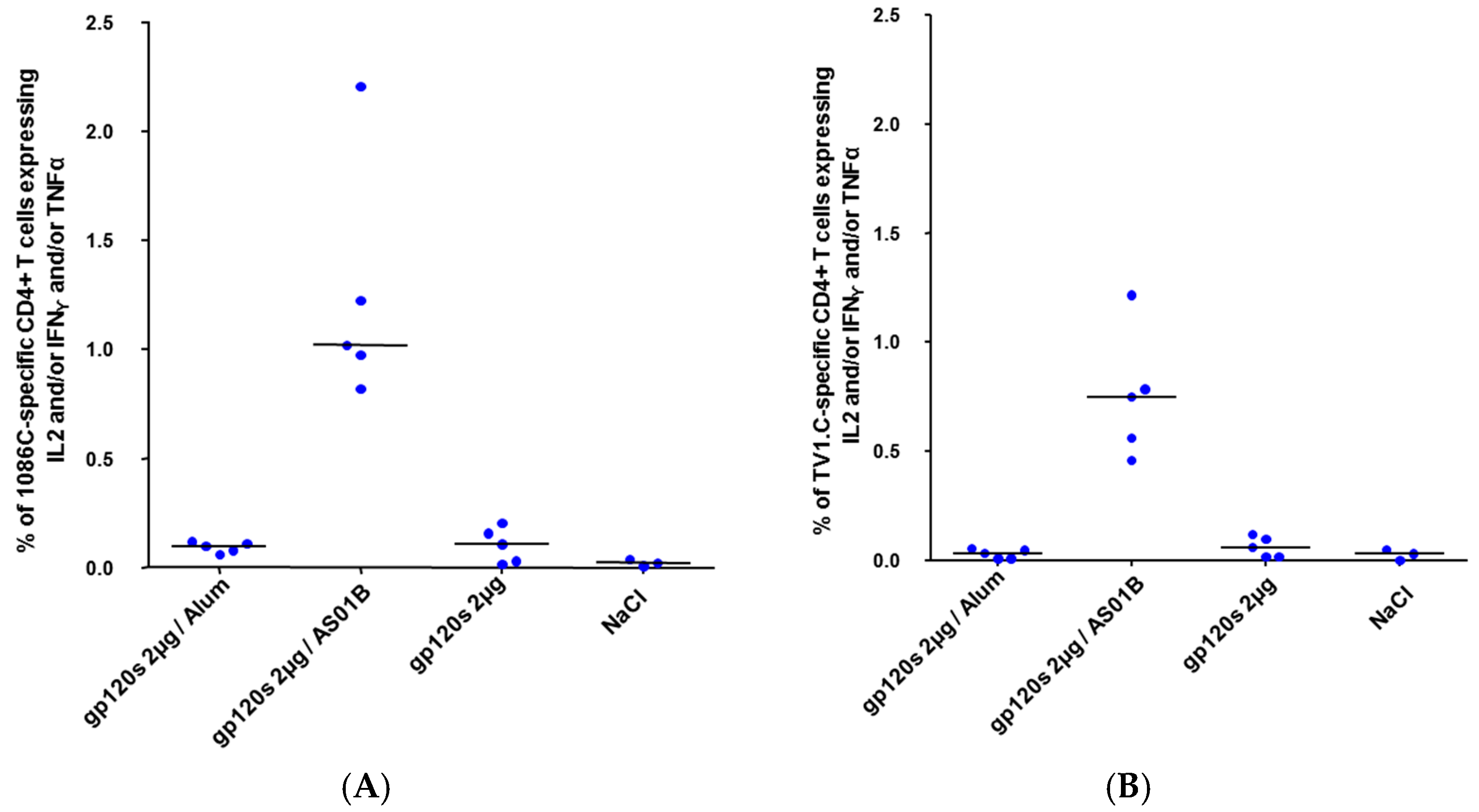
3.3. Immunogenicity of HIV-1 gp120 Clade C Envelopes
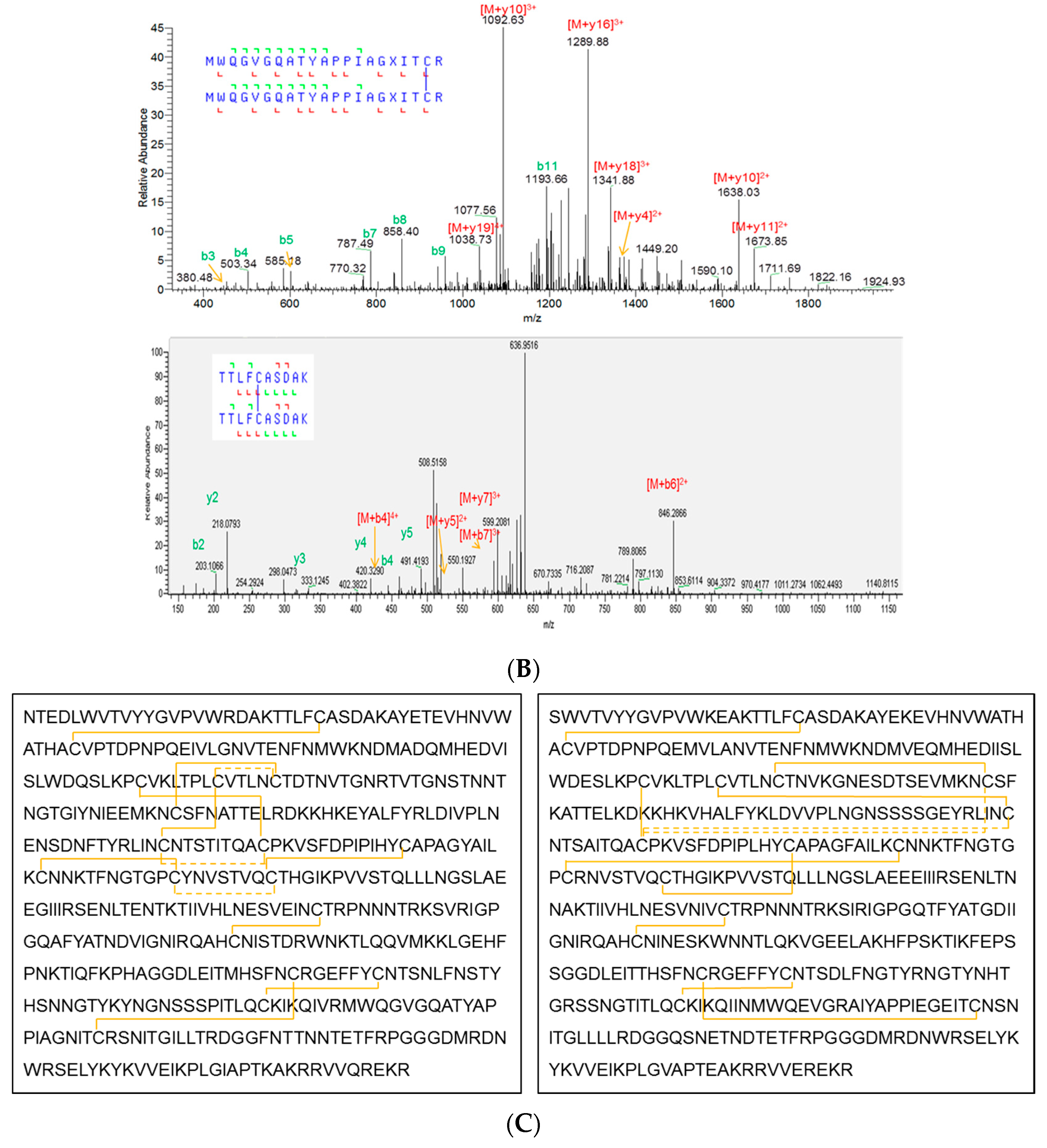
3.4. Primary Sequence and Peptide Mapping
3.5. O-Linked Glycosylation Characterization
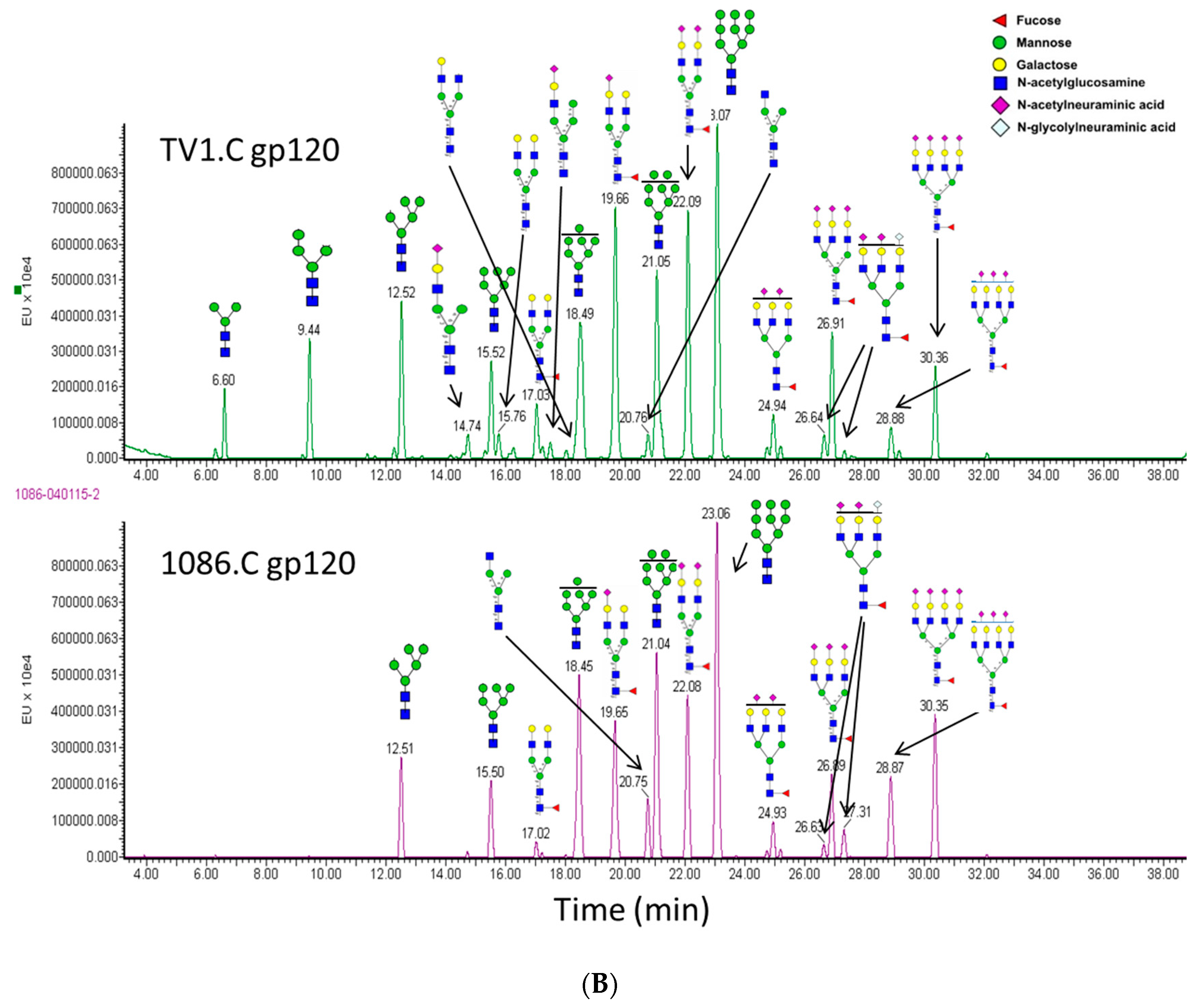
3.6. N-Linked Glycosylation Characterization
3.7. Disulfide Bond Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Robb, M.L.; Rerks-Ngarm, S.; Nitayaphan, S.; Pitisuttithum, P.; Kaewkungwal, J.; Kunasol, P.; Khamboonruang, C.; Thongcharoen, P.; Morgan, P.; Benenson, M.; et al. Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC-HIV (vCP1521) and AIDSVAX B/E: A post-hoc analysis of the Thai phase 3 efficacy trial RV 144. Lancet Infect. Dis. 2012, 12, 531–537. [Google Scholar] [CrossRef]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Ping, L.H.; Joseph, S.B.; Anderson, J.A.; Abrahams, M.R.; Salazar-Gonzalez, J.F.; Kincer, L.P.; Treurnicht, F.K.; Arney, L.; Ojeda, S.; Zhang, M.; et al. Comparison of viral Env proteins from acute and chronic infections with subtype C human immunodeficiency virus type 1 identifies differences in glycosylation and CCR5 utilization and suggests a new strategy for immunogen design. J. Virol. 2013, 87, 7218–7233. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.; Murray, D.; Justement, J.S.; Blazkova, J.; Hallahan, C.W.; Fankuchen, O.; Gittens, K.; Benko, E.; Kovacs, C.; Moir, S.; et al. Broadly neutralizing antibodies suppress HIV in the persistent viral reservoir. Proc. Natl. Acad. Sci. USA 2014, 111, 13151–13156. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Venturi, M.; Schiffner, L.; Kalyanaraman, R.; Katinger, H.; Lloyd, K.O.; Kwong, P.D.; Moore, J.P. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 2002, 76, 7293–7305. [Google Scholar] [CrossRef] [PubMed]
- Pejchal, R.; Doores, K.J.; Walker, L.M.; Khayat, R.; Huang, P.S.; Wang, S.K.; Stanfield, R.L.; Julien, J.P.; Ramos, A.; Crispin, M.; et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 2011, 334, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Shahzad-ul-Hussan, S.; Doria-Rose, N.A.; McLellan, J.S.; Bailer, R.T.; Dai, K.; Loesgen, S.; Louder, M.K.; Staupe, R.P.; Yang, Y.; et al. Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat. Struct. Mol. Biol. 2013, 20, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, L.; Dey, B.; Hessell, A.J.; Van Ryk, D.; Xiang, S.H.; Yang, X.; Zhang, M.Y.; Zwick, M.B.; Arthos, J.; et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 2007, 445, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Zambonelli, C.; Dey, A.K.; Hilt, S.; Stephenson, S.; Go, E.P.; Clark, D.F.; Wininger, M.; Labranche, C.; Montefiori, D.; Liao, H.; et al. GSK Vaccines. Unpublished data 2016. [Google Scholar]
- Totrov, M. Estimated secondary structure propensities within V1/V2 region of HIV gp120 are an important global antibody neutralization sensitivity determinant. PLoS ONE 2014, 9, e94002. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Fonseca, D.; O’Rourke, S.; Berman, P. Protease cleavage sites in HIV-1 gp120 recognized by antigen processing enzymes are conserved and located at receptor binding sites. J. Virol. 2010, 84, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Robert, F.; Bierau, H.; Rossi, M.; Agugiaro, D.; Soranzo, T.; Broly, H.; Mitchell-Logean, C. Degradation of an Fc-fusion recombinant protein by host cell proteases: Identification of a CHO cathepsin D protease. Biotechnol. Bioeng. 2009, 104, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.B.; Tucker, S.P.; Hunter, E.; Schutzbach, J.S.; Compans, R.W. Human immunodeficiency virus type 1 envelope glycoprotein is modified by O-linked oligosaccharides. J. Virol. 1994, 68, 463–468. [Google Scholar] [PubMed]
- Yang, W.; Shah, P.; Eshghi, S.T.; Yang, S.; Sun, S.; Ao, M.; Rubin, A.; Jackson, J.B.; Zhang, H. Glycoform analysis of recombinant and human immunodeficiency virus envelope protein gp120 via higher energy collisional dissociation and spectral-aligning strategy. Anal. Chem. 2014, 86, 6959–6967. [Google Scholar] [CrossRef] [PubMed]
- Go, E.P.; Liao, H.; Alam, S.M.; Hua, D.; Haynes, B.F.; Desaire, H. Characterization of host-cell line specific glycosylation profiles of early transmitted/founder HIV‑1 gp120 envelope proteins. J. Proteome Res. 2013, 12, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Bonomelli, C.; Doores, K.J.; Dunlop, D.C.; Thaney, V.; Dwek, R.A.; Burton, D.R.; Crispin, M.; Scanlan, C.N. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS ONE 2011, 6, e23521. [Google Scholar] [CrossRef] [PubMed]
- Go, E.P.; Zhang, Y.; Menon, S.; Desaire, H. Analysis of the disulfide bond arrangement of the HIV envelope protein CON-S gp140 ΔCFI shows variability in the V1 and V2 regions. J. Proteome Res. 2011, 10, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Finzi, A.; Pacheco, B.; Zeng, X.; Kwon, Y.D.; Kwong, P.D.; Sodroski, J. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J. Virol. Methods 2010, 168, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Coutu, M.; Finzi, A. HIV-1 gp120 dimers decrease the overall affinity of gp120 preparations for CD4-induced ligands. J. Virol. Methods 2015. [Google Scholar] [CrossRef] [PubMed]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Lorin, C.; Koutsoukos, M.; Franco, D.; Bayat, B.; Zhang, Y.; Carfi, A.; Barnett, S.W.; Porter, F. Comprehensive Characterization of Reference Standard Lots of HIV-1 Subtype C Gp120 Proteins for Clinical Trials in Southern African Regions. Vaccines 2016, 4, 17. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4020017
Wang Z, Lorin C, Koutsoukos M, Franco D, Bayat B, Zhang Y, Carfi A, Barnett SW, Porter F. Comprehensive Characterization of Reference Standard Lots of HIV-1 Subtype C Gp120 Proteins for Clinical Trials in Southern African Regions. Vaccines. 2016; 4(2):17. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4020017
Chicago/Turabian StyleWang, Zihao, Clarisse Lorin, Marguerite Koutsoukos, David Franco, Babak Bayat, Ying Zhang, Andrea Carfi, Susan W. Barnett, and Frederick Porter. 2016. "Comprehensive Characterization of Reference Standard Lots of HIV-1 Subtype C Gp120 Proteins for Clinical Trials in Southern African Regions" Vaccines 4, no. 2: 17. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines4020017