Tumor-Infiltrating Lymphoid Cells in Colorectal Cancer Patients with Varying Disease Stages and Microsatellite Instability-High/Stable Tumors

 ,
,  ,
, 
Abstract
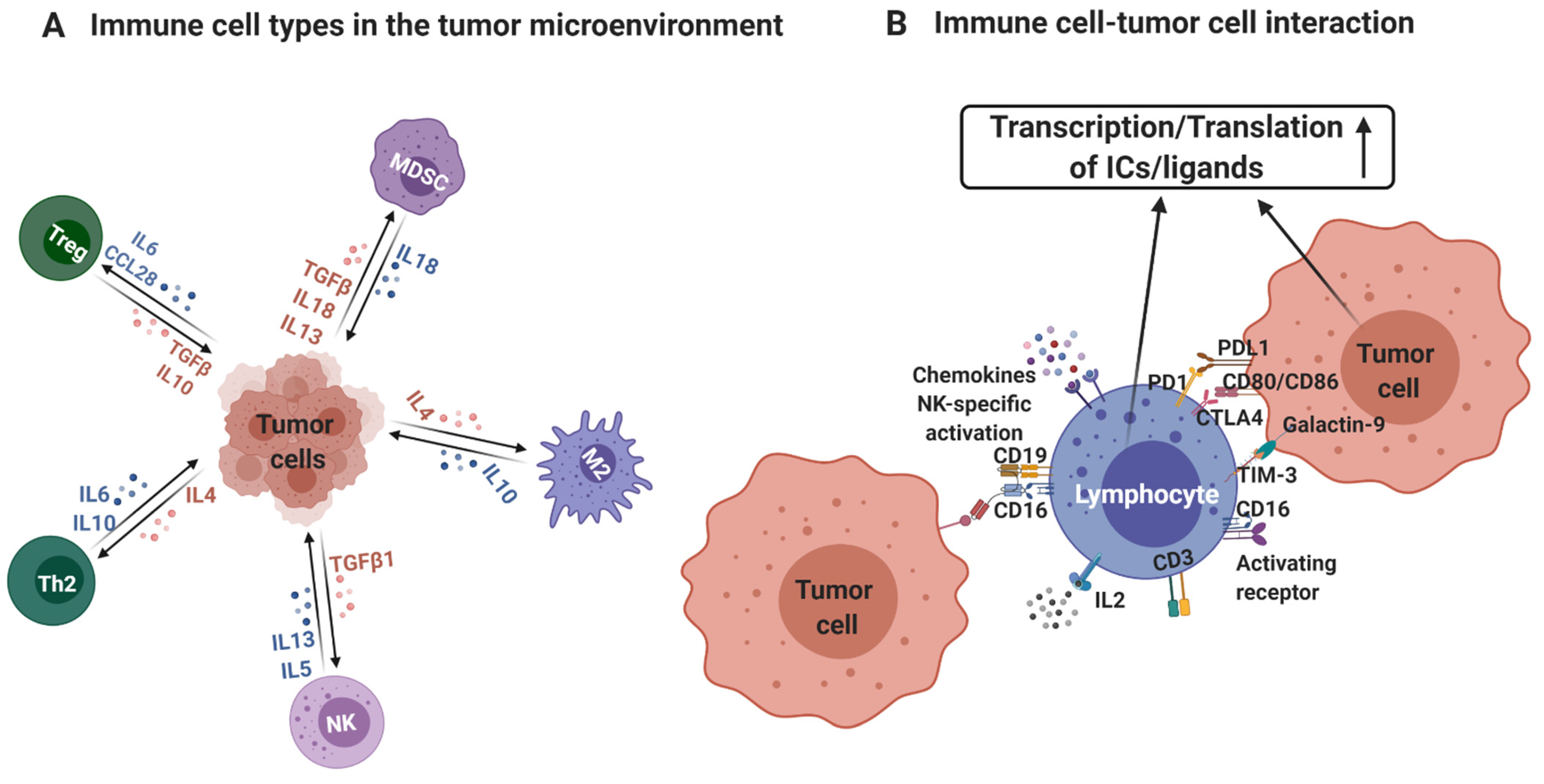
:1. Introduction
2. Materials and Methods
2.1. Sample Acquisition and Storage
2.2. Cell Segregation from Colorectal Tumor Tissues and Normal Colon Tissues
2.3. Flow Cytometric Analyses
2.4. Analyses Using the Cancer Genome Atlas (TCGA) Colorectal Cancer
2.5. Statistical Analyses
3. Results
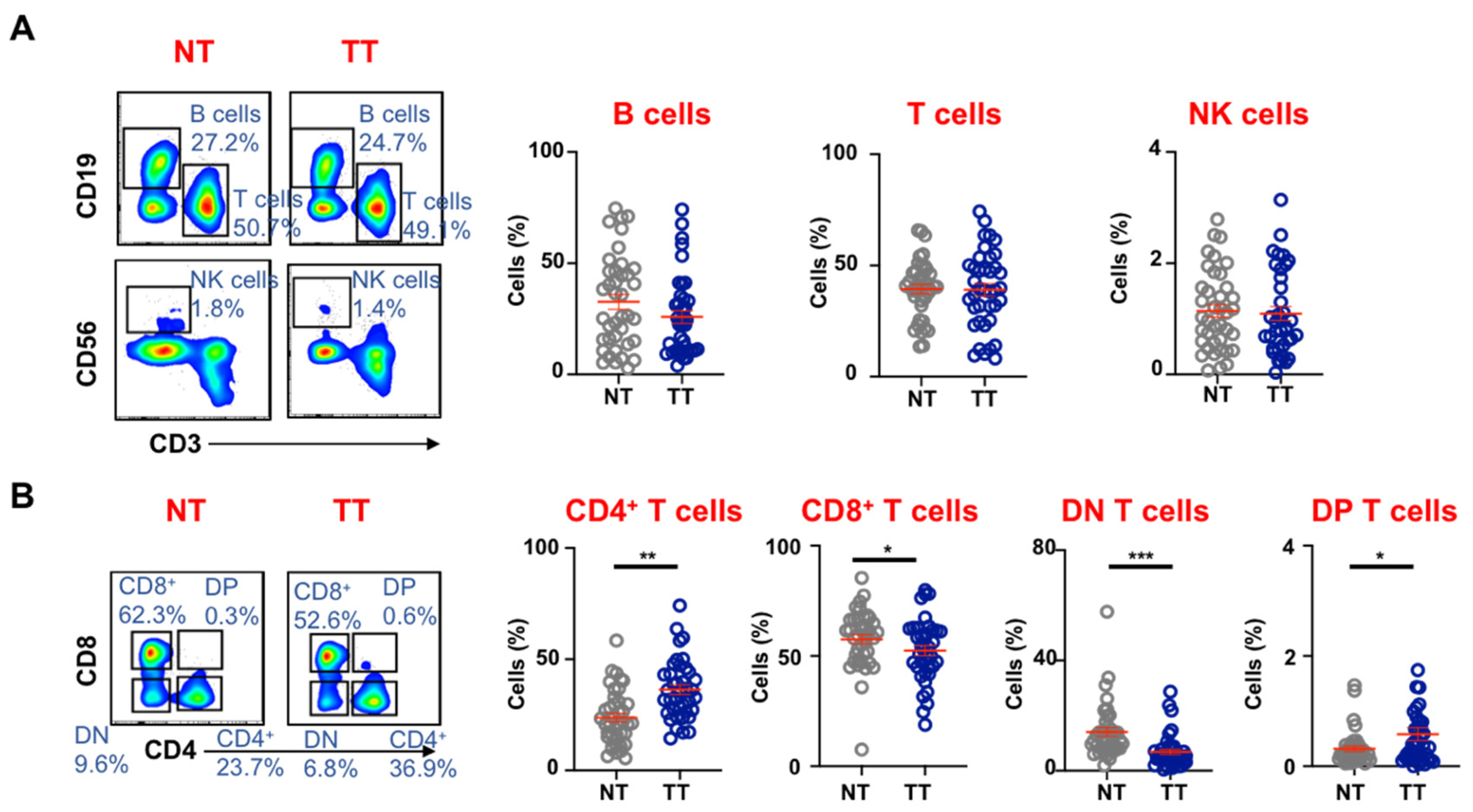
3.1. Tumor-Infiltrating Immune Cells in Colorectal Tumors
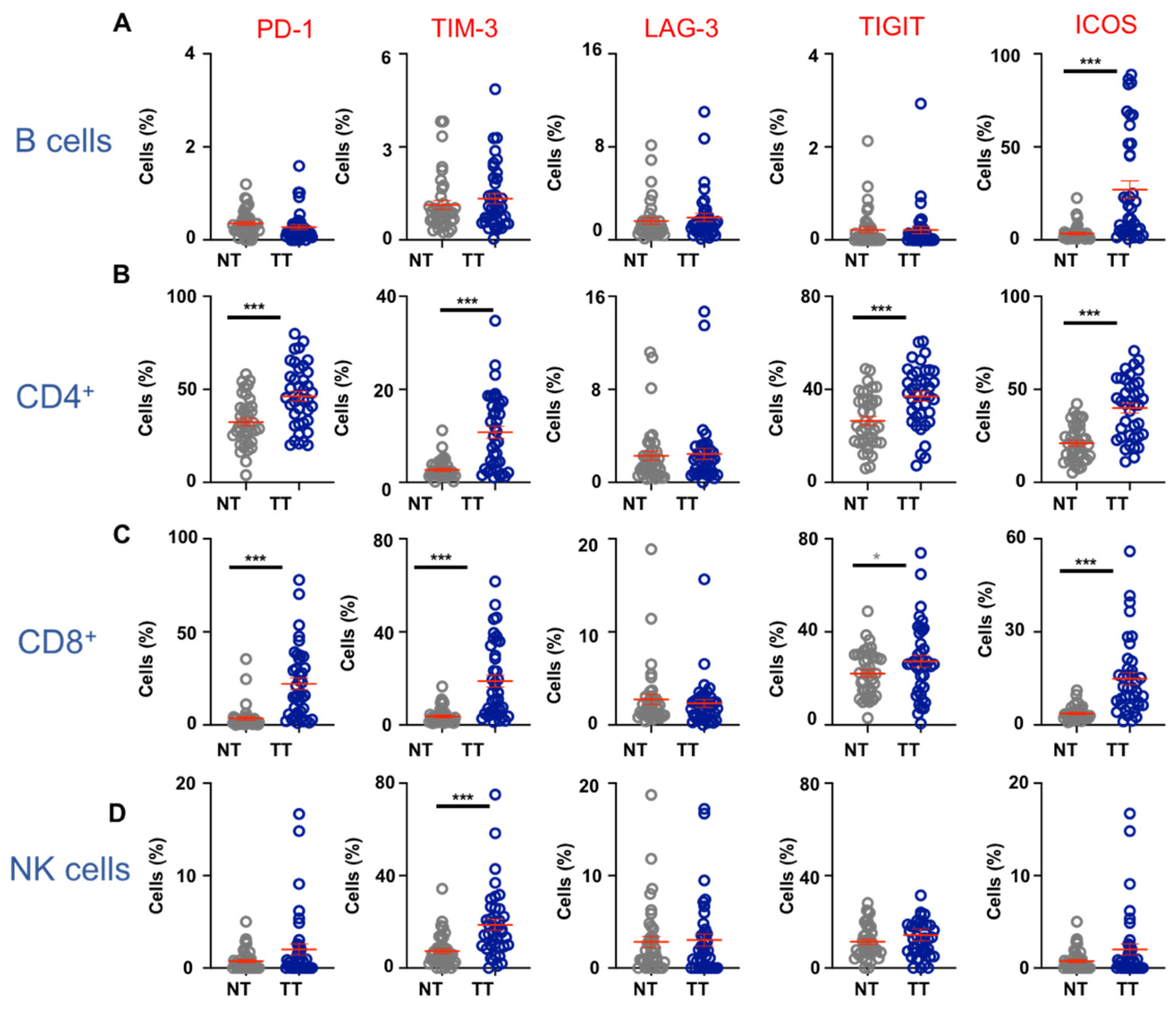
3.2. Immune Checkpoint Expression on Lymphoid Cell Populations in CRC TME
3.3. Immune Checkpoint Co-Expression on T Cells in the CRC TME
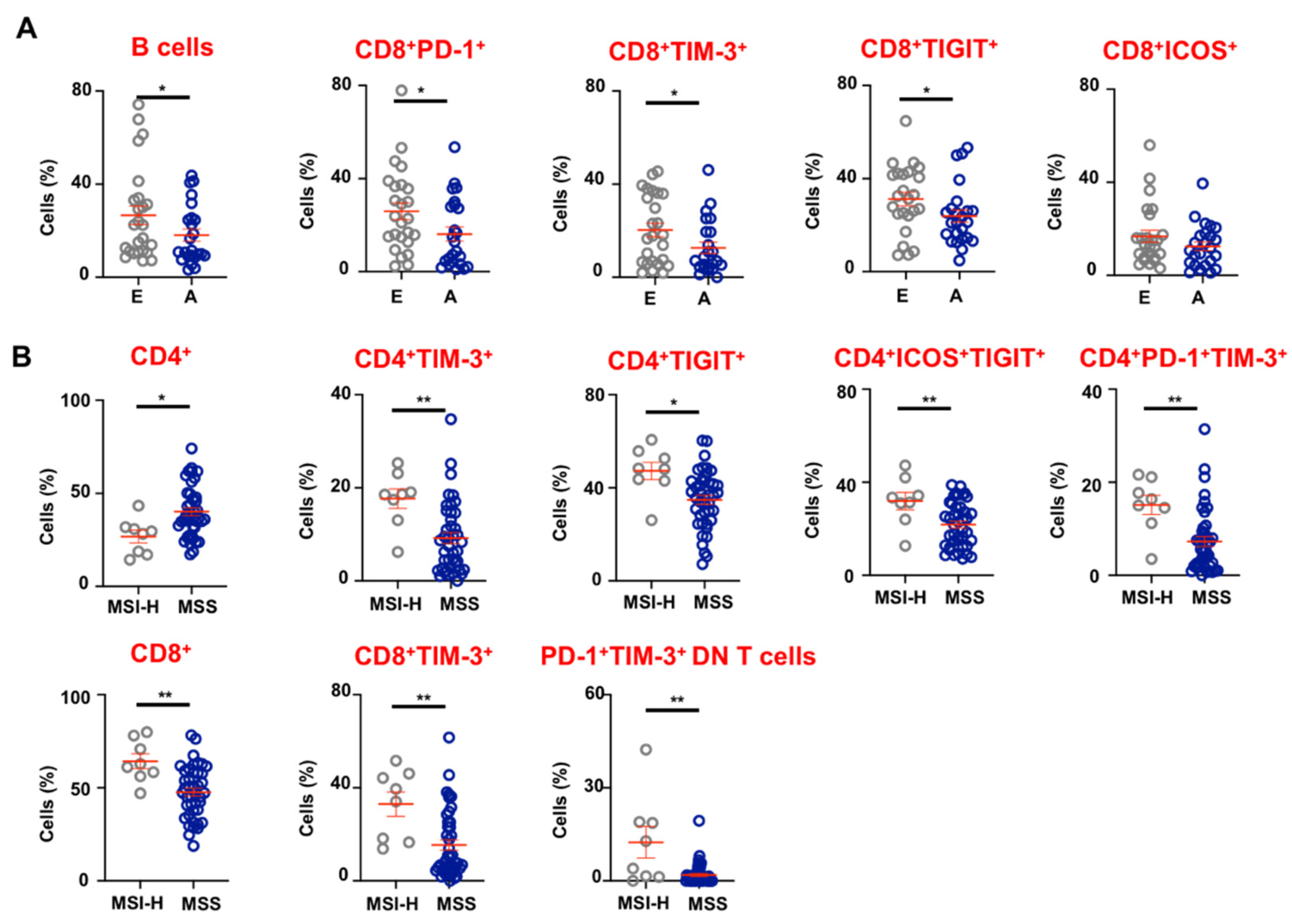
3.4. Tumor-Infiltrating Immune Cells in Advanced and Early Stage CRC Patients
3.5. CRC Patients with dMMR/MSI-H Tumors Have Higher Levels of CD8+ TILs and Exhibit Elevated Expression of Immune Checkpoints
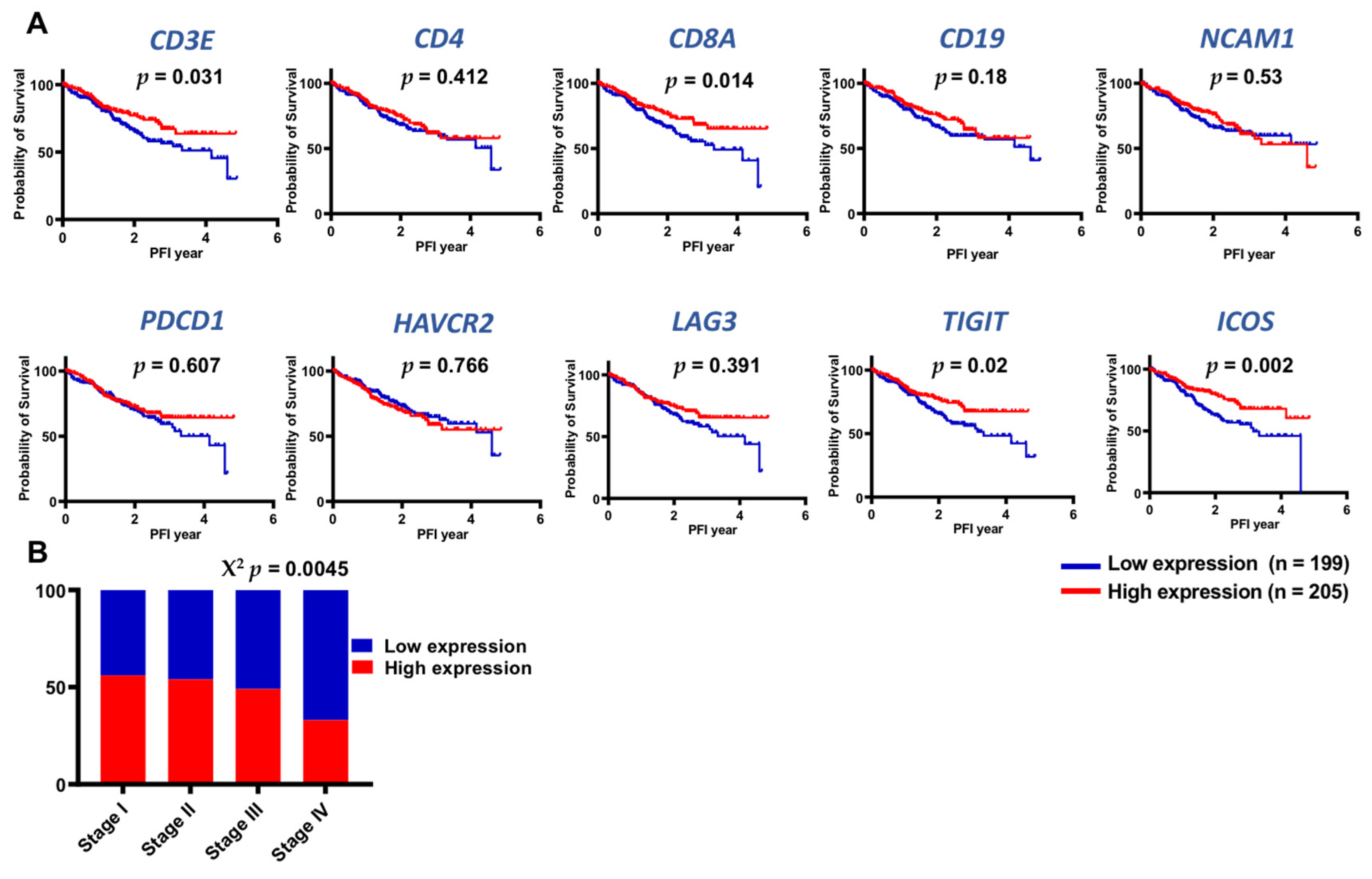
3.6. TCGA Datasets Showed Higher Expression of Lymphoid-Related and IC Genes in Early Stages of CRC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Schlom, J. Tumor-infiltrating immune cells and prognosis: The potential link between conventional cancer therapy and immunity. Exp. Biol. Med. (Maywood) 2011, 236, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Bindea, G.; Pages, F.; Galon, J. Tumor immunosurveillance in human cancers. Cancer Metastasis Rev. 2011, 30, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Sasidharan Nair, V.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2020, 65, 1–12. [Google Scholar] [CrossRef]
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune Checkpoint Modulation in Colorectal Cancer: What’s New and What to Expect. J. Immunol. Res. 2015, 2015, 158038. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Murshed, K.; Al-Dhaheri, M.; Khawar, M.; Abu Nada, M.; Elkord, E. Immune Checkpoints in Circulating and Tumor-Infiltrating CD4(+) T Cell Subsets in Colorectal Cancer Patients. Front. Immunol. 2019, 10, 2936. [Google Scholar] [CrossRef]
- Kitsou, M.; Ayiomamitis, G.D.; Zaravinos, A. High expression of immune checkpoints is associated with the TIL load, mutation rate and patient survival in colorectal cancer. Int. J. Oncol. 2020, 57, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite instability testing and its role in the management of colorectal cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S.; et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Kim, H.J.; Wu, H.; Jalali, S.; Tang, X.; Krull, J.E.; Ding, W.; Novak, A.J.; Ansell, S.M. TIGIT Expression Is Associated with T-cell Suppression and Exhaustion and Predicts Clinical Outcome and Anti-PD-1 Response in Follicular Lymphoma. Clin. Cancer Res. 2020, 26, 5217–5231. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; De Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 195–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atreya, I.; Neurath, M.F. Immune cells in colorectal cancer: Prognostic relevance and therapeutic strategies. Expert Rev. Anticancer Ther. 2008, 8, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimabukuro-Vornhagen, A.; Schlosser, H.A.; Gryschok, L.; Malcher, J.; Wennhold, K.; Garcia-Marquez, M.; Herbold, T.; Neuhaus, L.S.; Becker, H.J.; Fiedler, A.; et al. Characterization of tumor-associated B-cell subsets in patients with colorectal cancer. Oncotarget 2014, 5, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, A.M.; Henriksson, M.L.; Van Guelpen, B.; Stenling, R.; Oberg, A.; Rutegard, J.; Palmqvist, R. Colorectal cancer prognosis depends on T-cell infiltration and molecular characteristics of the tumor. Mod. Pathol. 2011, 24, 671–682. [Google Scholar] [CrossRef]
- Sherwood, A.M.; Emerson, R.O.; Scherer, D.; Habermann, N.; Buck, K.; Staffa, J.; Desmarais, C.; Halama, N.; Jaeger, D.; Schirmacher, P.; et al. Tumor-infiltrating lymphocytes in colorectal tumors display a diversity of T cell receptor sequences that differ from the T cells in adjacent mucosal tissue. Cancer Immunol. Immunother. 2013, 62, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Syed Khaja, A.S.; Toor, S.M.; El Salhat, H.; Ali, B.R.; Elkord, E. Intratumoral FoxP3(+)Helios(+) Regulatory T Cells Upregulating Immunosuppressive Molecules Are Expanded in Human Colorectal Cancer. Front. Immunol. 2017, 8, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrabayrouse, G.; Corvaisier, M.; Ouisse, L.H.; Bossard, C.; Le Mevel, B.; Potiron, L.; Meurette, G.; Gervois, N.; Jotereau, F. Tumor-reactive CD4+ CD8alphabeta+ CD103+ alphabetaT cells: A prevalent tumor-reactive T-cell subset in metastatic colorectal cancers. Int. J. Cancer 2011, 128, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Scurr, M.; Ladell, K.; Besneux, M.; Christian, A.; Hockey, T.; Smart, K.; Bridgeman, H.; Hargest, R.; Phillips, S.; Davies, M.; et al. Highly prevalent colorectal cancer-infiltrating LAP(+) Foxp3(−) T cells exhibit more potent immunosuppressive activity than Foxp3(+) regulatory T cells. Mucosal Immunol. 2014, 7, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Yang, L.; Young, K.J.; DuTemple, B.; Zhang, L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat. Med. 2000, 6, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar]
- Coppola, A.; Arriga, R.; Lauro, D.; Del Principe, M.I.; Buccisano, F.; Maurillo, L.; Palomba, P.; Venditti, A.; Sconocchia, G. NK Cell Inflammation in the Clinical Outcome of Colorectal Carcinoma. Front. Med. (Lausanne) 2015, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Saleh, R.; Taha, R.Z.; Toor, S.M.; Sasidharan Nair, V.; Murshed, K.; Khawar, M.; Al-Dhaheri, M.; Petkar, M.A.; Abu Nada, M.; Elkord, E. Expression of immune checkpoints and T cell exhaustion markers in early and advanced stages of colorectal cancer. Cancer Immunol. Immunother. 2020, 69, 1989–1999. [Google Scholar] [CrossRef]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macri, E.; Fornasarig, M.; Boiocchi, M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Nosho, K.; Baba, Y.; Tanaka, N.; Shima, K.; Hayashi, M.; Meyerhardt, J.A.; Giovannucci, E.; Dranoff, G.; Fuchs, C.S.; Ogino, S. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: Cohort study and literature review. J. Pathol. 2010, 222, 350–366. [Google Scholar] [CrossRef] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, L.; Diao, T.; Chen, Y.; Cao, C.; Zhang, X. Analysis of immune-related signatures of colorectal cancer identifying two different immune phenotypes: Evidence for immune checkpoint inhibitor therapy. Oncol. Lett. 2020, 20, 517–524. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRC Patients | |
|---|---|
| Number | 50 |
| Age (range) | 59 † (18–83) |
| Gender (Male:Female) | 32:18 |
| TNM stage | |
| I | 4 |
| II | 21 |
| III | 16 |
| IV | 9 |
| Tumor histological grade | |
| G2 (Moderately differentiated) | 45 |
| G3 (Poorly differentiated) | 5 |
| dMMR/MSI-H | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toor, S.M.; Sasidharan Nair, V.; Murshed, K.; Abu Nada, M.; Elkord, E. Tumor-Infiltrating Lymphoid Cells in Colorectal Cancer Patients with Varying Disease Stages and Microsatellite Instability-High/Stable Tumors. Vaccines 2021, 9, 64. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9010064
Toor SM, Sasidharan Nair V, Murshed K, Abu Nada M, Elkord E. Tumor-Infiltrating Lymphoid Cells in Colorectal Cancer Patients with Varying Disease Stages and Microsatellite Instability-High/Stable Tumors. Vaccines. 2021; 9(1):64. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9010064
Chicago/Turabian StyleToor, Salman M., Varun Sasidharan Nair, Khaled Murshed, Mohamed Abu Nada, and Eyad Elkord. 2021. "Tumor-Infiltrating Lymphoid Cells in Colorectal Cancer Patients with Varying Disease Stages and Microsatellite Instability-High/Stable Tumors" Vaccines 9, no. 1: 64. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9010064